

Abstract
Expression of ErbB2 protein is inversely correlated with the prognosis in cancer patients. Consequently, strategies targeting ErbB2 remain an attractive option in treating several types of malignancies, including oral cancer. In addition, many studies have shown that emodin and emodin derivatives are able to inhibit growth of ErbB2-overexpressing tumor cells. In this study, a series of computer modeling-generated emodin analogues were synthesized and tested for their antiproliferative activity against oral cancer cell lines overexpressing ErbB2. Among these analogues, em08red (1,8-dihydroxy-9(10H)-anthracenone) demonstrated potent antiproliferative activity against all three tested ErbB2-overexpressing cell lines, ie, FaDu, HSC3, and OECM1. Treatment with em08red significantly downregulated activation of ErbB2 as well as the ErbB2 protein expression level in the tested cell lines and induced G2 arrest. Antiapoptosis protein (Bcl-xl and Bcl-2) expression levels were also downregulated, and active caspase-3 and caspase-9 was detected in cells after treatment with em08red. Moreover, treatment with em08red stimulated production of cytotoxic reactive oxygen species in treated cells, and this could be partially reversed by pretreatment with N-acetylcysteine. Overall, we demonstrated inhibition of ErbB2 function and induction of reactive oxygen species in tumor cells by em08red, which prevented proliferation of tumor cells and induced apoptotic cell death.
Keywords: ErbB2, emodin, em08red, reactive oxygen species, structure-activity relationship
Introduction
Oral cancer, the sixth most common cancer worldwide, is becoming a serious issue in global cancer epidemiology, especially in South and Southeast Asia and in Pacific regions.1 Most prevalent malignancies affecting the oral cavity are squamous cell carcinomas, and the incidence of oral cancer is higher in men than in women in many countries.2 Heavy smoking, alcohol abuse, and chewing of betel nut contribute to the high occurrence of oral cancer in most countries. Among the risk factors, chewing of betel nut is a widespread habit among adult men aged 25–55 years in Taiwan, and is the major element contributing to the oral cancer incidence in men of 29.2 per 100,000.3–6 In clinic, surgical removal or radiotherapy alone is frequently used to treat early (stage I and II) oral cancer, but surgery combined with radiotherapy and follow-up chemotherapy are often required for advanced (stage III and IV) oral cancers.7 However, the overall treatment of oral cancers is unsatisfactory and the 5-year survival rate for advanced oral cancers under multimodal therapy is as low as ~12%.8 The high incidence and mortality associated with oral cancers has a heavy social and economic impact, and these cancers have become a major health issue for the government and health care agencies in Taiwan. Further, due to the high prevalence of oral cancers that become chemoresistant and radioresistant, new effective antitumor agents are urgently required.
Aggregation of genomic alterations leads to aberrant activation of oncogenes and/or inactivation of tumor suppressors, contributing to the development of cancer. Recently, ErbB2 gene amplification and elevated ErbB2 function was observed in clinical oral cancer specimens or patient serum,9–11 and increased ErbB2 expression is strongly correlated with the progression of cancer and a poor prognosis.12 The phosphorylation level of ErbB2 also serves as a poor prognostic indicator of survival in patients with cancer and is associated with resistance to gefitinib in cancer cells.13,14 Therefore, suppression of ErbB2 signaling has become an attractive therapeutic target when treating cancer patients with overactivated ErbB2, and many ErbB2-targeted drugs have been used in the clinic or are under active development.
Emodin, a well known anthraquinone and abundant in many foods and medicinal herbs, has been reported to have a variety of biological functions, including laxative, antimicrobial, redox regulatory, and antitumor properties.15 Several studies have shown that emodin enhances the therapeutic effects of several chemotherapeutic agents, including cisplatin, doxorubicin, etoposide, and paclitaxel, in ErbB2-overexpressing cancer cells.16 Moreover, an emodin azide methyl anthraquinone derivative has been shown to disturb the binding between ErbB2 and heat shock protein 90, resulting in proteasomal degradation of ErbB2;17 however, the detailed molecular mechanisms are still unknown. With computer modeling, a series of emodin analogues capable of occupying the ErbB2-ATP binding pocket were synthesized and evaluated for their antiproliferative efficacy against several oral cancer cell lines. A small molecule, em08red (1,8-dihydroxy-9(10H)-anthracenone), showed a low IC50 value (<10 μM) in three ErbB2-overexpressing oral cancer cell lines and dramatically decreased ErbB2 protein levels. Treatment with Em08red impeded cell cycle progression at G2 phase, with downregulated expression of Cdk1 and cyclin B. Moreover, signatures of apoptosis, including the sub-G1 population, phosphatidylserine exposure, caspase activation, and loss of antiapoptosis molecules (Bcl-xl and Bcl-2), were also observed after treatment with em08red. Further experiments also demonstrated that treatment with em08red induced reactive oxygen species (ROS), resulting in cell death. Overall, em08red, an emodin analogue, represents a novel ErbB2-targeting drug lead. Further more potent compounds of the em08red series for ErbB2-overexpressing oral cancers are under active investigation.
Materials and methods
Cell culture
Human tongue carcinoma (HSC3), human hypopharyngeal carcinoma (FaDu), and human oral squamous carcinoma (OECM1) cell lines were purchased from the Bioresource Collection and Research Center (Hsinchu, Taiwan). HSC3 and FaDu cells were maintained in minimum essential medium containing 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, and 1 mM sodium pyruvate, and OECM1 cells were cultured in Roswell Park Memorial Institute-1640 medium with the same supplements as mentioned above. All cells were cultured in a 5% CO2 humidified incubator set at 37°C.
Antibodies and chemicals
Antibodies against Bcl-2, Bak, Bcl-xl, and phosphor-MPM2 were obtained from Millipore (Billerica, MA, USA). Caspase antibodies and antibodies against phosphor-ErbB2-Y1221/1222, phosphor-ErbB2-Y1248, and total ErbB2 were purchased from Cell Signaling Technology (Beverly, MA, USA). Actin antibody was acquired from Santa Cruz (Dallas, TX, USA). All chemicals used were of molecular or analytical grade and were purchased from Sigma-Aldrich (St Louis, MO, USA) or USB Corporation (Santa Clara, CA, USA).
General procedure for preparation of emodin analogues
Anhydrous AlCl3 and prebaked NaCl were mixed in a 2:1 molar ratio and heated to 110°C until molten. After phthalic anhydride/substituted phenol mixtures were gradually added to the AlCl3/NaCl melt, the reaction temperature was gradually increased to and maintained at 160°C for 5 hours and then cooled to 0°C in an ice bath. Next, 20 mL of 10% HCl was added, stirred at 0°C for 15 minutes, and then refluxed at 100°C for 30 minutes. The reaction mixture was cooled to room temperature and extracted with ethyl acetate. The final product was purified by silica gel column chromatography (EtOAc/n-Hex =1:1).
Determination of antiproliferative efficacy
A modified MTT (3-(4,5-cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) method was used to determine the efficacy of antitumor activity, and the IC50 value was calculated.18 Briefly, tumor cells (3,000 cells/well) were seeded into 96-well plates and then treated with vehicle or various concentrations of the test compounds for 72 hours. Two hours before the end of incubation, MTT at a final concentration of 5 μg/mL was added. Afterwards, solubilization buffer (40% dimethylformamide and 20% sodium dodecyl sulfate in H2O) was added to the wells to dissolve the violet formazan precipitation overnight at 37°C. Absorbance at 570 nm was then detected by a microplate reader (Molecular Devices, Sunnyvale, CA, USA) and the IC50 value was calculated by linear regression analysis.
Cell cycle analysis
Em08red-treated and control cells were trypsinized, collected, and fixed with 70% ice-cold ethanol in phosphate-buffered saline. After overnight incubation at −20°C, the cells were collected, permeabilized with 0.1% Triton X-100 in phosphate-buffered saline for 15 minutes at room temperature, and then stained with propidium iodide 40 μg/mL and RNase A (0.1 mg/mL) in phosphate-buffered saline for 30 minutes at 37°C. After the free propidium iodide was removed by centrifugation, the samples were analyzed immediately on a Canto II flow cytometer (BD Biosciences, San Jose, CA, USA) and the proportion of each cell cycle phase was analyzed by FCS express software (De Novo, Los Angeles, CA, USA).
Apoptotic cell death detection
An annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (Enzo, Farmingdale, NY, USA) was used to monitor apoptotic cells according to the manufacturer’s manual. Briefly, samples were collected and resuspended in staining buffer containing 1 mg/mL propidium iodide and 0.025 mg/mL annexin V-FITC at room temperature for 15 minutes in the dark. The samples were then immediately analyzed on a Canto II flow cytometer and the proportion of each population was calculated using FCS express software. The annexin V-positive and annexin V/propidium iodide double-positive populations were considered as apoptotic cells.
Caspase activity measurement
Activation of caspase-3, caspase-8, and caspase-9 in living cells was evaluated by the CaspGLOW™ fluorescein active caspase staining kit (BioVision, Milpitas, CA, USA) according to the manufacturer’s protocols. Briefly, treated cells were incubated with medium containing caspase fluorescent substrates for 2 hours at 37°C. After trypsinization and centrifugation, samples were resuspended in wash buffer and analyzed on a Canto II cytometer. The acquired data were then analyzed using the FCS express software.
Intracellular ROS level measurement
To determine the amount of intracellular H2O2, the treated cells were incubated with 20 μM of 2′,7′-dichlorodihydrofluorescein diacetate for 30 minutes at 37°C prior to analysis. Cells were then trypsinized, resuspended in phosphate-buffered saline, and analyzed on a Canto II cytometer. The acquired data were then analyzed by the FCS express software.
Western blot analysis
Cells were collected and lysed with cell lysis buffer (150 mM NaCl, 20 mM Tris-Cl pH 8.0, 0.5% NP-40, 1 mM phenyl-methanesulfonyl fluoride, 1 mM NaF, 1 mM Na3VO4, and 20 μg/mL aprotinin in distilled H2O) and the total protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Scientific, Swedesboro, NJ, USA). Equal amounts of total protein (30–40 μg) were resolved in 10%–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels and then transferred to polyvinylidene difluoride membranes. After blocking with 5% bovine serum albumin/TBS-Tween for one hour at room temperature, the membrane was incubated with primary antibodies overnight at 4°C. Subsequently, horseradish peroxidase-conjugated secondary antibodies were reacted with the membrane for one hour at room temperature. After washing, signals were acquired on a LAS-4000 biomolecular imager (Fujifilm, Tokyo, Japan).
Statistical analysis
All experiments were carried out at least three times independently and the results are presented as the mean ± standard deviation. The Student’s t-test was used to determine the significance of the differences. A P-value less than 0.05 was considered to be statistically significant.
Results
Synthesis of emodin analogues
Anthraquinones 1–9 was synthesized by reaction of selected phthalic anhydrides with substituted phenols in the presence of AlCl3/NaCl (Figure 1A). Compound 7 and emodin were then treated with SnCl2 in the presence of HCl to generate compounds 10 and 11, respectively (Figure 1B).
Figure 1.

Synthesis of anthraquinone derivatives.
Notes: (A) (i) AlCl3/NaCl, 165°C; (ii)10% HCl; (B) (i) SnCl2, HCl, HOAc/reflux.
Antiproliferative activity of emodin analogues
To identify emodin analogues with potent antiproliferative activity against oral cancers, eleven compounds were synthesized and purified, and their structures were determined. The IC50 value for each compound was determined in three ErbB2-overexpressing oral cancer cell lines (FaDu, HSC3, and OECM1 cells) using the MTT assay. Compounds with an IC50 value over 50 μM were considered not to be cytotoxic towards oral cancer cells. As shown in Table 1, compounds with no hydroxyl substitution (compound 1) or with three hydroxyl substitutions (compound 9) displayed very weak antiproliferative activity. Compounds 5 and 7 had an IC50 value of ~15 μM in FaDu cells, but both compounds showed very weak cytotoxicity towards the other two tested cell lines. However, compounds with two hydroxyl substitutions (compounds 4, 6, and 8) showed very little antiproliferative activity against oral cancers. Compound 7 demonstrated the most potent cytotoxicity of all the compounds synthesized and was chosen for further reduction to generate compound 10 (em08red). The antiproliferative assay results for em08red showed potent antiproliferative activity towards all three oral cancer cell lines, with an IC50 value below 10 μM. Therefore, this compound was chosen for further study.
Table 1.
IC50 value (μM) of emodin analogues in three oral cancer cell lines
| Compound | FaDu | HSC3 | OECM1 |
|---|---|---|---|
| 1 | 30.0±14.7 | >50 | >50 |
| 2 | 39.5±7.3 | >50 | >50 |
| 3 | >50 | >50 | >50 |
| 4 | >50 | >50 | >50 |
| 5 | 14.7±7.3 | 48.4±5.9 | 47.6±4.9 |
| 6 | 38.2±11.1 | >50 | >50 |
| 7 | 14.9±7.3 | 43.1±6.6 | 45.5±2.2 |
| 8 | >50 | >50 | >50 |
| 9 | 40.9±18.1 | >50 | >50 |
| 10 (em08red) | 8.7±3.2 | 9.8±5.7 | 9.8±2.1 |
| 11 | >50 | >50 | >50 |
| Emodin | 35.8±4.7 | 46.0±0.3 | >50 |
Treatment with em08red decreased ErbB2 protein levels
Overactivation of the ErbB2 signaling pathway has a critical role in the development of oral cancer and survival. Previous reports indicate that emodin may serve as an adjuvant treatment for ErbB2-overexpressing cancer cells.9,19 Therefore, all compounds with antiproliferative activity were tested for their influence on ErbB2 activity. Phosphor-ErbB2 protein levels and total-ErbB2 expression were determined by Western blot analysis (data not shown). Among the analogues synthesized, only em08red showed significant inhibition of ErbB2 activity. As shown in Figure 2A and B, after 36 hours of treatment, ErbB2 expression levels were dramatically reduced and the level of ErbB2 phosphorylation at tyrosine 1221/1222 and 1248, which both account for activation of ErbB2 receptors,20 was below the level of detection in FaDu and HSC3 cells. The downregulatory effect of em08red on ErbB2 protein was also observed in the treated OECM1 cells (data not shown). Our results demonstrated effective inhibition of ErbB2 activity by em08red, warranting further investigation.
Figure 2.
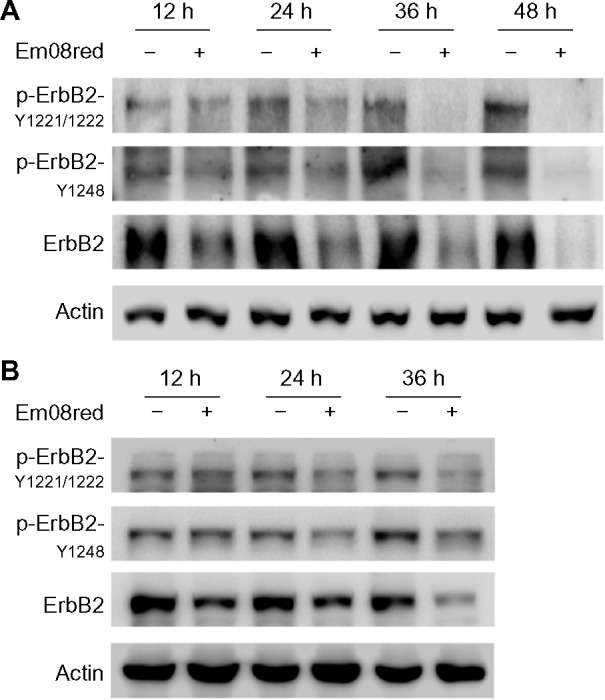
Treatment with em08red downregulates phosphorylation and expression of ErbB2.
Notes: Treated FaDu (A) and HSC3 (B) cells were harvested at the indicated times and total cell lysates were subjected to Western blot analysis. Antibodies specifically against total ErbB2 and phosphorylation sites of ErbB2 at tyrosine 1221/1222 or tyrosine 1248 were applied to detect protein expression after treatment. Actin expression served as the loading control.
Em08red induced G2/M phase arrest and intrinsic apoptosis
A previous study showed that downregulation of ErbB2 protein expression by treatment with neu differential factor could induce arrest of tumor cells in G2/M phase in cells overexpressing ErbB2.21 To determine if em08red has a similar pharmacological effect on cell cycle progression, em08red-treated cells were subjected to flow cytometry analysis. The data indicated significant G2/M accumulation after 24 hours of treatment with em08red in both FaDu and HSC3 cell lines (Figure 3A). G2/M phase-associated proteins, Cdk1 and cyclin B,22 were downregulated after treatment with em08red and phosphor-MPM2 signals were also reduced after em08red, indicating that the treated cells were blocked at G2 phase (Figure 3B). Additionally, we observed a population of sub-G1 em08red-treated HSC3 and FaDu cells after 24 and 48 hours of treatment, respectively, indicative of G2 phase arrest and cell death (Figure 3A). To determine if the molecular mechanisms of em08red-induced cell death occurred via apoptosis or necrosis, the phosphatidylserine level (a marker of apoptosis) on the outer membrane of em08red-treated cells was examined by annexin V staining. As shown in Figure 4A, the percentage annexin V-positive population in em08red-treated FaDu and HSC3 cells increased in a time-dependent manner and correlated with our observations on sub-G1 cell cycle analysis.
Figure 3.

Treatment with em08red induced G2 phase cell cycle arrest.
Notes: (A) FaDu (left panel) and HSC3 (right panel) cells were treated with vehicle (0.1% dimethyl sulfoxide) or em08red (10 μM) for 24, 48, and 72 hours and then harvested for fixation. After staining with propidium iodide, cell cycle analysis was carried out using a Canto II cytometer. The data are representative of three independent experiments. (B) Treated FaDu cells were harvested at 12, 24, 36, and 48 hours, and total cell lysates were subjected to Western blot analysis. Antibodies specific for phosphor-Cdk1, total Cdk1, cyclin B, and phosphor-MPM2 were used to detect expression of each protein. Actin expression served as the loading control.
Figure 4.

Treatment with em08red triggered the intrinsic apoptotic pathway and suppressed the antiapoptosis molecules.
Notes: (A) Cells were treated for 24, 48, and 72 hours and then harvested for detection of the annexin V-positive population by flow cytometry. (B) FaDu cells were treated with em08red (10 μM) for 24 hours, and intracellular caspase-3, caspase-8, and caspase-9 activity was detected using a CaspGLOW™ fluorescein active caspase staining kit. Control cells were incubated with vehicle only (0.1% dimethyl sulfoxide). The data are presented as the fold increase in treated samples and the asterisk indicates a significant difference between the treated group and the control group (P<0.05). (C) Treated FaDu cells were harvested and lyzed at the indicated times. Equal amounts of total cell lysates were subjected to Western blot analysis. Antibodies specific for cleaved caspase-3, caspase-8, or caspase-9 were used in the experiment. Actin expression served as the loading control. (D) Total lysates extracted from treated FaDu cells were resolved by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis. After transferring to the membranes, antibodies specific for Bcl-2, Bcl-x, Bak, and actin were reacted with the indicated membranes. Actin expression served as the loading control. All Western blotting images were acquired on an LAS-4000 biomolecular imager.
Activation of caspase-8 and caspase-9 is a hallmark of extrinsic and intrinsic apoptotic cascade pathways, respectively.23 To identify which apoptotic signaling is involved, caspase activation in em08red-treated FaDu cells was monitored using caspase-specific fluorogenic substrates and Western blots. As shown in Figure 4B, both caspase-3 and caspase-9 were activated, whereas no activation of caspase-8 was detected. Matching Western blot analysis results also demonstrated that em08red could induce cleavage and activation of caspase-3 and caspase-9 but not caspase-8 (Figure 4C). Moreover, Bcl-2 and Bcl-x, which are antiapoptosis molecules involved in the intrinsic apoptosis pathway, were decreased in the oral cancer cell line after treatment with em08red (Figure 4D). Collectively, our observations indicate that treatment with em08red induced cell cycle arrest at G2 phase and subsequently triggered the intrinsic apoptosis pathway in ErbB2-overexpressing oral cancer cells.
Em08red caused cell death via production of ROS
Many anthraquinones have been reported to cause ROS-mediated apoptosis,24,25 and it has also been shown that perturbation of ErbB2 activity in non-small-cell lung cancers can contribute to elevation of ROS and cell death.26 Hence, whether ROS production is also involved in em08red-mediated cytotoxicity was investigated to clarify the mechanism of action further. As shown in Figure 5A, treatment with em08red significantly increased the intracellular ROS level. To determine whether em08red-mediated cytotoxicity is ROS-dependent, cells were pretreated with N-acetylcysteine, a free radical scavenging agent, for 2 hours followed by treatment with em08red for 24 hours. Our results showed that pretreatment with N-acetylcysteine could partially inhibit the cytotoxic effects of em08red (Figure 5B), suggesting that production of ROS accounts for a portion of em08red-induced cell apoptosis.
Figure 5.
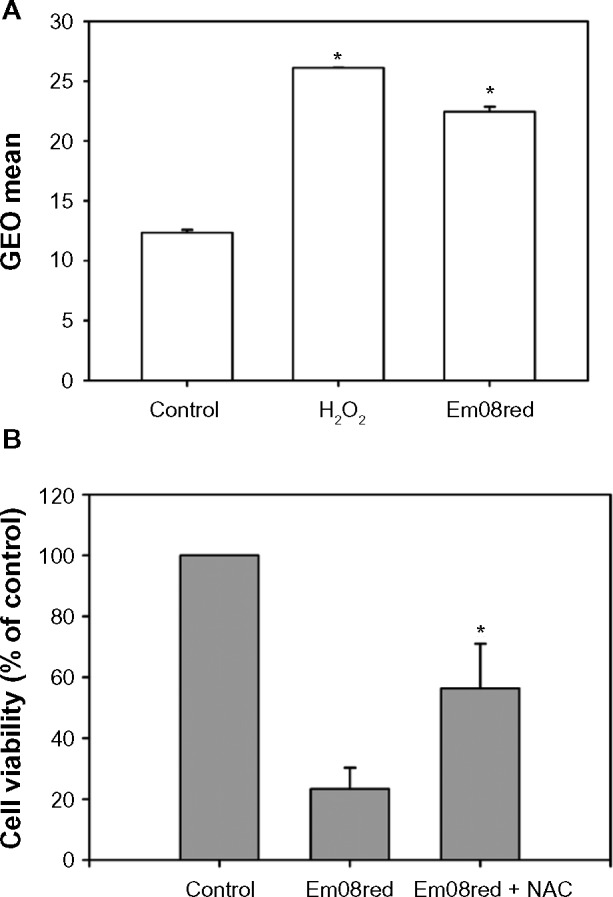
Intracellular H2O2 production accounted for em08red-mediated cytotoxicity.
Notes: (A) FaDu cells were treated with em08red (10 μM) or H2O2 (500 μM) for 24 and 16 hours, respectively, and 2′,7′-dichlorodihydrofluorescein diacetate staining was used to detect the intracellular H2O2 level. The data are presented as the geometric mean for each group and the asterisk (*) indicates a significant difference between the treated group and the control group (P<0.05). (B) The cotreatment group was preincubated with N-acetylcysteine (2 mM) for 2 hours, and em08red (10 μM) was then added to each treatment group for a further 24 hours of treatment. The control group was treated with 0.1% dimethyl sulfoxide only. The MTT assay was used to determine relative cell viability and the results are presented as the mean ± standard deviation. The asterisk (*) indicates a significant difference between the emo8red-treated group and the cotreatment group (P<0.05).
Em08red induced proteasomal degradation of ErbB2 protein
To clarify further whether em08red-induced ErbB2 downregulation may also occur via proteasomal regulation, MG132, a proteasome inhibitor, was used to hinder proteasome activity in the presence of em08red. As shown in Figure 6, treatment with MG132 could restore ErbB2 expression in em08red-treated cells, indicating that em08red-induced ErbB2 downregulation could occur in a proteasome-dependent manner.
Figure 6.
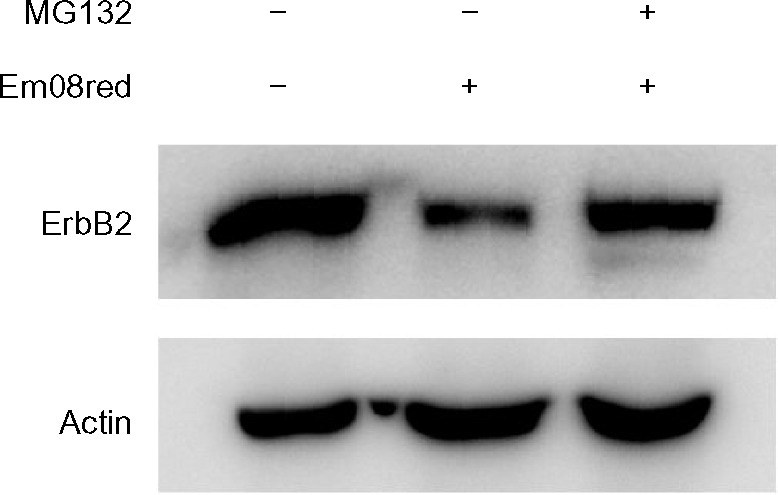
MG132, a proteasome inhibitor, abolished em08red-mediated ErbB2 protein downregulation.
Notes: In the cotreatment group, HSC3 cells were preincubated with MG132 (10 μM) for one hour, and em08red (10 μM) was then added to each treatment group for a further 16 hours of incubation. Cells were harvested at the indicated times and the total cell lysates were subjected to Western blot analysis. Antibodies specific for total ErbB2 and actin were used to detect protein expression after treatment. Actin expression served as the loading control.
Discussion
Extensive investigation and progressive clinical application of target-specific small molecule inhibitors of kinase activity has now been well documented in many malignancies. Gefitinib and erlotinib are two effective pioneer drugs with activity against cancers overexpressing epidermal growth factor receptor, a member of the ErbB family.27 Overactivation of ErbB2 is believed to participate in the development and progression of oral cancers and many other types of malignancy, so is an attractive therapeutic target.10 Kinase inhibitors targeting ErbB2 or the epidermal growth factor receptor/ErbB2 signaling pathway have become a top research priority in the treatment of cancer.28
Previously, emodin has been reported to induce apoptosis or growth inhibition in a number of cancer cell lines,29,30 including some ErbB2-overexpressing breast cancer cells. Treatment with emodin sensitizes cells to treatment with paclitaxel,19 but the details of the molecular mechanism involved remain elusive. Recently, studies have also shown that an azide methyl containing emodin could promote proteasomal degradation of ErbB2 and induce cell death.17 Consequently, emodin is an attractive pharmaceutical reagent for use to disrupt ErbB2 activity in oral cancers. In this study, a computer-aided design approach was used and molecules that best fitted the ATP-binding pocket of ErbB2 were synthesized and evaluated for antiproliferative activity in ErbB2-overexpressing oral cancer cell lines. Initially, the antiproliferative effects of analogues with hydroxyl substitutions at different sites on the anthraquinone were examined (Figure 1 and Table 1). A preliminary structure-activity relationship study suggested compounds with no hydroxyl substitution (compound 1) or more than three hydroxyl substitutions (compound 9, emodin) displayed very weak antiproliferative activity. The compounds containing one or two hydroxyl groups showed increased antiproliferative activity; however, the results indicated that hydroxyl substitution at the R1 position would be necessary for antiproliferative activity (compounds 2, 5, 6, and 7) but hydroxyl substitution at position 2 seemed to reduce the potency of the analogues (compound 3, 4, 8 and 9). Additional hydroxyl substitutions at position 4 or 8 augmented the potency of compounds (5 and 7). As shown in Figure 1 and Table 2, it is speculated that the number of hydroxyl substitutions might govern the potency of emodin analogues with regard to anticancer activity in oral malignancies (2>0>3≥1). Conversely, simply reduction of ketone at position 10 of emodin (compound 11) did not improve the potency of antiproliferative activity, but reduction of ketone at position 10 of the two hydroxyl-substituted analogue (compound 7) for compound 10, designated as em08red, showed greatly enhanced antiproliferative activity towards all three oral cancer cell lines, with an IC50 value below 10 μM. Em08red was therefore chosen for further study.
Our results clearly show that treatment with em08red significantly blocks activation of ErbB2 by reducing ErbB2 protein levels via proteasomal degradation (Figures 2 and 6), implying a shared characteristic of some anthraquinones in affecting ErbB2 function or stability and contributing to retardation of G2. Our observation was similar to previous findings of downregulation of ErbB2 and G2/M arrest via neu differential factor in an ErbB2-overexpressing breast cancer cell line.21 Other studies have shown that generation of ROS is an essential product for anthraquinone-mediated cell death in several cancer cell lines.24,25,31 Further investigation showed that treatment with em08red increased intracellular ROS levels and that induction of ROS had a causative role in em08red-induced cell death (Figure 5). In addition, our results demonstrate that em08 red-mediated apoptosis reduces ErbB2 protein expression and is accompanied by induction of ROS (Figures 2 and 5A). Both mechanisms have previously been shown to induce apoptosis.30,31 To the best of our knowledge, em08red is the first anthraquinone that can simultaneously downregulate expression of ErbB2 protein and efficiently induce production of ROS (Figure 2). Whether the molecular mechanisms of ErbB2 downregulation by treatment with em08red occurs via reactive oxygen species (ROS)-involved lysosomal degradation or enhanced ubiquitination process of ErbB2 is still under active investigation.
A fascinating and efficient pharmacore will be established and further development of potential analogues has been launched for further study. In conclusion, our data provide a novel scheme of emodin analogue effectively targeted ErbB2 protein and imply a developmental future of anthraquinones on target therapy.
Supplementary materials
Chemistry
1,4-dihydroxyanthraquinone (compound 1)
Yield 82%. Melting point 194°C–195°C. MS: 240 (M+). FTIR: 3,065, 1,630, 1,590 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 12.67 (s, 1H), 8.23–8.25 (m, 2H), 7.95–7.97 (m, 2H), 7.47 (s, 2H). Anal. Calcd for C14H8O4: C, 70.00; H, 3.36. Found: C, 69.98; H, 3.35.
1,8-dihydroxyanthraquinone (compound 2)
Yield 77%. Melting point 194°C −187°C (decomposes). MS: 240 (M+). FTIR: 1,676, 1,668, 1,627 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 11.88 (s, 2H), 7.75 (d, 2H, J=7.6 Hz), 7.68 (t, 2H, J=7.6 Hz), 7.36 (d, 2H, J=7.6 Hz). Anal. Calcd for C14H8O4: C, 70.00; H, 3.36. Found: C, 70.01; H, 3.37.
1,5-dihydroxyanthraquinone (compound 3)
Yield 82%. Melting point 94°C–96°C. MS: 240 (M+); FTIR: 1,637, 1,604, 1,627 cm−1. 1H NMR (CDCl3, 400 MHz): δ 12.01 (s, 2H), 7.82 (d, 2H, J=7.5 Hz), 7.66 (dd, 2H, J=7.6, 8.5 Hz), 7.28 (d, 2H, J=8.5 Hz). Anal. Calcd for C14H8O4: C, 70.00; H, 3.36. Found: C, 70.02; H, 3.37.
2,6-dihydroxyanthraquinone (compound 4)
Yield 82%. Melting point 348°C–349°C. MS: 240 (M+). FTIR: 3,322, 1,666, 1,566 cm−1. 1H NMR (DMSO, 400 MHz): δ 10.97 (s, 2H), 8.05 (d, 2H, J=8.5 Hz), 7.48 (d, 2H, J=2.4 Hz), 7.19 (dd, 2H, J=2.4, 8.5 Hz). Anal. Calcd for C14H8O4: C, 70.00; H, 3.36. Found: C, 69.98; H, 3.33.
1,2-dihydroxyanthraquinone (compound 5)
Yield 57%. Melting point 173°C–174°C. MS: 240 (M+). FTIR: 3,426, 1,664, 1,634 cm−1. 1H NMR (DMSO-d6, 400 MHz): δ 12.57 (s, 1H), 10.86 (s, 1H), 8.11–8.18 (m, 2H), 7.88–7.89 (m, 2H), 7.60 (d, 1H, J=8.24), 7.20 (d, 1H, J=8.24). Anal. Calcd for C14H8O4: C, 70.00; H, 3.36. Found: C, 69.99; H, 3.37.
1,2,4-trihydroxyanthraquinone (compound 6)
Yield 76%. Melting point 372°C–375°C. MS: 256 (M+). FTIR: 3,426, 1,622, 1,586 cm−1. 1H NMR (DMSO, 400 MHz): δ 13.31 (s, 1H), 13.06 (s, 1H), 12.62 (s, 1H), 8.12–8.16 (m, 2H), 7.85–7.90 (m, 2H), 6.60 (s, 1H). Anal. Calcd for C14H8O5: C, 65.63; H, 3.15. Found: C, 65.66; H, 3.16.
1-hydroxyanthraquinone (compound 7)
Yield 83%. Melting point 150°C (decomposes). MS: 224 (M+). FTIR: 1,670, 1,637 cm−1. 1H NMR (DMSO, 400 MHz) 12.40 (s, 1H), 8.24 (dd, 1H, J=1.17, 8.26 Hz), 8.17 (dd, 1H, J=1.50, 7.63 Hz), 8.12–8.16 (m, 2H), 7.92–7.97 (m, 2H), 7.81 (t, 1H, J=7.60, 7.60 Hz), 7.71 (d, 1H, J=7.60 Hz), 7.38 (d, 1H, J=7.60 Hz). Anal. Calcd for C14H8O3: C, 75.00; H, 3.60. Found: C, 75.02; H, 3.61.
2-hydroxyanthraquinone (compound 8)
Yield 79%. Melting point 195°C–196°C. MS: 224 (M+); FTIR: 3,370, 1,680 cm−1; 1H NMR (DMSO, 400 MHz) 11.40 (s, 1H), 8.08–8.11 (m, 2H), 8.03 (d, 1H, J=8.56 Hz), 7.78–7.88 (m, 2H), 7.43 (d, 1H, J=2.40 Hz), 7.18 (dd, 1H, J=2.40, 8.56 Hz). Anal. Calcd for C14H8O3: C, 75.00; H, 3.60. Found: C, 75.01; H, 3.62.
Anthraquinone (compound 9)
Yield 85%. Melting point 284°C–286°C. MS: 208 (M+); FTIR: 1,686, 1,592 cm−1. 1H NMR (CDCl3, 400 MHz) 8.32 (d, 1H, J=8.56 Hz), 7.81 (d, 1H, J=8.56 Hz). Anal. Calcd for C14H8O2: C, 80.76; H, 3.87. Found: C, 80.77; H, 3.89.
1,8-dihydroxy-10H-anthracene-9-one (compound 10, emd08red)
SnCl2 solution (13.19 mmol in 37% HCl) was added dropwise over a 3-hour period into refluxed 1,8-dihydroxyanthracene-9,10-dione (0.7 mmole in glacial acetic acid). The solution was then cooled, and the resulting crystals were collected by filtration. Purification by column chromatography (EtOAc/n-Hex =1:1) provided the yellow platelets: 71% yield; melting point 144°C; MS: 226 (M+); FTIR: 1,616, 1,597 cm−1; 1H NMR (400 MHz, CDCl3): δ 12.26 (1H, s, 1,8-OH), 7.47 (2H, dd, J=15.6, 15.6, H-3,6), 6.88 (4H, d, J=15.6, H-2,4,5,7), 4.33 (2H, s, H-10); Anal. Calcd for C14H10O3: C, 74.33; H, 4.46. Found: C, 74.37; H, 4.45.
1,3,8-trihydroxy-6-methyl-10H-anthracene- 9-one (compound 11)
Emodin (1.35 g, 5 mmol) was reacted with SnCl2 (5.0 g, 26 mmol) as described for the preparation of 11 to afford 12 (1.02 g, 80%) in the form of yellow plates. MS: 256 (M+). FTIR: 1,611, 1,590 cm−1. Melting point 250°C (decomposes). 1H-NMR (DMSO-d6, 400 MHz) δ 12.33 (s, 1H), 12.18 (s, 1H), 10.79 (s, 1H), 6.74 (s, 1H), 6.64 (s, 1H), 6.38 (s, 1H), 6.18 (s, 1H), 4.27 (s, 2H), 2.28 (s, 3H). Anal. Calcd for C15H12O4: C, 70.31; H, 4.72. Found: C, 70.33; H, 4.73.
Acknowledgments
This research was supported by the Ministry of Science and Technology of Taiwan (NSC 99-2320-B-039-011-MY3, NSC 99-2320-B-039-010-MY3, NSC101-2320-B-039-011 and NSC 102-2320-B-039-010). We thank Mr Shih-Wei Wayne Juang for his scientific and writing expertise.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45(4–5):309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin. 2002;52(4):195–215. doi: 10.3322/canjclin.52.4.195. [DOI] [PubMed] [Google Scholar]
- 3.Johnson NW, Warnakulasuriya S, Gupta PC, et al. Global oral health inequalities in incidence and outcomes for oral cancer: causes and solutions. Adv Dental Res. 2011;23(2):237–246. doi: 10.1177/0022034511402082. [DOI] [PubMed] [Google Scholar]
- 4.Zini A, Czerninski R, Sgan-Cohen HD. Oral cancer over four decades: epidemiology, trends, histology, and survival by anatomical sites. J Oral Pathol Med. 2010;39(4):299–305. doi: 10.1111/j.1600-0714.2009.00845.x. [DOI] [PubMed] [Google Scholar]
- 5.Shiu MN, Chen TH, Chang SH, Hahn LJ. Risk factors for leukoplakia and malignant transformation to oral carcinoma: a leukoplakia cohort in Taiwan. Br J Cancer. 2000;82(11):1871–1874. doi: 10.1054/bjoc.2000.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saman DM. A review of the epidemiology of oral and pharyngeal carcinoma: update. Head Neck Oncol. 2012;4:1. doi: 10.1186/1758-3284-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang SH, O’Sullivan B. Oral cancer: current role of radiotherapy and chemotherapy. Med Oral Patol Oral Cir Bucal. 2013;18(2):e233–e240. doi: 10.4317/medoral.18772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsantoulis PK, Kastrinakis NG, Tourvas AD, Laskaris G, Gorgoulis VG. Advances in the biology of oral cancer. Oral Oncol. 2007;43(6):523–534. doi: 10.1016/j.oraloncology.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Chu F, Feng Q, Qian Y, Zhang C, Fang Z, Shen G. ERBB2 gene amplification in oral squamous cell malignancies: a correlation with tumor progression and gene expression. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(1):90–95. doi: 10.1016/j.tripleo.2011.01.026. [DOI] [PubMed] [Google Scholar]
- 10.Andolfo I, Petrosino G, Vecchione L, et al. Detection of erbB2 copy number variations in plasma of patients with esophageal carcinoma. BMC Cancer. 2011;11:126. doi: 10.1186/1471-2407-11-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin-Ezquerra G, Salgado R, Toll A, et al. Multiple genetic copy number alterations in oral squamous cell carcinoma: study of MYC, TP53, CCDN1, EGFR and ERBB2 status in primary and metastatic tumours. Br J Dermatol. 2010;163(5):1028–1035. doi: 10.1111/j.1365-2133.2010.09947.x. [DOI] [PubMed] [Google Scholar]
- 12.Werkmeister R, Brandt B, Joos U. Clinical relevance of erbB-1 and -2 oncogenes in oral carcinomas. Oral Oncol. 2000;36(1):100–105. doi: 10.1016/s1368-8375(99)00069-x. [DOI] [PubMed] [Google Scholar]
- 13.Cicenas J, Urban P, Kung W, et al. Phosphorylation of tyrosine 1248-ERBB2 measured by chemiluminescence-linked immunoassay is an independent predictor of poor prognosis in primary breast cancer patients. Eur J Cancer. 2006;42(5):636–645. doi: 10.1016/j.ejca.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Erjala K, Sundvall M, Junttila TT, et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12(13):4103–4111. doi: 10.1158/1078-0432.CCR-05-2404. [DOI] [PubMed] [Google Scholar]
- 15.Srinivas G, Babykutty S, Sathiadevan PP, Srinivas P. Molecular mechanism of emodin action: transition from laxative ingredient to an antitumor agent. Med Res Rev. 2007;27(5):591–608. doi: 10.1002/med.20095. [DOI] [PubMed] [Google Scholar]
- 16.Wang SC, Zhang L, Hortobagyi GN, Hung MC. Targeting HER2: recent developments and future directions for breast cancer patients. Semin Oncol. 2001;28(6 Suppl 18):21–29. doi: 10.1053/sonc.2001.29724. [DOI] [PubMed] [Google Scholar]
- 17.Yan YY, Zheng LS, Zhang X, et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol Pharm. 2011;8(5):1687–1697. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- 18.Shia CS, Suresh G, Hou YC, Lin YC, Chao PD, Juang SH. Suppression on metastasis by rhubarb through modulation on MMP-2 and uPA in human A549 lung adenocarcinoma: an ex vivo approach. J Ethnopharmacol. 2011;133(2):426–433. doi: 10.1016/j.jep.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Lau YK, Xia W, Hortobagyi GN, Hung MC. Tyrosine kinase inhibitor emodin suppresses growth of HER-2/neu-overexpressing breast cancer cells in athymic mice and sensitizes these cells to the inhibitory effect of paclitaxel. Clin Cancer Res. 1999;5(2):343–353. [PubMed] [Google Scholar]
- 20.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 21.Daly JM, Jannot CB, Beerli RR, Graus-Porta D, Maurer FG, Hynes NE. Neu differentiation factor induces ErbB2 downregulation and apop-tosis of ErbB2-overexpressing breast tumor cells. Cancer Res. 1997;57(17):3804–3811. [PubMed] [Google Scholar]
- 22.Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9(12):1287–1293. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- 23.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351(1–2):41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 24.Su YT, Chang HL, Shyue SK, Hsu SL. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol. 2005;70(2):229–241. doi: 10.1016/j.bcp.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 25.Lee HZ, Lin CJ, Yang WH, Leung WC, Chang SP. Aloe-emodin induced DNA damage through generation of reactive oxygen species in human lung carcinoma cells. Cancer Lett. 2006;239(1):55–63. doi: 10.1016/j.canlet.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 26.Dogan I, Cumaoglu A, Aricioglu A, Ekmekci A. Inhibition of ErbB2 by herceptin reduces viability and survival, induces apoptosis and oxidative stress in Calu-3 cell line. Mol Cell Biochem. 2011;347(1–2):41–51. doi: 10.1007/s11010-010-0610-7. [DOI] [PubMed] [Google Scholar]
- 27.Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol. 2005;23(11):2556–2568. doi: 10.1200/JCO.2005.07.799. [DOI] [PubMed] [Google Scholar]
- 28.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9(7):463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 29.Schutze C, Dorfler A, Eicheler W, et al. Combination of EGFR/HER2 tyrosine kinase inhibition by BIBW 2992 and BIBW 2669 with irradiation in FaDu human squamous cell carcinoma. Strahlenther Onkol. 2007;183(5):256–264. doi: 10.1007/s00066-007-1696-z. [DOI] [PubMed] [Google Scholar]
- 30.Park SJ, Kim MJ, Kim YK, Kim SM, Park JY, Myoung H. Combined cetuximab and genistein treatment shows additive anti-cancer effect on oral squamous cell carcinoma. Cancer Lett. 2010;292(1):54–63. doi: 10.1016/j.canlet.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Srinivas G, Anto RJ, Srinivas P, Vidhyalakshmi S, Senan VP, Karunagaran D. Emodin induces apoptosis of human cervical cancer cells through poly(ADP-ribose) polymerase cleavage and activation of caspase-9. Eur J Pharmacol. 2003;473(2–3):117–125. doi: 10.1016/s0014-2999(03)01976-9. [DOI] [PubMed] [Google Scholar]