

1. Introduction
Progressive neurological diseases of the human central nervous system (CNS) are often characterized in part by the occurrence of insoluble, pro-inflammatory lesions whose major component is amyloid. These include the age-related CNS disorders Alzheimer’s disease (AD) and age-related macular degeneration (AMD), two insidious, incapacitating diseases that currently represent the most common progressive degenerations of the human brain and retina in the industrialized world [1–19]. The family of major insoluble, pro-inflammatory lipoprotein-enriched lesions that characterize AD and AMD - the amyloid/senile plaques of AD and the drusen of the aged retina of AMD - are highly enriched in beta amyloid precursor protein (βAPP)-derived amyloid beta (Aβ) peptides 42 amino acids in length (Aβ42) [11–23]. The ubiquity of Aβ42 peptides as major constituents of AD and AMD lesions not only suggests an important underlying commonality in disease pathology but implicates a complex and shared plasma membrane-mediated βAPP processing mechanism involving the actions of membrane-associated alpha, beta and gamma secretases (α-, β- and γ-secretases), accessory enzymes and membrane-structural proteins which are required for Aβ42 peptide generation [1–18]. Common ancillary components of the AD senile plaque and AMD drusen including complement proteins such as complement factor H (CFH), the βAPP-associated proteins sortilin (SORL1), TSPAN12 proteins, additional membrane-associated lipids and proteins and pro-inflammatory microRNAs (miRNAs) in the pathogenic brain neocortex and retina further suggest the shared participation of neuro-immune, amyloidogenic and pro-inflammatory pathways. While multiple classifications of amyloidosis exist, this ‘research perspective’ will focus on what is currently known about the contribution of βAPP–derived Aβ42-peptides to the formation of senile plaques and retinal drusen in sporadic AD and dry AMD, and how the progressive aggregation of neural- and retinal-toxic constituents suggests an underlying commonality in the pathological mechanisms for each disease. Importantly, our understanding of the molecular genetics of each of these progressive diseases may shed light on similar pathogenic mechanisms in the other, and successful pharmacological approaches to one disease may be useful in the clinical management of both of these age-related human disorders.
2. Historical – the drusen (amyloid ‘esenile’ plaques) of AD
The French neuropathologists Paul Oscar Blocq (1860–1896) and Gheorghe Marinesco (1863–1938) in 1892 first described the presence of ‘sclerotic plaques of neuroglia’, a name which Emil Redlich changed to ‘miliary sclerosis’ or ‘drusen’ in the aging human brain neocortex [1–9]. The term ‘amyloid’ originated from an early mistaken identification by the German neuroanatomist Rudolf Virchow (1821–1902) of a neural and CNS substance resembling starch (‘amylum’ in Latin) – since it has been subsequently acknowledged that amyloids are, in fact, deposits of ‘proteinaceous albumoid’ material [1–16]. The German psychiatrist/neuropathologist Aloysius ‘Alois’ Alzheimer (1864–1915) first made the connection between neocortical amyloid-containing drusen and senile dementia, and first described these ‘senile plaque’ deposits in the neocortex of a 51 year old psychiatric patient in his original, now classical paper entitled “Uber eine eigenartige Erkankung der Hirnrinde” (“A characteristic disease of the human cerebral cortex”). Here Alzheimer described these lesions as ‘senile drusen, containing amyloid’ [10]. The roughly spherical senile plaques of AD represent a very dense, insoluble aggregation of Aβ42 peptides and other amyloidogenic material in the extracellular space surrounding CNS neurons, and particularly in the neocortex of the brain, hence the classification of AD as a ‘dense deposit disease’ [8–11]. Aβ42 peptide deposition is primarily perineuronal and is shed into the extracellular space, however βAPP holoprotein may be located within internal neuronal membranes, so at least some Aβ42 peptide generation may be intra-neuronal [1–10,11]. A two amino acid shorter, less hydrophobic Aβ40 peptide deposition commonly generated along with Aβ42 peptide is primarily perivascular [10–14]. Interestingly, lack of connective tissue in the human neocortex places virtually no restriction on the size of neocortical amyloid-dense senile plaques which can reach in excess of 100 μm in diameter, although most average only one-third to one-half of that size [11–18]. The extreme compactness and insolubility of senile plaque deposits made these lesions extremely difficult to analyze until George Glenner (1928–1995) found a way to solubilize them by heating purified human senile plaque cores in 5 M guanidine-HCl/1 N acetic acid [5–8]. Glenner was subsequently able to characterize and sequence their high content of Aβ42 peptides, especially at the senile plaque core [7–10]. Once when asked at an Alzheimer meeting what was the concentration of Aβ42 peptide(s) at the senile plaque core Glenner answered without hesitation ‘at least 5 molar’ [authors personal note]. The term ‘senile drusen of AD’ has since been renamed ‘senile plaques’ in part to distinguish them from the drusen of AMD (see below). Mature AD senile plaque amyloid is now known to adopt complex cross-beta sheet quaternary structures at their cores and the gold test for the presence of AD amyloid is by (i) staining for birefringence using the secondary diazo dye Congo red [the sodium salt of 3,3′-([1,1′-biphenyl]-4,4′-diyl)bis(4-aminonaphthalene-1-sulfonic acid] which gives these lesions a ‘glittering geode-like appearance’ under light microscopy; or (ii) by positron emission tomography (PET) imaging using various amyloid-binding ligands [11–19]. AD senile plaque amyloids are thought to impact AD pathology by at least 4 mechanisms: (i) through progressive distortion of neocortical architecture both physically (structurally) and functionally; (ii) by promoting mitochondrial dysfunction with the resultant generation of reactive oxygen species (ROS) leading to the oxidation of neuronal components and apoptosis; (iii) by leading to the activation of microglial cells, the chief innate-immune ‘phagocytosis’ and ‘scavenging’ cells of the brain and CNS; and/or (iv) by activating microglial-mediated pathological pathways that have subsequent cellular and genetic pro-inflammatory consequences [1–15]. Note that in AD that all four of the pathological effects (i–iv) of senile plaques may occur concurrently, or come to the forefront at different stages as AD progresses from mild to more severe and terminal stages of the disease [16–19]. It is further noteworthy to point out that AD occurs in two major types, a genetic or familial early onset form, accounting for only about 5% of all AD cases, and a sporadic or idiopathic late-onset form of unknown origin, accounting for about 95% of all AD cases, in which environmental and genetic differences may act as AD risk factors. Importantly, Aβ42 peptide and senile plaque generation, accumulation and aggregation is characteristic of the neuropathology of both the familial and sporadic forms of AD [1–8].
3. Historical – the drusen of AMD
As for AD, AMD is divided into two major types: (i) an exudative or ‘wet’ form exhibiting choroidal neovascularization with subretinal exudation (accounting for about 5% of all AMD cases) and (ii) a non-exudative ‘dry’ form accompanied by the formation of retinal drusen (accounting for about 95% of all AMD cases) [15,16]. Drusen in the aging human retina were first described by the Dutch ophthalmologist Francis Donders (1818–1889) who initially referred to them as ‘colloid spheres’ (‘colloidkugeln’ in German) and subsequently by the German comparative anatomist Heinrich Muller (1820–1864) who renamed them, based on their ‘glittering’ spheroidal appearance with ‘drusen’, the German word for ‘geode’ (from the Greek ‘geodes, ‘earthlike’; essentially hollow masses consisting of a cryptocrystalline quartz shell lined internally by various cystalline minerals). In view of their anatomical location between the retinal pigment epithelium (RPE) and the choriocapillis (the vascular supply of the retina), retinal drusen, similarly to AD amyloid, deprive RPE, photoreceptor cells and the retinal ganglion of oxygen and nutrients, processes that lead to their atrophy and death. Interestingly, confocal microscopic analyses of middle-to-aged eyes indicate that drusen always seem to form non-randomly between two choroidal microvessels and immediately above the ‘pillars of choriocapillaris’, a layer of microcapillaries immediately adjacent to Bruch’s membrane in the choroid plexus [14–16]. Interestingly, and analogous to the amyloid senile plaques of AD, retinal drusen typically range in size from ~30-to-100 μm in diameter and are sandwiched between the connective tissue of Bruch’s membrane and the RPE cell layer which may restrict them from becoming any larger [14,15,36]. The source of the proteolipid or ‘albumoid’ material of the retinal drusen may have potential contributions from both the RPE cells and the choroid, the presence of amyloid, cholesterol, complement factor proteins and other inflammatory components of the retinal drusen suggests that these lesions may be products of the innate-immune system delivered at least in part from the systemic circulation. To this end patients with hyperlipidemia have increased incidence of retinal drusen and increased risk of developing AMD [15,17,18]. As is the case for AD senile plaque amyloids, drusen are thought to impact AMD pathology: (i) through progressive distortion of retinal architecture both structurally and functionally; (ii) by promoting mitochondrial dysfunction with the resultant generation of ROS leading to the oxidation of retinal cellular components and apoptosis; (iii) by leading to the activation of microglial cells, again the chief innate-immune ‘phagocytosis’ and ‘scavenging’ cells of the retina; and/or (iv) by activating microglial-mediated pathological pathways that have subsequent cellular and genetic pro-inflammatory and innate-immune consequences [1–16]. Again, as in AD, all four of the pathological effects (i–iv) triggered by retinal drusen may occur concurrently during the onset and propagation of the AMD process [14,15].
4. Characteristics and processing of the beta amyloid precursor protein (βAPP)
Because Aβ42 peptides common to senile plaque amyloid and retinal drusen are derived from βAPP holoprotein we may gain insight into the common pathological mechanisms shared by AD and AMD by reviewing what is known about the membrane-embedded βAPP holoprotein (Figure 1). Because of its central role in the amyloid cascade hypothesis for AD, βAPP is one of the most thoroughly studied CNS proteins in all of neurobiology [16–18]. βAPP, encoded at chr21q21.3, undergoes alternative splicing to yield eight possible isoforms, three of which (the 695, 751 and 770 amino acid isoforms) predominate in the brain and retina [1–6]. βAPP is an evolutionary ancient and highly conserved transmembrane receptor-type glycoprotein with homologous single-pass, integral membrane proteins in Drosophila melanogaster, the nematode Caenorhabditis elegans and most mammals including homo sapiens (Drosophila-Homo sapiens evolutionary divergence ~850 million years) [1–5,22–26]. Interestingly the species-specific physiological environment of βAPP has been suggested as being as critical as the primary peptide sequence to Aβ42 peptide formation and the generation of other βAPP-derived cleavage products [22,23]. Expressed in neurons throughout the human CNS and enriched in the synapse and neuronal membranes the ~770 amino acid βAPP consists of a large N-terminal ~600 amino acid extracellular domain containing heparin- and metal-binding sites, a short ~70 amino acid hydrophobic transmembrane domain, and a short ~100 amino acid C-terminal intracellular domain [22,23]. While the structure of the βAPP holoprotein has been fairly well characterized by multiple laboratories the function of βAPP has remained elusive. Currently there is evidence for at least 14 functions for βAPP, and these include βAPP as: (i) a structural regulator of synapse formation [24,25]; (ii) a facilitator of synaptic activity, dendritic spine density and inter-neuronal signaling [25,26]; (iii) a neuronal membrane- and synapse-associated factor in neural plasticity and memory [26,27]; a regulator of metal metabolism in the brain including copper and iron export [28,29]; (iv) a mediator of human hormonal regulation, including chorionic gonadotrophin and other reproductive hormones [30]; (v) a cellular adhesion and neuronal migration protein involved in neuronal architecture during development [31–34]; (vi) a key signaling protein involved in growth cone guidance [34–36]; (vii) a mediator of anterograde neuronal transport [37]; (viii) a neuronal regulator of oxidative stress [38]; (ix) a CNS trophic factor involved in neural stem cell development, neuronal survival, neurite outgrowth and neuro-repair [39]; (x) a cell surface growth factor (as the N-terminal domain of βAPP is similar in structure to cysteine-rich growth factors and contributes to neuronal growth and mobility) [39,40]; (xi) as a kinesin I membrane receptor that directs β-secretase (BACE1) and presenilin 1 (PS1) transport [41,42]; (xii) as a modulator of apoptosis-inducing pathways including iron- and copper-mediated neuronal death [28,29,43]; (xiii) as a signal transducer and regulator of transcription (the C-terminal intracellular domain appears to be involved in transcriptional regulation and βAPP can promote gene activation through binding to APBB1/Tip60 and/or the iron adaptor protein FE65 to transactivate a wide variety of CNS promoters) [35,38,44] and (xiv) as a membrane-resident holoprotein precursor to multiple peptide fragments each with neuronal modulatory and sensing activities that include neurotrophic and transcriptional effects [39–45]. Interestingly, βAPP holoprotein associates with a group of membrane-integral or membrane-peripheral proteins clustered in neuronal lipid raft domains [40,42,44]. These include tetraspanin-12 (TSPAN12), presenilin-1 (PS1) and other βAPP-associated secretases; for example TSPAN12 has been implicated in directing homeostatic βAPP processing and PS1 has been implicated in oxygen sensing and as a regulator of glial-specific gene expression [45–49]. Taken together, the functions of βAPP can be summarized as being either ‘neurotrophic’ and supportive of healthy neuronal function or as ‘amyloidogenic’ and pathological in both the brain and retina. The natural and pathological catabolism of βAPP and homeostatic phagocytic-clearance pathways for Aβ42 peptide in multiple, interactive brain- and retinal-relevant pathways is further summarized in Figure 1.
Figure 1. βAPP signaling via the neurotrophic, amyloidogenic or phagocytic (clearance) pathways.
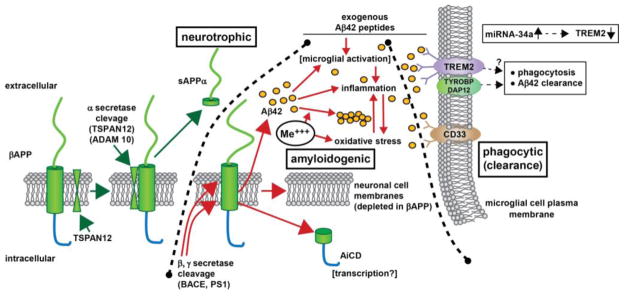
– beta amyloid precursor protein (βAPP) is a glycosylated integral trans-membrane protein of ~695–770 amino acids (~80 kDa MW) that is acted upon by a series of secretases to generate a range of neuroactive peptides. For example, α-secretase activity in association with tetraspanin (TSPAN12) proximity, distintegrin and metalloproteinase protein ADAM10, and other accessory proteins cleave βAPP to generate a ~621 amino acid neurotrophic sAPPα peptide via the neurotrophic signaling pathway. Interestingly sAPPα is very abundant in the vitreous of the eye and in healthy brain cells, particularly in the extracellular space (ECF) [Bhattacharjee, Zhao and Lukiw; unpublished observations]. Alternate processing of βAPP by tandem beta- and gamma secretase (β, γ secretase) cleavage (in the presence of ancillary membrane proteins such as presenilin 1 (PS1) and other proteins can result in the generation of 37–43 amino acid Aβ peptides, the most common of which are the peri-vascular Aβ40 and peri-neuronal Aβ42; the Aβ40 and especially Aβ42 peptides self-aggregate to form dense senile plaque and retinal drusen deposits (yellow ovals); the natural accumulation and aggregation of Aβ40 and especially Aβ42 peptides are strongly amyloidogenic; certain neurotoxic trivalent elements such as aluminum and iron (Me+++) and perhaps other genotoxic metals in our environment appear to stimulate this aggregation while also promoting oxidative stress, inflammation and other neurotoxic effects [84–90]. Importantly, Aβ42 peptides added exogenously and acutely may activate different cellular responses than the more chronic endogenous generation of these peptides [69,91–93,99,106]. Normally Aβ42 peptides are efficiently removed by a phagocytosis clearance system involving in part the triggering receptor for myeloid/microglial cells (TREM2), a variably glycosylated transmembrane sensor-receptor known to be enriched in the microglial cell plasma membrane. Signaling via the tyrosine kinase-binding protein (DNAX activation protein 12) [TYROBP (DAP12)] accessory receptor results in phagocytosis and ultimately, removal of Aβ42 peptides (yellow ovals) from the extracellular space [78–81]; interestingly, TREM2 knockout/knockdown mice have attenuated immunological and inflammatory responses and/or increases in age-related neuroinflammatory markers and cognitive deficiency [33,78,82]; TYROBP knockout mice exhibit immune system deficits and an impairment in microglial cell differentiation [68,78]; insufficient TREM2 may be in part responsible for the inability to adequately phagocytose Aβ42 peptides, resulting in their buildup and self-aggregation in the extracellular space. Aβ42 peptide sensing and clearance may also be accomplished in part through the microglial-enriched transmembrane CD33/Siglec protein and others (see text); interestingly, like TREM2, CD33/Siglec exhibits decreased expression in peripheral mononuclear cells in AD [99,100]. Upper right inset: miRNA-34a has been found to be significantly increased in AD hippocampal CA1, superior temporal lobe, in stressed microglial cells and in the retina of transgenic AD murine models overexpressing Aβ42 peptides [63,77; Bhattacharjee, Zhao, Lukiw, unpublished observations); miRNA-34a targeting of the TREM2 mRNA 3′-UTR and TREM2 down-regulation appears to be in part responsible for this defect [45,77–81,83,90,102,110]. Because miRNA-34a is encoded on an NF-kB-sensitive transcript, natural or synthetic anti-NF-kB and/or anti-miRNA strategies may be clinically useful in the restoration of homeostatic phagocytosis and the clearance of excessive Aβ42 peptides from the brain, CNS and retina in AD and AMD [39,74,75,111]. Black hatched lines between ‘neurotrophic’ and ‘amyloidogenic’ and ‘amyloidogenic’ and ‘phagocytic (clearance)’ are drawn to delineate specific βAPP cleavage product pathways, however, in brain and retinal physiology these pathways are most likely to be highly integrated to achieve homeostasis in the βAPP-sAPPα-Aβ42 trafficking system (see text).
5. Involvement of complement factor H (CFH) in AD and AMD
Complement factor H (CFH) is a key, soluble, internally repetitive glycoprotein inhibitor of the innate-immune response in the brain and retina, and mutations, deletions, duplications or rearrangements in the individual CFH gene are associated with a number of neuroimmune CNS diseases [50–55]. In one study of 2065 Scandinavian patients a CFH Y402H loss-of-function polymorphism appeared to increase the risk for AMD and predisposed patients for co-morbidity in AD [52], but this observation and analysis may not hold true for other human populations [53–55]. Low CFH abundance and deficits have been reported in sporadic AD neocortex and hippocampus and these correlate with up-regulated pro-inflammatory signaling in the AD brain [56,57]. Interestingly, loss of function in CFH as a result of the Y402H mutation may have the same end effect as an insufficiency in a functional CFH with the consequence driving innate-immune alterations and pro-inflammatory signaling. CFH deficits in sporadic AD and AMD may be driven by the significant up-regulation of the inducible, NF-kB-regulated pro-inflammatory microRNA-146a (miRNA-146a) in both the brain and retina [56,57].
6. Common epigenetic mechanisms in AD and AMD; microRNA
The human family of microRNAs (miRNAs) constitute a group of about 2200 ~22 nucleotide non-coding, single-stranded RNAs that recognize, via base-pair complementarity and hydrogen bonding, ribonucleotide sequences in the 3′ un-translated region (3′-UTR) of their target brain messenger RNAs (mRNAs) [56–62]. A major action of up-regulated miRNAs is to down-regulate the expression of their mRNA targets, and hence down-regulate expression of that target’s mRNA [56–62]. Interestingly, the human neocortex and retina share a specific family of about 25–40 high abundance miRNAs suggesting that a similar set of genes are regulated in the brain and retina by miRNA-mediated effects [61–64]. This small family of miRNAs are all inducible, NF-kB regulated and pro-inflammatory in nature and consist of the miRNA-9, miRNA-34a, miRNA-125b, miRNA-146a and miRNA-155; other miRNAs common to the brain and retina may also be involved in neuronal-based signaling pathways [56,57,62]. Interestingly, in the late stages of AD the entire primary visual cortex-thalamic-retinal circuit exhibits pro-inflammatory pathology suggesting the intriguing possibility that AD-relevant mechanisms can spread to the retina via soluble extracellular miRNA signaling [56,57,62]. In support of this idea is the fact that miRNAs are highly soluble and mobile, and the same pathogenic miRNAs found within neurons and astroglia are also found in the extracellular space and in the circulating cerebrospinal fluid (CSF) [60,62].
7. Transgenic murine models for AD (Tg-AD) exhibit accumulation of Aβ42 in both brain and retina
Transgenic murine models of AD (Tg-AD) have been useful in the analysis of the contribution of βAPP, Aβ42 peptides and the pro-inflammatory and altered innate-immune signaling mechanisms that characterize AD- and AMD-type neuropathology. For example, increases in the abundance of Aβ42 peptides, miRNA-146a and progressive decreases in the expression of complement factor H (CFH) in the aging retina of several different Tg-AD models and in the aging AD-affected brain indicate a surprisingly maintained inflammatory pathology across the antero-posterior-retinal cortical axis [46,63–69]. CFH, a glycoprotein and an important repressor protein known to contribute to the regulation of the brain’s innate-immune and inflammatory response, is found to be consistently down-regulated not only in Alzheimer neocortex but also across the entire visual pathway in late-stage Alzheimer’s disease (Table 1). In one study the 5xFAD Tg-AD model that overexpresses 5 mutated familial amyloid AD genes exhibited (i) the highest CNS-specific expression of amyloid; (ii) the greatest extent of synaptic pathology; (iii) the highest concentration of Aβ42 peptides; and (iv) the lowest levels of CFH in the brain and retina [63]. The fact that five pathogenic human βAPP genes drive AD-like pathology in the 5xFAD Tg-AD system may explain the greater brain and retinal pathology in this particular transgenic model when compared to other transgenic models used for progressive neurodegeneration studies [46,63,67–69].
Table 1.
Common structural and pathogenic components associated with AD senile plaques and AMD retinal drusen
| feature | AD | AMD | reference |
|---|---|---|---|
| location of lesions (early stage) | hippocampus, temporal lobe neocortex | retinal ganglion cells (RGC), photoreceptors | [1–16] |
| location of lesions (late stage) | hippocampus, temporal lobe neocortex, primary visual cortex | RGC, photoreceptors, subretina between Bruch’s membrane and the choroid | [1–16] |
| development of pathology | age-related | age-related | [1–20] |
| lesions - nature | dense, insoluble aggregates; misfolded proteins | dense, insoluble aggregates misfolded proteins | [15–19] |
| lesions - type | senile plaques and neurofibrillary tangles (NFT) | retinal drusen | [15–19] |
| lesions-classification | ‘dense deposit disease’ | ‘dense deposit disease’ | [16–18] |
| lesions - pathology | disruptive to brain structure and function; pro-inflammatory | disruptive to retinal structure and function; pro-inflammatory | [12–18] |
| shape of lesions | roughly spherical | oblong to roughly spherical | [8–12] |
| size of lesions | 20 to >100 um (diameter) | 20–100 um (diameter) | [8–15; unpublished] |
| major component of lesions | βAPP derived Aβ peptides; Aβ40, Aβ42 peptide | βAPP derived Aβ peptides; Aβ40, Aβ42 peptide | [23,26,39,51,91–95] |
| other components of lesions | complement proteins, CFH, C-reactive protein (CRP), apolipoprotein E (ApoE), angiogenic markers (VEGF), TREM2 | complement proteins, CFH, CRP, apolipoprotein E (ApoE), angiogenic markers (VEGF), TREM2 | [34–37,39, 91–95; unpublished] |
| lesion stain | stain with thioflavin S and Congo Red | stain with thioflavin T, not Congo Red | [16–19] |
| evidence of oxidative stress and ROS | surrounding senile plaques | surrounding retinal drusen | [38,86,87,95] |
| evidence of innate-immune deficits and inflammation | CFH and TREM2 down-regulation | CFH and TREM2 down-regulation | [46,54,55,62–64,74–83] |
| glial reactivity | activated microglia around senile plaques and NFT | activated microglia in subretinal space and around drusen | [46,48,98–103,111] |
| common complement factor involvement | complement factor H (CFH) | complement factor H (CFH) | [50–57] |
| common miRNA involvement | miRNA-9, miRNA-34a, miRNA-125b, miRNA-146a, miRNA-155, others | miRNA-9, miRNA-34a, miRNA-125b, miRNA-146a, miRNA-155, others | [62,107] |
| risk factors – lifestyle and environment | education, systolic blood pressure, total cholesterol level, diabetes mellitus, smoking status, apolipoprotein E genotype | education, systolic blood pressure, total cholesterol level, diabetes mellitus, smoking status, apolipoprotein E genotype | [69–71, 91–93] |
| genetic linkage - genes | CFH deficits in sporadic AD | CFH (Y402H) deficits in familial AMD | [50–55,62–68] |
| genetic linkage – population and linkage studies | Logistic regression; controlling for age, sex, race, education, systolic blood pressure, total cholesterol level, diabetes mellitus, smoking status, and ApoE genotype, persons with low levels of cognition were more likely to have AMD (OR of 2.00; 95% confidence interval, 1.29–3.10) | rate of mild cognitive impairment (MCI) was higher in AMD patients than in controls (52.4% vs 26.8%; p<0.001), with an odds ratio (OR) of 3.127 (95% confidence interval, 1.855–5.271) after adjustment for age, education, and visual acuity; | [62–72,107–112] |
8. Co-occurrence of cognitive impairment and AMD in human populations
Genetic- and population-based studies relating the prevalence of cognitive impairment and AMD (and vice versa) can be informative when linking the potential occurrence of a common disease mechanism. For example, one population-based study evaluated the association of cognitive function and dementia with early AMD in 2088 elderly individuals aged 69–97 years [69–71]. Using logistic regression and controlling for age, sex, race, education, systolic blood pressure, total cholesterol level, diabetes mellitus, smoking status, and ApoE genotype, persons with low levels of cognition were more likely to have AMD (OR of 2.00; 95% confidence interval, 1.29–3.10). This study concluded that especially in older populations, cognitive impairment may share common age-related pathogenesis and risk factors with early AMD [69–71]. In another complementary Korean study involving 360 AMD patients and controls it was found that the rate of mild cognitive impairment (MCI) was higher in AMD patients than in controls (52.4% vs 26.8%; p<0.001), with an odds ratio (OR) of 3.127 (95% confidence interval, 1.855–5.271) after adjustment for age, education, and visual acuity [71]. The results suggested that patients with AMD, especially those with the geographic atrophy subtype (i.e. degeneration of the deepest cells of the retina in the most advanced (late) forms of dry AMD), are at greater risk for cognitive impairment than are non-AMD control subjects [71]. While these studies need to be expanded, much recent, independently derived data support the contention that different human populations have variable risk for disease due to different genetic backgrounds, lifestyle and other environmental factors; however the emerging epidemiological links between cognitive impairment and AMD in these early studies are remarkable [68–75].
9. Other inflammatory markers associated with drusen and senile plaques
Table 1 shows common constituents of AD senile plaque and AMD drusen. Other common components include complement proteins such as complement factor H (CFH), vitronection, the βAPP-associated proteins nicastrin, sortilin, TSPAN proteins, additional membrane-associated lipids and membrane proteins involved in Aβ42 sensing, the innate-immune response and inflammatory signaling [56,57,60,62,67].
10. Environmental neurotoxins in AD and AMD – the possible involvement of aluminum
As explained below, it is tempting to speculate that the ubiquitous environmental neurotoxin aluminum may contribute to the neuropathology of both AD and AMD. Aβ42 peptide monomers appear to be homeostatically cleared by microglial cells in part through a TREM2-TYROBP(DAP12)-mediated phagocytic mechanism [75–83]. When Aβ42 peptide monomers become overabundant or assume higher order dimeric or oligomeric structures, microglial cells appear to become limited in their ability to phagocytose [34–37,39,82,83]. Hence, any factors that promote Aβ42 peptide aggregation might be expected to promote amyloidogenesis. Environmentally abundant neurotoxic metals such as aluminum (at ~8% of the earth’s crust, an average of 3.3 M, the most abundant neurotoxic metal in the biosphere) are capable of aggregating Aβ42 peptide monomers into higher order structures at physiologically realistic concentrations, and aluminum is found at significantly higher concentrations within senile plaque cores in the AD brain [84–86]. Interestingly, combinatorial chelation strategies targeted to remove trivalent metals such as aluminum and iron (3+) from the CNS have been shown to be effective in reducing aluminum in the brain, both in in vitro studies using human brain cells in primary culture [86] and in early clinical trials [87,88]. The metal abundance and aluminum content of AMD retinal drusen is currently not known. It is noteworthy that aluminum is also capable of inducing the NF-kB sensitive, pro-inflammatory miRNA-34a that in turn down-regulates the expression of TREM2, and impairs the ability of normal microglial cells to effectively phagocytose Aβ42 peptides, resulting in their aggregation [89,90].
11. Relationships between AD and AMD – roles for Aβ42 and sAPPα
A number of studies have demonstrated that Aβ42 peptides accumulate in the drusen of AMD patients and that this accumulation is associated with a functional reduction of vision [69,91–93]. Aβ42 accumulation is also seen in the retina of AD patients and in transgenic murine models of AD, and is currently being developed as an early AD biomarker [49,63,94]. Proteome analysis of drusen show accumulation of at least 36 proteins with considerable overlap between normal and AMD subjects suggesting that drusen formation in AMD may be an amplification or intensification of a naturally occurring process [95]. Interestingly, certain drusen from normally aging subjects (that slowly accumulate as a natural consequence of aging) do not exhibit extensive Aβ42 peptide abundance, suggesting that significant Aβ42 accumulation may be more characteristic of the pathology of AMD [3,96–99]. Animal models of AMD generated by high fat-cholesterol (HF-C) diets also accumulate amyloid and impaired retinal function can be partially mitigated by anti-amyloid therapies [95–97]. Deficits of Aβ42-generating BACE1 have been reported to result in another type of retinal degeneration involving vascular dysregulation and accumulation of age pigment [98]. Aβ42 is present in the vitreous and aqueous humors at significantly higher levels than in blood plasma suggesting that there are considerable amounts of this peptide generated within the eye itself [98,99]. Consistent with this finding, high levels of α-, β- and γ-secretase processing of βAPP is seen in both the retina and retinal pigment epithelial (RPE) cells [98,99]. In addition, Aβ42 levels are two-fold higher in the vitreous humor than in the aqueous humor consistent with the gradient of generation of Aβ42 in the retina and secretion into the vitreous humor followed by its clearance via Schlemm’s canal [99]. Levels of secreted βAPP cleavage products generated by α- and β-secretase activities are very low in the aqueous humor, but are extremely high in the vitreous humor, suggesting that βAPP precursor is present at high levels in this compartment and that, unlike Aβ42, there may be a strong physiological barrier for its transport into the aqueous humor.
Further evaluation of the secreted βAPP catabolites have shown that there are high amounts of sAPPα generated by the non-amyloidogenic pathway and that sAPPα was mostly generated from the neuronal 695 amino acid form of βAPP [39,99]. However, the function of such high levels of sAPPα in the vitreous humor remains unclear; there is evidence that sAPPα in the brain is neuritogenic and neurotrophic [16–19,22,23,49]. Interestingly, βAPP-knockout mice present a normal electroretinogram (ERG) pattern, suggesting that a virtual total loss of βAPP is well tolerated in this system (Prakasam and Sambamurti, unpublished observations). Further, high levels of Aβ42 peptides in the eye fluids and its efficient clearance suggest that multiple mechanisms of protein turnover or removal via microglial-cell mediated phagocytosis failure can induce its accumulation [45,77–83]. If the accumulating Aβ42 peptides become excessive and toxic, it could be directly damaging to the retina and could serve as a useful pathological marker for the failure of Aβ42 peptide clearance. Excessive Aβ42 peptides may induce neurodegeneration due to the co-accumulation of several potentially neurotoxic proteins that are cleared by common pathways. Some of these Aβ42 clearance pathways are initiated by several microglial-enriched Aβ42 recognition and sensor transmembrane glycoproteins including the triggering receptor expressed in myeloid/microglial cells (TREM2) and the cluster of differentiation protein/sialic acid-binding immunoglobulin-like lectin CD33/Siglec [43,76–83,100–104]. Deficits in TREM2 and CD33 leading to an impairment in Aβ42 peptide phagocytosis have been observed in both AD and AMD and as such have been proposed to contribute to amyloidogenesis, heightened inflammatory response, synaptic dysfunction and related aberrant cell membrane and innate-immune processes [49,77–79,100–104; Zhao, Bhattacharjee and Lukiw; unpublished observations]. Interestingly, a number of studies using transgenic βAPP mice have failed to detect widespread retinal toxicity of Aβ42 peptides (Prakasam and Sambamurti; unpublished observations), but vitreal injection studies lead to rapid retinal cell death - leaving open the question of endogenous versus direct and indirect, acute and chronic mechanisms of Aβ42 peptide toxicity [12,99–106].
12. Summary
It is clear that the senile plaque amyloids of AD and the retinal drusen of AMD share several basic compositional features with lesion cores significantly enriched in Aβ42 peptides and related amyloidogenic molecules. Whether the progressive deposition of Aβ42 peptides in AD and AMD is cause or effect is still somewhat open to speculation by many researchers. However, (i) gain-of-function gene mutations in the β- and γ-secretases that drive Aβ42 generation in familial AD; (ii) the progressive, pro-inflammatory nature of senile plaque and drusen formation; (iii) temporal studies on Aβ42 abundance, maturation, speciation and conformation; and (iv) the fact that synthetic Aβ42 peptides, when added to healthy control neurons instill a robust pro-inflammatory response, suggest that the buildup of Aβ42 peptides in the brain and retina is pathologically linked to each disease process [1,2,49,86–90]. The involvement of not only Aβ42 peptides, but also the complex enzymatic systems necessary to generate them suggests that the progressively deposited lesions of both AD amyloid and AMD drusen possess a common disease mechanism involving multiple membrane-associated secretases and the ancillary proteins required to make these secretases functional [3,4,107–110]. Indeed, the membrane proteolipid-mediated mechanisms of Aβ42 peptide generation from βAPP holoproteins are complex, involving multiple βAPP processing enzymes and ancillary lipid raft domain membrane proteins including TREM2 [3,4,45–47,76]. It is further informative to point out (i) that late stages of AD exhibit significant retinal pathway involvement including inflammatory neuropathology, altered innate-immune signaling and visual disturbances [82,110,111]; (ii) that Tg-AD mice engineered to overexpress Aβ42 peptide in the brain cortex also show significant and progressive Aβ42 peptide increases in their retina [63]; (iii) that deficiencies in CFH in both sporadic and genetic forms of AD and AMD are implicated in aberrant innate-immune and pro-inflammatory signaling in both AD and AMD [64–69]; (iv) that both the AD brain and the AMD retina exhibit highly selective increases in the same family of NF-kB-sensitive, inducible miRNAs including miRNA-9, miRNA-34a, miRNA-125b, miRNA-146a and miRNA-155, with known regulatory roles in pro-inflammatory signaling and the innate-immune response [56,57,62,63]; (v) that patients with low levels of cognition are at increased risk for AMD [52,69]; and (vi) that patients with AMD may be at increased risk for mild cognitive impairment (MCI), widely thought to be the clinical precursor to AD, although it is still not entirely clear if persons with AMD are at increased risk for co-occurring AD [52,68–71]. These fundamental links underscore multiple common mechanisms between Aβ42 peptide neurobiology in AD and AMD at the cellular, physiological, genetic, epigenetic and epidemiological levels. Lastly, a clearer understanding of the amyloidogenic, pro-inflammatory and altered innate-immune mechanisms of AD may have strong relevance to AMD retinal biology with the potential implementation of common pharmacological strategies directed toward the treatment and alleviation of each of these progressive, age-related diseases [62,94,95,109–112].
Acknowledgments
The work in this research perspective was presented in part at the Alzheimer Association International Conference 2013 (AAIC 2013) Annual Meeting held in Boston MA, USA and at the AAIC 2014 held in Copenhagen, Denmark. Sincere thanks are extended to Drs. PN Alexandrov, F Culicchia, C Eicken and C Hebel for short post-mortem interval (PMI) human brain tissues or extracts, miRNA array work and initial data interpretation, and to D Guillot and J Lockwood for expert technical assistance. Additional thanks are extended to the physicians and neuropathologists of Canada and the USA who have provided high quality, short postmortem interval human brain and retinal tissues for study. Additional human control and AD brain tissues were provided by the Memory Impairments and Neurological Disorders (MIND) Institute and the University of California, Irvine Alzheimer’s Disease Research Center (UCI-ADRC; NIA P50 AG16573). Research on miRNA in the Lukiw laboratory involving the innate-immune response in AD, amyloidogenesis and neuro-inflammation was supported through a COBRE III Pilot Project NIH/NIGMS Grant P30-GM103340, an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN) and NIH grants NEI EY006311, NIA AG18031 and NIA AG038834. Research on AD, Down’s Syndrome and amyloidosis in the Sambamurti laboratory are supported by the Alzheimer’s Association IIRG 10-173180 and NIH NIA AG046200.
References
- 1.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 2.Jellinger KA. Interaction between pathogenic proteins in neurodegenerative disorders. J Cell Mol Med. 2012;16:1166–83. doi: 10.1111/j.1582-4934.2011.01507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Sun GY, Eckert GP, Lee JC. Cellular membrane fluidity in amyloid precursor protein processing. Mol Neurobiol. 2014 Feb 20; doi: 10.1007/s12035-014-8652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukiw WJ. Alzheimer’s disease (AD) as a disorder of the plasma membrane. Front Physiol. 2013;4:24. doi: 10.3389/fphys.2013.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorey E, Chang N, Liu QY, Yang Z, Zhang W. Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci Bull. 2014;30:317–330. doi: 10.1007/s12264-013-1422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glenner GG, Henry JH, Fujihara S. Congophilic angiopathy in the pathogenesis of Alzheimer’s degeneration. Ann Pathol. 1981;1:120–9. [PubMed] [Google Scholar]
- 7.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;425:534–9. doi: 10.1016/j.bbrc.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 8.Glenner GG, Murphy MA. Amyloidosis of the nervous system. J Neurol Sci. 1989;94:1–28. doi: 10.1016/0022-510x(89)90214-1. [DOI] [PubMed] [Google Scholar]
- 9.Buda O, Arsene D, Ceausu M, Dermengiu D, Curca GC. Georges Marinesco and the early research in neuropathology. Neurology. 2009;72:88–91. doi: 10.1212/01.wnl.0000338626.93425.74. [DOI] [PubMed] [Google Scholar]
- 10.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–31. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong RA. Spatial correlations between beta-amyloid (Abeta) deposits and blood vessels in familial Alzheimer’s disease. Folia Neuropathol. 2008;46:241–248. [PubMed] [Google Scholar]
- 12.Furumoto S, Okamura N, Iwata R, Yanai K, Arai H, Kudo Y. Recent advances in the development of amyloid imaging agents. Curr Top Med Chem. 2007;7:1773–89. doi: 10.2174/156802607782507402. [DOI] [PubMed] [Google Scholar]
- 13.Mathis CA, Mason NS, Lopresti BJ, Klunk WE. Development of positron emission tomography β-amyloid plaque imaging agents. Semin Nucl Med. 2012;42:423–32. doi: 10.1053/j.semnuclmed.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lengyel I, Tufail A, Hosaini HA, Luthert P, Bird AC, Jeffery G. Association of drusen deposition with choroidal intercapillary pillars in the aging human eye..Association of drusen deposition with choroidal intercapillary pillars in the aging human eye. Invest Ophthalmol Vis Sci. 2004;45:2886–92. doi: 10.1167/iovs.03-1083. [DOI] [PubMed] [Google Scholar]
- 15.Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol. 2011;95:14–25. doi: 10.1016/j.pneurobio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:38338–8. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 17.Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 2013;587:2046–2054. doi: 10.1016/j.febslet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Sorrentino P, Iuliano A, Polverino A, Jacini F, Sorrentino G. The dark sides of amyloid in Alzheimer’s disease pathogenesis. FEBS Lett. 2014;588:641–652. doi: 10.1016/j.febslet.2013.12.038. [DOI] [PubMed] [Google Scholar]
- 19.Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog Retin Eye Res. 2011;30:217–238. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Huang EJ, Wu SH, Lai CH, Kuo CN, Wu PL, Chen CL, Chen CY, King YC, Wu PC. Prevalence and risk factors for age-related macular degeneration in the elderly Chinese population in south-western Taiwan: the Puzih eye study. Eye (Lond) 2014 Mar 14; doi: 10.1038/eye.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moon BG, Joe SG, Hwang JU, Kim HK, Choe J, Yoon YH. Prevalence and risk factors of early-stage age-related macular degeneration in patients examined at a health promotion center in Korea. J Korean Med Sci. 2012;27:537–541. doi: 10.3346/jkms.2012.27.5.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tharp WG, Sarkar IN. Origins of amyloid-β. BMC Genomics. 2013;14:290. doi: 10.1186/1471-2164-14-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Tang BL. The amyloid precursor protein and postnatal neurogenesis/ neuroregeneration. Biochemical and Biophysical Research Communications. 2006;341:1–5. doi: 10.1016/j.bbrc.2005.12.150. [DOI] [PubMed] [Google Scholar]
- 24.Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyan SH, Shih AY, Walsh JJ, Maruyama H, Sarsoza F, Ku L, Eggert S, Hof PR, Koo EH, Dickstein DL. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci. 2012;51:43–52. doi: 10.1016/j.mcn.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoe HS, Lee HK, Pak DT. The upside of APP at synapses. CNS Neurosci Ther. 2012;18:47–56. doi: 10.1111/j.1755-5949.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 28.Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem. 2003;278:17401–7. doi: 10.1074/jbc.M300629200. [DOI] [PubMed] [Google Scholar]
- 29.Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porayette P, Gallego MJ, Kaltcheva MM, Meethal SV, Atwood CS. Amyloid-beta precursor protein expression and modulation in human embryonic stem cells: a novel role for human chorionic gonadotropin. Biochem Biophys Res Commun. 2007;364:522–527. doi: 10.1016/j.bbrc.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Sheng B, Song B, Zheng Z, Zhou F, Lu G, Zhao N, Zhang X, Gong Y. Abnormal cleavage of APP impairs its functions in cell adhesion and migration. Neurosci Lett. 2009;450:327–331. doi: 10.1016/j.neulet.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Yang L, Zheng H. Role of APP and Aβ in synaptic physiology. CurrAlzheimer Res. 2012;9:217–226. doi: 10.2174/156720512799361691. [DOI] [PubMed] [Google Scholar]
- 33.Yin RH, Yu JT, Tan L. The role of SORL1 in Alzheimer’s disease. Mol Neurobiol. 2014 May 16; doi: 10.1007/s12035-014-8742-5. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 34.Allsop D, Ikeda S, Glenner GG. Evidence for the derivation of a peptide ligand from the amyloid beta-protein precursor of Alzheimer’s disease. Prog Clin BiolRes. 1989;317:893–902. [PubMed] [Google Scholar]
- 35.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 36.Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12:1–12. doi: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satpute-Krishnan P, Degiorgis JA, Conley MP, Jang M, Bearer EL. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proceedings of the National Academy of Sciences. 2006;103:16532. doi: 10.1073/pnas.0607527103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swomley AM, Förster S, Keeney JT, Triplett J, Zhang Z, Sultana R, Butterfield DA. Abeta, oxidative stress in Alzheimer disease: Evidence based on proteomics studies. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbadis.2013.09.015. pii: S0925-4439(13)00297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dawkins E, Small DH. Insights into the physiological function of the β-amyloid precursor protein: beyond Alzheimer’s disease. J Neurochem. 2014;129:756–769. doi: 10.1111/jnc.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW. Crystal structure of theN-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol. 1999;6:327–31. doi: 10.1038/7562. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein LS. Kinesin molecular motors: transport pathways, receptors, and human disease. Proc Natl Acad Sci U S A. 2001;98:6999–7003. doi: 10.1073/pnas.111145298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taru H, Suzuki T. Regulation of the physiological function and metabolism of AbetaPP by AbetaPP binding proteins. J Alzheimers Dis. 2009;18:253–65. doi: 10.3233/JAD-2009-1148. [DOI] [PubMed] [Google Scholar]
- 43.Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, osMJ Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Müller T, Schrötter A, Loosse C, Pfeiffer K, Theiss C, Kauth M, Meyer HE, Marcus K. A ternary complex consisting of AICD, FE65, and TIP60 down-regulates stathmin1. Biochim Biophys Acta. 2013;1834:387–94. doi: 10.1016/j.bbapap.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 45.Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem Pharmacol. 2013;88:495–8. doi: 10.1016/j.bcp.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li YY, Cui JG, Dua P, Pogue AI, Bhattacharjee S, Lukiw WJ. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci Lett. 2011;499:109–113. doi: 10.1016/j.neulet.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lukiw WJ. NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp Neurol. 2012;235:484–490. doi: 10.1016/j.expneurol.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui JG, Fraser PE, St George-Hyslop P, Westaway D, Lukiw WJ. Potential roles for presenilin–1 in oxygen sensing and in glial-specific gene expression. Neuroreport. 2004;15:2025–2028. doi: 10.1097/00001756-200409150-00006. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, Bazan NG. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS One. 2011;6:e15816. doi: 10.1371/journal.pone.0015816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs) Mol Immunol. 2013;56:170–180. doi: 10.1016/j.molimm.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014 Apr 16; doi: 10.1146/annurev-genom-090413-025610. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zetterberg M, Landgren S, Andersson ME, Palmér MS, Gustafson DR, Skoog I, Minthon L, Thelle DS, Wallin A, Bogdanovic N, Andreasen N, Blennow K, Zetterberg H. Association of complement factor H Y402H gene polymorphism with Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147:720–726. doi: 10.1002/ajmg.b.30668. [DOI] [PubMed] [Google Scholar]
- 53.Le Fur I, Laumet G, Richard F, Fievet N, Berr C, Rouaud O, Delcourt C, Amouyel P, Lambert JC. Association study of the CFH Y402H polymorphism with Alzheimer’s disease. Neurobiol Aging. 2010;31:165–166. doi: 10.1016/j.neurobiolaging.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Proitsi P, Lupton MK, Dudbridge F, Tsolaki M, Hamilton G, Daniilidou M, Pritchard M, Lord K, Martin BM, Johnson J, Craig D, Todd S, McGuinness B, Hollingworth P, Harold D, Kloszewska I, Soininen H, Mecocci P, Velas B, Gill M, Lawlor B, Rubinsztein DC, Brayne C, Passmore PA, Williams J, Lovestone S, Powell JF. Alzheimer’s disease and age-related macular degeneration have different genetic models for complement gene variation. Neurobiol Aging. 2012;33:1843.e9–17. doi: 10.1016/j.neurobiolaging.2011.12.036. [DOI] [PubMed] [Google Scholar]
- 55.Liu MM, Chan CC, Tuo J. Genetic mechanisms and age-related macular degeneration: common variants, rare variants, copy number variations, epigenetics, and mitochondrial genetics. Hum Genomics. 2012;6:13. doi: 10.1186/1479-7364-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lukiw WJ, Zhao Y, Cui JG. An NF-kB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283:31315–22. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lukiw WJ, Alexandrov PN. Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol Neurobiol. 2012;46:11–19. doi: 10.1007/s12035-012-8234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 59.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 60.Alexandrov PN, Dua P, Hill JM, Bhattacharjee S, Zhao Y, Lukiw WJ. microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF) Int J Biochem Mol Biol. 2012;3:365–373. [PMC free article] [PubMed] [Google Scholar]
- 61.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lukiw WJ, Surjyadipta B, Dua P, Alexandrov PN. Common micro RNAs (miRNAs) target complement factor H (CFH) regulation in Alzheimer’s disease (AD) and in age-related macular degeneration (AMD) Int J Biochem Mol Biol. 2012;3:105–116. [PMC free article] [PubMed] [Google Scholar]
- 63.Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. Neuroreport. 2011;22:623–627. doi: 10.1097/WNR.0b013e3283497334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quan YL, Zhou AY, Feng ZH. Association between complementary factor H Y402H polymorphisms and age-related macular degeneration in Chinese: Systematic review and meta-analysis. Int J Ophthalmol. 2012;5:242–246. doi: 10.3980/j.issn.2222-3959.2012.02.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kondo N, Bessho H, Honda S, Negi A. Complement factor H Y402H variant and risk of age-related macular degeneration in Asians: a systematic review and meta-analysis. Ophthalmology. 2011;118:339–344. doi: 10.1016/j.ophtha.2010.06.040. [DOI] [PubMed] [Google Scholar]
- 67.Ermilov VV, Tiurenkov IN, Nesterova AA, Zagrebin VA. Alzheimer’s disease and geriatric eye diseases in the aspect of amyloid genesis] Arkh Patol. 2013;75:37–42. [PubMed] [Google Scholar]
- 68.Waring SC, Rosenberg RN. Genome-wide association studies in Alzheimer disease. Arch Neurol. 2008;65:329–334. doi: 10.1001/archneur.65.3.329. [DOI] [PubMed] [Google Scholar]
- 69.Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baker ML, Wang JJ, Rogers S, Klein R, Kuller LH, Larsen EK, Wong TY. Early age-related macular degeneration, cognitive function, and dementia: the Cardiovascular Health Study. Arch Ophthalmol. 2009;127:667–73. doi: 10.1001/archophthalmol.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Woo SJ, Park KH, Ahn J, Choe JY, Jeong H, Han JW, Kim TH, Kim KW. Cognitive impairment in age-related macular degeneration and geographic atrophy. Ophthalmology. 2012;119:2094–101. doi: 10.1016/j.ophtha.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 72.Raj A, Rifkin SA, Andersen E, van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olson MV. Human genetic individuality. Annu Rev Genomics Hum Genet. 2012;13:1–27. doi: 10.1146/annurev-genom-090711-163825. [DOI] [PubMed] [Google Scholar]
- 74.Lukiw WJ. Antagonism of NF-κB-up-regulated micro RNAs (miRNAs) in sporadic Alzheimer’s disease (AD): anti-NF-κB vs. anti-miRNA strategies. Front Genet. 2013a;4:77–79. doi: 10.3389/fgene.2013.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lukiw WJ. Variability in micro RNA (miRNA) abundance, speciation and complexity amongst different human populations and potential relevance to Alzheimer’s disease (AD) Front Cell Neurosci. 2013b;7:133. doi: 10.3389/fncel.2013.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang T, Yu JT, Zhu XC, Tan MS, Gu LZ, Zhang YD, et al. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014;35:1243–1251. doi: 10.1016/j.neurobiolaging.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 77.Zhao Y, Lukiw WJ. TREM2 signaling, miRNA-34a and the extinction of phagocytosis. Front Cell Neurosci. 2013;7:131–135. doi: 10.3389/fncel.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8(1):e52982. doi: 10.1371/journal.pone.0052982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao Y, Bhattacharjee S, Jones BM, Dua P, Alexandrov PN, Hill JM, Lukiw WJ. Regulation of TREM2 expression by an NF-κB-sensitive miRNA-34a. Neuroreport. 2013;24:318–323. doi: 10.1097/WNR.0b013e32835fb6b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forabosco P, Ramasamy A, Trabzuni D, Walker R, Smith C, Bras J, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013 doi: 10.1016/j.neurobiolaging.2013.05.001. S0197-4580(13)00199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hickman SE, El Khoury J. TREM2 and the neuro-immunology of Alzheimer’s disease. Biochem Pharmacol. 2014;88:495–8. doi: 10.1016/j.bcp.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cui JG, Hill JM, Zhao Y, Lukiw WJ. Expression of inflammatory genes in the primary visual cortex of late-stage Alzheimer’s disease. Neuroreport. 2007;18:115–119. doi: 10.1097/WNR.0b013e32801198bc. [DOI] [PubMed] [Google Scholar]
- 83.Jones BM, Bhattacharjee S, Dua P, Hill JM, Zhao Y, Lukiw WJ. Regulating amyloidogenesis through the natural triggering receptor expressed inmyeloid/microglial cells 2 (TREM2) Front Cell Neurosci. 2014;8:94. doi: 10.3389/fncel.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Exley C. The aluminum-amyloid cascade hypothesis of Alzheimer’s disease. Subcell Biochem. 2005;38:225–234. doi: 10.1007/0-387-23226-5_11. [DOI] [PubMed] [Google Scholar]
- 85.Walton JR. Aluminum involvement in the progression of Alzheimer’s disease. J Alzheimers Dis. 2013;35:7–43. doi: 10.3233/JAD-121909. [DOI] [PubMed] [Google Scholar]
- 86.Kruck TP, Cui JG, Percy ME, Lukiw WJ. Molecular shuttle chelation: the use of ascorbate, desferrioxamine and Feralex-G in combination to remove nuclear bound aluminum. Cell Mol Neurobiol. 2004;24:443–459. doi: 10.1023/B:CEMN.0000022773.70722.b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 88.Percy ME, Kruck TP, Pogue AI, Lukiw WJ. Towards the prevention of potential aluminum toxic effects and an effective treatment for Alzheimer’s disease. J Inorg Biochem. 2011;105:1505–1512. doi: 10.1016/j.jinorgbio.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bhattacharjee S, Zhao Y, Hill JM, Percy ME, Lukiw WJ. Aluminum and its potential contribution to Alzheimer’s disease (AD) Front Aging Neurosci. 2014;6:62. doi: 10.3389/fnagi.2014.00062. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alexandrov PN, Zhao Y, Jones BM, Bhattacharjee S, Lukiw WJ. Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-induced miRNA-34a. J Inorg Biochem. 2013;128:267–269. doi: 10.1016/j.jinorgbio.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Invest. 2006;116:378–385. doi: 10.1172/JCI25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dentchev T, Milam AH, Lee VM, Trojanowski JQ, Dunaief JL. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol Vis. 2003;9:184–190. [PubMed] [Google Scholar]
- 93.Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–256. doi: 10.1016/j.exer.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 94.Frost S, Kanagasingam Y, Sohrabi H, Vignarajan J, Bourgeat P, Salvado O, Villemagne V, Rowe CC, Macaulay SL, Szoeke C, Ellis KA, Ames D, Masters CL, Rainey-Smith S, Martins RN, Group AR. Retinal vascular biomarkers for early detection and monitoring of Alzheimer’s disease. Transl Psychiatry. 2013;3:e233. doi: 10.1038/tp.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ding JD, Lin J, Mace BE, Herrmann R, Sullivan P, Bowes Rickman C. Targeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an age-related macular degeneration mouse model. Vision Res. 2008;48:339–345. doi: 10.1016/j.visres.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ding JD, Johnson LV, Herrmann R, Farsiu S, Smith SG, Groelle M, Mace BE, Sullivan P, Jamison JA, Kelly U, Harrabi O, Bollini SS, Dilley J, Kobayashi D, Kuang B, Li W, Pons J, Lin JC, Bowes Rickman C. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci USA. 2011;108:E279–287. doi: 10.1073/pnas.1100901108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cai J, Qi X, Kociok N, Skosyrski S, Emilio A, Ruan Q, Han S, Liu L, Chen Z, Bowes Rickman C, Golde T, Grant MB, Saftig P, Serneels L, de Strooper B, Joussen AM, Boulton ME. beta-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med. 2012;4:980–991. doi: 10.1002/emmm.201101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Prakasam A, Muthuswamy A, Ablonczy Z, Greig NH, Fauq A, Rao KJ, Pappolla MA, Sambamurti K. Differential accumulation of secreted AbetaPP metabolites in ocular fluids. J Alzheimers Dis. 2010;20:1243–1253. doi: 10.3233/JAD-2010-100210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Neumann H, Daly MJ. Variant TREM2 as risk factor for Alzheimer’s disease. N Engl J Med. 2013;368:182–184. doi: 10.1056/NEJMe1213157. [DOI] [PubMed] [Google Scholar]
- 101.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 102.Jones BM, Bhattacharjee S, Dua P, Hill JM, Zhao Y, Lukiw WJ. Regulating amyloidogenesis through the natural triggering receptor expressed in myeloid/microglial cells 2 (TREM2) Front Cell Neurosci. 2014;8:94. doi: 10.3389/fncel.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu N, Tan MS, Sun L, Jiang T, Wang YL, Tan L, Zhang W, Yu JT, Tan L. Decreased expression of CD33 in peripheral mononuclear cells of Alzheimer’s disease patients. Neurosci Lett. 2014;563:51–54. doi: 10.1016/j.neulet.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 105.Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefes Arch Clin Exp Ophthalmol. 2009;247:1213–1221. doi: 10.1007/s00417-009-1060-3. [DOI] [PubMed] [Google Scholar]
- 106.Aruoma OI, Jen SS, Watts HR, George J, Gentleman SM, Anderson PJ, Jen LS. Acute and chronic effects of intravitreally injected beta-amyloid on the neurotransmitter system in the retina. Toxicology. 2009;256:92–100. doi: 10.1016/j.tox.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sivak JM. The aging eye: common degenerative mechanisms between the Alzheimer’s brain and retinal disease. Invest Ophthalmol Vis Sci. 2013;54:871–880. doi: 10.1167/iovs.12-10827. [DOI] [PubMed] [Google Scholar]
- 108.Chiu K, Chan TF, Wu A, Leung IY, So KF, Chang RC. Neurodegeneration of the retina in mouse models of Alzheimer’s disease: what can we learn from the retina? Age (Dordr) 2012;34:633–649. doi: 10.1007/s11357-011-9260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog Retin Eye Res. 2011;30:217–38. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 110.Jiang T, Yu JT, Zhu XC, Tan L. TREM2 in Alzheimer’s disease. Mol Neurobiol. 2013;48:180–185. doi: 10.1007/s12035-013-8424-8. [DOI] [PubMed] [Google Scholar]
- 111.Lukiw WJ. Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer’s disease (AD) Expert Opin Emerg Drugs. 2012 Mar 23; doi: 10.1517/14728214.2012.672559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Logue MW, Schu M, Vardarajan BN, Farrell J, Lunetta KL, Jun G, Baldwin CT, Deangelis MM, Farrer LA. A search for age-related macular degeneration risk variants in Alzheimer disease genes and pathways. Neurobiol Aging. 2014;35:1510.e7–18. doi: 10.1016/j.neurobiolaging.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]