

Abstract
Owing to the persistence of tuberculosis (TB) as well as the emergence of multidrug-resistant and extensively drug-resistant (XDR) forms of the disease, the development of new antitubercular drugs is crucial. Developing inhibitors of shikimate kinase (SK) in the shikimate pathway will provide a selective target for antitubercular agents. Many studies have used in silico technology to identify compounds that are anticipated to interact with and inhibit SK. To a much more limited extent, SK inhibition has been evaluated by in vitro methods with purified enzyme. Currently, there are no data on in vivo activity of Mycobacterium tuberculosis shikimate kinase (MtSK) inhibitors available in the literature. In this review, we present a summary of the progress of SK inhibitor discovery and evaluation with particular attention toward development of new antitubercular agents.
Keywords: Mycobacterium tuberculosis, shikimate kinase inhibitors, antitubercular
Introduction
Tuberculosis (TB) remains a global health concern, with about 8.6 million cases in 2012.1 WHO also estimated that there were 450,000 cases of multidrug-resistant tuberculosis (MDR-TB) in 2012 compared to 62,000 cases in 2011.1 Of the diagnoses in 2012, 9.6% of these cases estimated to be extensively drug-resistant tuberculosis (XDR-TB).1 With the growing number of MDR-TB and XDR-TB cases, there is a need for the discovery of new antitubercular agents.
The only FDA-approved TB drug since the 1960s has been recently announced, Bedaquiline Sirturo™ for the treatment of MDR-TB.2 Sirturo specifically inhibits mycobacterial ATP (adenosine 5’-triphosphate) synthase, an enzyme that is essential for the final step in ATP production by oxidative phosphorylation.3 Its associated risk of potentially lethal heart problems has emphasized the unmet and urgent need for the development of safer antitubercular drugs with novel targets and mechanisms of action to treat resistant forms of the disease.
One favorable mechanism for antitubercular agents is the inhibition of shikimate kinase (SK) in the shikimate pathway. The shikimate pathway is used in a variety of bacteria, including Mycobacterium tuberculosis, for the production of chorismate, a precursor for aromatic amino acids and other aromatic compounds.4 Mammals do not have the shikimate pathway enzymes necessary for de novo synthesis of these amino acids but rather obtain them from the diet.4 Consequently, inhibitors of SK are anticipated to be selective antitubercular drugs. SK is the fifth of seven enzymes involved in the shikimate pathway. SK is responsible for catalyzing phosphoryl transfer from ATP to shikimate to form shikimate-3-phosphate and ADP. M. tuberculosis shikimate kinase (MtSK) is encoded by aroK and is essential for the survival of M. tuberculosis.5
MtSK Structure and Substrate Binding Sites
MtSK belongs to the nucleoside monophosphate (NMP) kinase family.6 The core of its structure (Fig. 1, PDB accession number 1ZYU, resolution 2.90 Å) is a five-stranded parallel β-sheet.6 Consistent with other family members, MtSK has a lid (residues G112–D124) and NMP-binding domain (T33–E61).6 The latter is a major contributor to shikimate binding in MtSK, and as such, is also referred to as the shikimate-binding domain.6 In addition, there is the P-loop (also referred to as the Walker A-motif) (G9–S16).7 This forms part of the binding site for ATP and ADP,8 particularly by interaction with their β- and γ-phosphates, respectively.6,9 Additionally, ATP/ADP is associated with SK through the adenine-binding loop (V148–P155). Finally, there is the Walker-B motif (V75–G80), which lies near the interface between shikimate and ATP-binding sites.7 Similar to other NMP kinases, MtSK undergoes a large conformational change upon substrate binding with the largest shifts in structure occurring in the shikimate-binding and lid domains.6,10
Figure 1.

Structure of MtSK bound to shikimate and the ATP analog, 5′-adenosyl-methylene-triphosphate (ADPCP).
Notes: The shikimate-binding (red) and lid (blue) domains are both shown as are the Walker-B motif (cyan), the P-loop (magenta), and the adenine-binding loop (yellow). The structures of shikimate (right) and ADPCP (left) are shown with carbons in green. PDB accession number, 1ZYU.6
The shikimate-binding domain is made up of residues 33–61 and consists of α-helices α2 and α3 and the N-terminal region of α4 following strand β2.7 The carboxyl group of shikimate interacts with R58 and R136.8 The 3-hydroxyl group interacts with D34 and G80, while the 2-hydroxyl group interacts with D34.8
Mg2+ and ADP interact with residues in the P-loop, lid domain, and adenine-binding loop. Mg2+ interacts with the β-phosphate oxygen of ADP, S16 (P-loop), and four water molecules. O1B (phosphate) binds S13, G14, and K15 (P-loop). O2B (phosphate) binds S16 (P-loop), and O3B (phosphate) binds G12 (P-loop) and R117 (lid domain). R117 also binds O1A. O2A binds S16 and T17. O3A binds G14, and N6 of adenine binds R153 within the adenine-binding loop.7,10
To date, 21 crystal structures of MtSK have been solved and coordinates are deposited in the protein data bank (http://www.rcsb.org/pdb/home/home.do) (Table 1).
Table 1.
Crystal structures of MtSK.
| STRUCTURE ID | LIGAND | RESOLUTION (Å) | REFERENCE |
|---|---|---|---|
| 2IYZ | Shikimate-3-phosphate and ADP | 2.30 | 11 |
| 2IYS | Shikimate | 1.40 | 11 |
| 2IYQ | Shikimate and ADP | 1.80 | 11 |
| 2IYR | Shikimate | 1.98 | 11 |
| 2IYY | Shikimate-3-phosphate and SO4 | 1.62 | 11 |
| 2IYX | Shikimate and SO4 | 1.49 | 11 |
| 2IYT | Unliganded, open LID | 1.47 | 11 |
| 2IYU | ADP, open LID | 1.85 | 11 |
| 2IYW | MgATP, open LID | 1.85 | 11 |
| 2IYV | ADP | 1.35 | 11 |
| 3BAF | AMP-PNP | 2.25 | 12 |
| 2DFN | ADP and shikimate | 1.93 | 13 |
| 4BQS | ADP and a shikimic acid derivative | 2.15 | 14 |
| 1U8A | Shikimate and ADP | 2.15 | 15 |
| 2G1K | Shikimate | 1.75 | 6 |
| 1ZYU | Shikimate and AMPPCP | 2.85 | 6 |
| 2G1J | No ligand | 2.00 | 6 |
| 2DFT | ADP and Mg | 2.80 | 13 |
| IWE2 | MgADP and shikimic acid | 2.30 | 7 |
| 1L4Y | MgADP | 2.00 | 10 |
| 1L4U | MgADP and Pt(II) | 1.80 | 10 |
SK Inhibitors
Computational approaches
ZINC database compounds
Using MOLDOCK, a molecular docking algorithm,15 Vianna and de Azevedo16 screened a total of 4579 molecules from the ZINC database for interaction with MtSK. A part of the validation of the results was the calculation of root-mean-square deviation (RMSD). This measures the differences in the predicted values by a model and the observed values. The desired RMSD is <2.0 Å, and in this docking, simulation was found to be between 1 and 2 Å. The calculation of the enrichment factor (EF) of 52.6 further validated the virtual screening protocol. EF indicates the number of active compounds found within the virtual screening strategy with larger EFs indicating a large number of active molecules.17
Based on favorable MOLDOCK scores, those representing lower energy required for binding,15 a total of 20 compounds were selected for further evaluation by FAF-Drugs18 to determine which fit Lipinski’s rule of five.19 Of the 20 compounds, 9 fit the requirements, including staurosporine, a known cyclin-dependent kinase inhibitor. That a known kinase inhibitor was among the positive results of the screen validated the algorithm utilized; however, staurosporine was excluded from further analysis because its known toxicity precludes its use as an antitubercular agent. The remaining eight compounds (MOLDOCK scores ranging from –144.208 to –151.943) (Fig. 2) were evaluated further using the LIGPLOT program to determine the interactions occurring between the compounds and enzyme.20 The top-scoring compound, ZINC15707201, formed hydrogen bonds with K15 (P-loop) and van der Waals interactions with S16 (P-loop), V116, and R117 (lid domain). The other top-scoring compounds also displayed interactions with these residues, suggesting that these residues will be important points of contact for potential MtSK inhibitors.16
Figure 2.

The top eight scoring compounds from ZINC database.
Using the same virtual screening protocol and ZINC database, 89,425 compounds were screened against the ATP-binding pocket of MtSK. Appropriate validation tests were applied, resulting in a RMSD of 0.48 Å and an EF of 24.57.
A total of 17 compounds displayed inhibitory properties, with MOLDOCK scores ranging from –190.173 to –201.689. Of these 17 compounds, 5 complied with filters and measurements of the drug-likeness Lipinski’s rule of five19 and Veber rules,21 indicating they would be orally bioavailable. An analysis of the pattern of hydrogen bonds in the docking results for SK at ATP-binding pocket of the inhibitors containing indole ring linked to a chromone ring by seven covalent bonds was carried out. This analysis indicated that residues G14, K15, and R117 participate in interactions with all this compound series. These residues show contacts with the PO43− group present in ADP in the crystallographic structure 1WE2 of MtSK. Additionally, van der Waals interactions were noted, which involved I18 (P-loop) and R110 (N-terminal to the lid domain), as well as P155 and V158 (adenine-binding loop). The binding of all five top-scoring compounds to R110 and P155 suggests that these binding interactions are crucial for ligand-binding affinity.22
In the crystallographic structure 1WE2 of MtSK, the hydrogen bond acceptor is the main chain carbonyl group of Arg 153 with compound ZINC02160785. Analogs of this compound can be synthesized by adding a group to participate in an intermolecular contact with Arg 154 to improve ligand–binding affinity (Fig. 3).
Figure 3.

Chemical structure of five compounds from ZINC database, which display inhibitory effects for MtSK.
Dipeptide inhibitor
Kumar et al.23 identified the dipeptide +H3 N-Arg-Asp-COO− (RD, compound 14) as a potential SK inhibitor because of its high-predicted binding affinity. Using the LigandFit24 protocol as the molecular docking method, both the open- and closed-lid conformations of the enzyme were analyzed.
RD maintains hydrogen bonding with D34, R58 (NMP-binding domain), and R136, which was seen in the binding of SK. In the closed-lid conformation, RD interacted with K15, S16 (P-loop), D32 (NMP-binding domain), L78 (Walker-B motif), V116, and R117 (lid domain), which was not seen with shikimate binding and resulted in KD = 5.49 nM. In the open-lid conformation, RD demonstrated additional interactions with K15 (p-loop), D32, F49 (NMP-binding domain), S77, and L78 (Walker-B motif) and resulted in KD = 26 nM (Table 2).23
Table 2.
Interacting residues for shikimate and dipeptide RD (hydrogen bonds are bold).
| CONFORMATION OF MTSK | LIGAND | INTERACTING RESIDUES |
|---|---|---|
| Closed LID | Shikimate | P11, D34, I45, F49, R58, E61, G79, G80, G81, P118, L119, R136 |
| Open LID | Shikimate | D34, I45, F57, R58, E61, G79, G80, G81, R136 |
| Closed LID | RD | P11, K15, S16, D32, D34, I45, R58, L78, G79, G80, G81, V116, R117, P118, L119, R136, ADP |
| Open LID | RD | K15, D32, D34, F49, F57, R58, E61, S77, L78, G79, G80, G81, R136 Arg136 |
FAF-drugs
Using the eHiTS® software25 (available at http://www.simbiosys.ca/ehits/), Segura-Cabrera and Rodríguez-Pérez26 analyzed 214,492 compounds from FAF-Drugs18 by high-throughput virtual screening. Each compound was given a binding score through eHiTS software, which then allowed the compounds to be compared to control shikimate and control 6-S-fluoroshikimate, which had eHiTS scores of –4.260 and –4.268, respectively. A total of 644 molecules were determined to have eHiTS scores, which were more negative, meaning the compound had better binding affinity than the control compounds, with the best score being –7.252 for compound AsXE1. It was reported that the best scoring compounds contained a mercapto group and a triazole or tetrazole ring in the scaffold.
The relevant moieties, triazole and tetrazole, most likely interact with R58, L119, and R136 residues and mimic key interactions of the enzyme–substrate complex, specifically the interaction of these residues with the carboxyl group of shikimate. Two of the nitrogen atoms of the azole system were hydrogen bonded to R58 and R136 and the proximity of the L119 with the heteroaromatic ring, suggesting the possibility of stabilizing CH–electron interactions.26 The mercapto group interacted with G80, R117, and L119 through hydrophobic forces and hydrogen bonds. The interaction with R117 is key for the phosphoryl transfer reaction catalyzed by MtSK.26
Of the 644 molecules, the 200 top-scoring compounds were analyzed to determine the main interactions that were occurring between the compound and the binding site. Hydrophobic forces were seen with residues P11, K15 (P-loop), D34, I45, F49, F57 (NMP-binding domain), G79, G80 (Walker-B motif), G81 (C-terminal to the Walker-B motif), V116, R117, P118, and L119 (lid domain). Additionally, hydrogen bonding with residues D34, R58 (NMP-binding domain), G80 (Walker-B motif), G81, V116, R117, L119 (lid domain), and R136 were observed (Table 3).26
Table 3.
eHiTS scores and molecular structures for controls and top compounds.
| Control shikimate (15) | −4.260 |
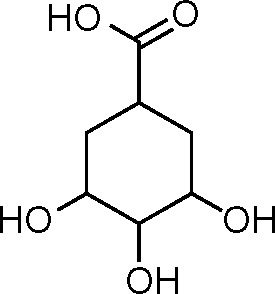
|
| Control 6-S-fluoroshikimate (16) | −4.268 |

|
| ASXE1 (17) | −7.252 |
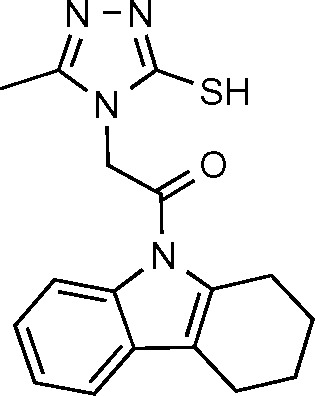
|
| MW1 (18) | −6.595 |
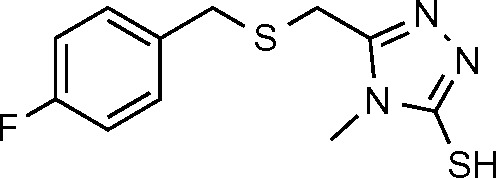
|
| DIVER1 (19) | −6.453 |
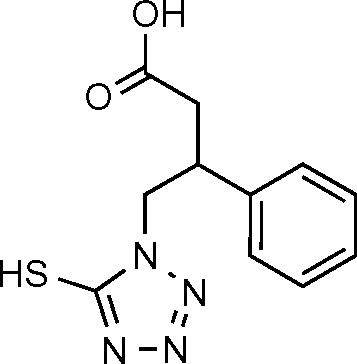
|
| ASXC1 (20) | −6.372 |

|
| SPCH1 (21) | −6.366 |
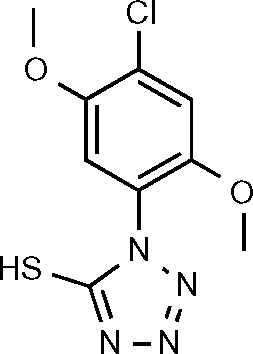
|
| ASXC2 (22) | −6.319 |

|
| SPCA1 (23) | −6.142 |
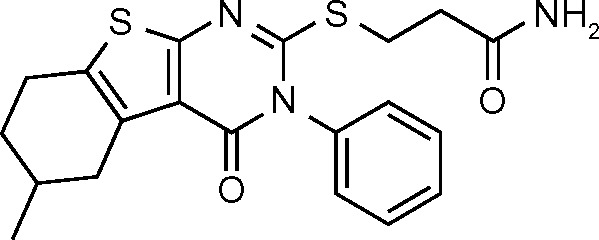
|
| SPCA2 (24) | −6.109 |
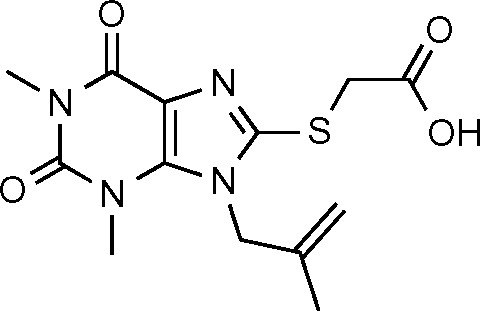
|
| KIN1 (25) | −6.08 |

|
| ASXB1 (26) | −6.041 |

|
Molecular docking of known inhibitors
A total of 33 known inhibitors of MtSK were analyzed against the 3D model of SK from ESyPred3D27 using Schrodinger docking software. The 3D model from ESyPred3D was used because when superimposed with the crystal structure of MtSK from Dali Pairwise Comparison, PDB ID: 2IYT, this predicted model had the lowest RMSD value, suggesting a highly accurate model. The docking scores for the top 10 compounds are given (Table 4) with the best docking score being –6.174338 for compound SPB01099 versus –6.138796 for control shikimate.28
Table 4.
Docking scores of the top 10 MtSK inhibitors for ESyPred3D model.
| COMPOUND NAME | DOCKING SCORE |
|---|---|
| SPB01099 (659) | −6.174338 |
| AG538 (660) | −6.151543 |
| Control shikimate (15) | −6.138796 |
| Control 6-S-fluoroshikimate (16) | −6.089551 |
| ZINC20464408 (7) | −5.777331 |
| ASXB1 (26) | −5.75549 |
| DIVER1 (19) | −5.538557 |
| ZINC20462780 (2) | −5.484212 |
| MW1 (18) | −5.354823 |
| RH00016 (661) | −5.231464 |
Shikimic acid analogs
Using structural and molecular dynamics simulation studies, Blanco et al.29 designed shikimic acid analogs that would bind to the shikimate binding site and determined experimental measurements of inhibition constants. Seven shikimic acid analogs (Fig. 4) were assayed and shown to be reversible competitive inhibitors and bind to the same binding site as shikimic acid. The KI values (Table 5), obtained by Dixon plots (1/v vs [I]), ranged from 46 μM to >4000 μM with compound 660 being the most potent inhibitor.
Figure 4.
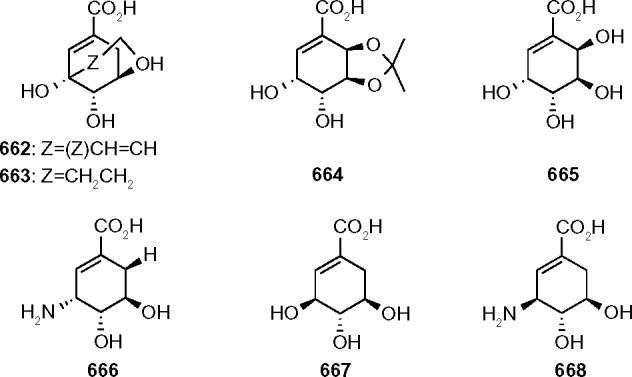
Molecular structures of shikimic acid analogues.
Table 5.
KI values of shikimic acid analogs obtained by Dixon plots.
| COMPOUND | KI (μM) |
|---|---|
| 662 | 62 ± 1 |
| 663 | 46 ± 2 |
| 664 | 1050 ± 17 |
| 665 | 792 ± 16 |
| 666 | 62 ± 1 |
| 667 | >4000 |
| 668 | 65 ± 1 |
The crystal structure of compound 662 interacting with MtSK and ADP was determined at 2.15 Å resolution and proved to bind in a similar manner as shikimic acid. Compound 662 binds to the same active site as shikimic acid via the formation of a salt-bridge between the carboxylate group and guanidinium group of R136. Additional hydrogen bonds are formed between the carboxylate group of D34 (NMP-binding domain) and C3 and C4 hydroxyl groups of compound 662. The C3 hydroxyl group forms an additional hydrogen bond with the amide nitrogen of G80 (Walker-B motif). Several bridging water molecules also have a role in the interaction between ligand and enzyme. The C4 hydroxyl group hydrogen bonds with the water molecules to form interactions with R58, E61 (NMP-binding domain), and G81. Additional bonds with bridging water molecules result in the interaction of K15 (P-loop) with C3 hydroxyl group.
Compounds 663–668 were analyzed by molecular dynamics simulation studies using GOLD 5.0.130 to determine the resulting interactions. Compound 663, only differing from compound 662 by a reduction of the double bond, binds to the shikimate binding site in a similar manner as compound 662. It was concluded that the reduction of the double bond allows for more lipophilic interactions with R117 and P118 (lid), and the C3–C5 bridge located closer to these residues therefore decreases the flexibility of the lid domain. Additionally, the NMP-binding domain seemed to close effectively; this combined with the reduced flexibility of the lid domain may account for the higher KI seen with compound 663.
Compounds 666 and 668 are both 3-aminoshikimic acids in which the cyclohexene ring binds similar to endogenous shikimate. Based on the presence of C3 amino groups, it was proposed that the amino group would interact with the γ-phosphate of ATP via electrostatic interactions. Owing to similar interactions between compounds 666 and 668 with the binding site, it is expected that their inhibitory effects will be similar.
Compound 665 is a hydroxyshikimic acid. The additional OH group at C6 causes changes in the water network and the recognition of the carboxylate group resulting in movement of α-helices α2 and α3. This results in decreased favorability of the closed shikimate binding domain, leading to lower inhibitory effects of compound 665.29
In vitro biological evaluation
Assays for known inhibitors staurosporine and pyrazolone analogs
Using an ultrafiltration-liquid chromatography/mass spectrometry (UF-LC/MS)-based MtSK binding assay and an LC/MS-based MtSK functional assay, Mulabagal and Calderón31 evaluated four compounds for inhibition of MtSK. The former addresses ligand–enzyme binding affinity, and the latter assesses the amount of shikimate-3-phosphate (S3P) formed in the presence of shikimic acid, SK, and a given SK inhibitor. Compound 669, staurosporine, is a known competitive inhibitor of protein kinases acting as a competitive inhibitor of ATP.32 Compounds 670–672 are pyrazolone analogs; pyrazolones and their analogs have previously been shown to be MtSK inhibitors.33
The UF-LC/MS assay was used to measure the extent of MtSK binding when 1 μM enzyme and 1 μM inhibitor were incubated together. The binding affinities inferred from the data were 671 > 672 > 670 > 669. The LC/MS functional assay was then used to measure the extent of inhibition each compound displayed. The compounds were tested at varying concentrations, ranging from 0.05 μM to 1 μM, with 1 μM MtSK and 2 μM shikimic acid. The amount of S3P formed was measured at each concentration, and EC50 values were determined. All four of the compounds showed complete inhibition at 1 μM of inhibitor. The EC50 values for compounds 669, 670, 671, and 672 were determined to be 0.03, 0.24, 0.07, and 0.18 μM, respectively (Fig. 5).31
Figure 5.
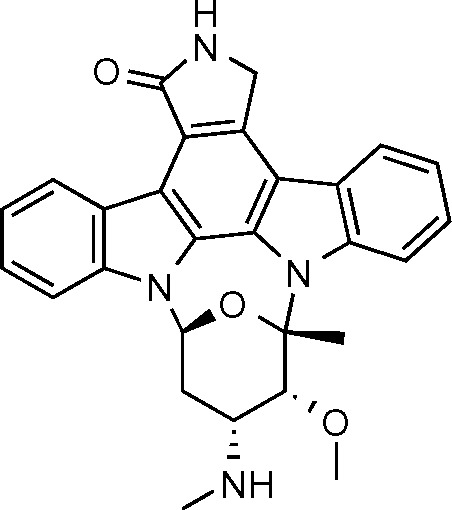
Staurosporine, compound 669.
The pyrazolone analogs displayed MtSK inhibition at IC50 values lower than 1 μM.31 Based on the structure–activity relationship in the series, the phenyl sulfonamide portion of the molecule is required for the activity, and a single substitution of a halogen at the Para position of the phenyl enhances the MtSK inhibition as for compound 671 (Fig. 6).
Figure 6.

Pyrazolone analogs.
Oxadiazole-amides and 2-aminobenzothiazoles
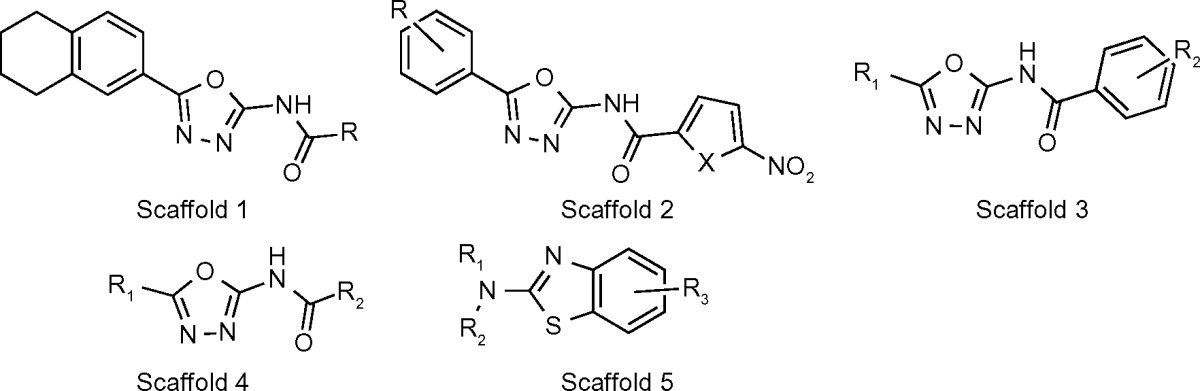
A total of 404 antitubercular compounds from the National Institute of Allergy and Infectious Diseases (NIAID) were tested in vitro using a LC/MS functional assay, of which 14 compounds displayed inhibition of MtSK by >90%. IC50 values (Table 6) were determined for these 14 compounds with the lowest IC50 = 1.94 ± 0.06 for compound 686.
Table 6.
Chemical structure of oxadiazole-amides and 2-aminobenzothiazoles.
| ||||||
|---|---|---|---|---|---|---|
| COMPOUND | SCAFFOLD NUMBER | R1 | R2 | R3 | X | IC50 (μM) |
| 673 | 1 | n-butyl | 32.87 ± 0.04 | |||
| 674 | 1 | Ph-trio-Ethyl | 13.81 ± 0.03 | |||
| 675 | 1 | Ph-CH=CH | 3.79 ± 0.02 | |||
| 676 | 2 | 2,4-diMe | O | 10.35 ± 0.12 | ||
| 677 | 2 | 2,5-diMe | S | 13.09 ± 0.06 | ||
| 678 | 3 | 2,4-diMe-Ph | 2-Cl | 36.04 ± 0.03 | ||
| 679 | 3 | 2-Cl-Ph | 3-O-Ph | 3.43 ± 0.04 | ||
| 680 | 4 | 4-Br-Ph | t-but | 28.36 ± 0.07 | ||
| 681 | 4 | 2-furanyl | 4-EtS-Ph | 20.73 ± 0.03 | ||
| 682 | 4 | 2,4-diMe-Ph | Ph-O-Ethyl | 15.14 ± 0.06 | ||
| 683 | 5 |
|

|
4-Ethyl | 10.43 ± 0.04 | |
| 684 | 5 |
|
5,7-dimethyl | 29.93 ± 0.05 | ||
| 685 | 5 |
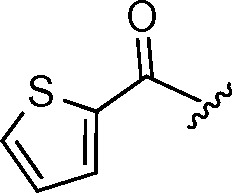
|

|
4,6-difluoro | 20.42 ± 0.06 | |
| 686 | 5 |
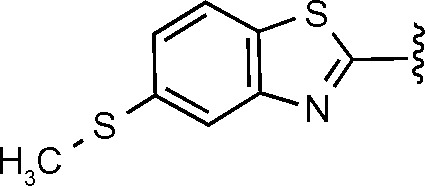
|
H | 6-Acetylamino | 1.94 ± 0.06 | |
In all, 10 of the 14 compounds, compounds 673–682, contain an oxadiazole-amide scaffold, while the remaining four compounds, compounds 683–686, contain a 2-aminobenzothiazole scaffold. Ligand docking, using Schrodinger software, was performed to determine the interactions between the different ligand scaffolds and the MtSK crystal structure (PDB code 2DFT, conformer B). Compounds containing the oxadiazole-amide structure interact with residues V35, R58 (NMP-binding domain), P118, and L119 (lid). Compounds containing the 2-aminobenzothiazole structure displays interactions with P11 (P-loop), V35, I37, I45, F57 (NMP-binding domain), and R136.34
All oxadiazole-amides and 2-aminobenzothiazoles in this series have H37Rv IC90 values below 5 μg/mL and SI values well over the threshold of 10.29 Based on molecular modeling described in Table 5 containing MtSK inhibitors, most compounds 675, 679, and 686 displayed IC50 values of ≤ 5 μM. Compounds 675 and 679 contain the 2,5-disubstituted-1,3, 4-oxadiazole system. Both compounds contain, as required, structural features, a phenyl at the 5-position of the oxadiazole ring and at the other end, the carboxamide is linked to a substituted phenyl.
Compounds containing the oxadiazole-amide structure prefer the following interactions: R58 (NMP-binding domain), an essential shikimate contact residue that forms a positive charge – π interaction with the aromatic ring of the tetrahydronaphthaline of the compounds, and the rest of the ring structure forms non-polar interactions with P118 and L119 (lid). The oxadiazole ring is aligned for favorable polar interactions with guanidinium group in R117. According to the crystal structure, the amide linker participates in hydrogen bonding with water.
While oxadiazole-amide scaffolds have been well documented as having potent antibacterial activity, the benzothiazole cores have not been well represented in the antibacterial literature. They are more prevalent in kinase inhibitor literature, having been identified as inhibitors of kinases such as SHP-2 and JNK kinases.34 The most active compound in this series, 686, contains a unique 6-methylthio-benzothiazole structure at R1/R2.34
Compounds containing the 2-aminobenzothiazole structure display interactions as follows: the benzothiazole ring forms favorable aromatic–aromatic interactions with F57 (NMP-binding domain), which orients R3 in close proximity to the side chain of I45 and also provides non-polar interactions with I37. I45 and V35 form steric interactions with thiophene. The R2 group participates in favorable polar interactions with R136.34
Inhibitors of M. tuberculosis and Helicobacter pylori
Owing to the similarity in conserved binding environments in orthologous proteins, Hsu et al.35 identified inhibitors of SK for both M. tuberculosis and H. pylori through the development of a core site-moiety map (CoreSiMMap). H. pylori shikimate kinase (HpSK) inhibitors have been identified by high-throughput screening36 as well as attempts to discover inhibitors of both shikimate dehydrogenase and SK in HpSK.37 Six consensus anchors, statistically significant subsite–moiety interactions, were identified from the site-moiety maps of the proteins (Table 7).
Table 7.
Consensus anchors and residues present.
| CONSENSUS ANCHOR | RESIDUES (MTSK) |
|---|---|
| H1 | Gly12, Ser13, Gly14, Lys15, Ser16, Arg117 |
| V1 | Pro11, Gly12, Ser13, Gly14, Lys15, Ser16, Arg117 |
| H2 | Lys15, Ser16, Asp32, Asp34 |
| H3 | Lys15, Asp34, Gly80, Thr115 |
| V2 | Phe49, Asp34, Gly79, Gly80, Gly81, Arg117 |
| E1 | Arg58, Arg136, Arg117 |
Following the identification of consensus anchors, the CoreSiMMap was used to screen inhibitors from the Maybridge and NCI databases. A total of 12 inhibitors were identified, 2 of which, AG538 and GW5074, were compounds derived from existing kinase inhibitors. Of the remaining 10 inhibitors, all showed IC50 values ≤100 μM for both MtSK and HpSK with 6 of these inhibitors more active, displaying IC50 values ≤10 μM (Table 8). The six compounds with IC50 values ≤10 μM were shown to fit into five of the six consensus anchors: H1, V1, H3, V2, and E1.
Table 8.
Chemical structure and IC50 (μM) for inhibitors of MtSK and HpSK.
| COMPOUND NAME | IC50 (μM) | STRUCTURE |
|---|---|---|
| NSC45611 (687) | 1.5 |
|
| NSC162535 (688) | 1.6 |
|
| NSC45612 (689) | 2.8 |

|
| NSC45174 (690) | 2.8 |

|
| NSC45547 (691) | 3.4 |
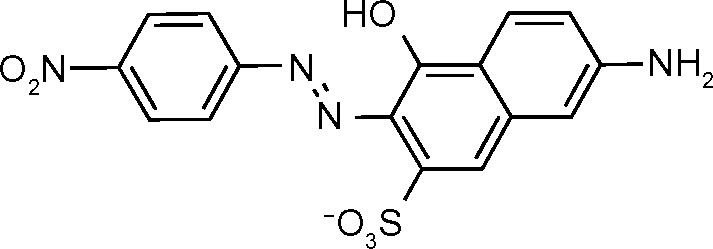
|
| NSC45609 (692) | 2.0 |

|
| RH00037 (693) | <100 |

|
| RH00016 (661) | <100 |

|
| GK01385 (694) | <100 |

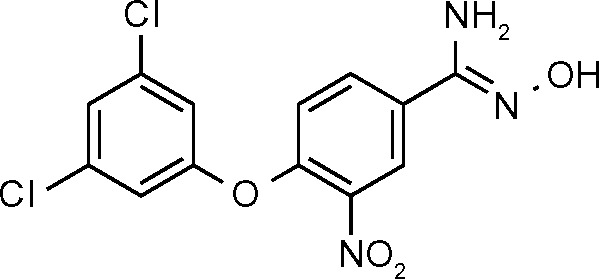
|
| SPB01099 (659) | <100 |
|
Further kinetic analysis revealed that NSC45611, NSC162535, and NSC45612 were competitive inhibitors with respect to ATP and shikimate. Owing to the low IC50 and αKI values of NSC45611, NSC162535, and NSC45612, the interactions of these inhibitors were analyzed using GEMDOCK.38
These three compounds fit well into the consensus anchors by having rings that could form van der Waals interactions at V1 (P11, G12, S13, G14, K15, S16 of P-loop, and R117 of lid domain) and V2 (D34 and F49 of NMP-binding domain, G79 and G80 of Walker-B motif, G81 COO− end of Walker-B motif, and R117 of lid domain) as well as having sulfonate groups to interact with H1 (G12, S13, G14, K15, S16 of P-loop, and R117 of lid domain). Additionally, these three inhibitors were able to form electrostatic interactions with R58 (NMP-binding domain), R117 (lid domain), and R136 residues in E1.35
Chemical Features Required for Selective Inhibitors
Inhibition of MtSK can be achieved by compounds that bind at either the shikimate binding site16,23,26,29,31,34 or ATP-binding site.22,35 The in silico and in vitro data collected infers that compounds with interactions with residues D34, R58 (NMP-binding domain), and R117 show inhibition of MtSK because of their similar interactions found with shikimate.23,26,29,34,35 Additionally, in silico data showed top-scoring compounds interacted with residues K15, S16 (P-loop) and R117 (lid domain),16 and R110 (N-terminal to lid domain) and P155 (adenine-binding loop),22 which were determined to be key interactions between protein and ligand. The in vitro data show that V35 (NMP-binding domain), R117, and P118 (lid domain) may be important interactions.29,34
Structurally, inhibitors toward MtSK have differed. The shikimic acid analogs, being structurally similar to endogenous shikimate, bind similarly as shikimate; however, they do not allow for the transfer of phosphate from ATP to shikimate to form shikimate 3-phosphate.29 Additional MtSK inhibitors presented have contained an indole ring linked to a chromene ring by seven covalent bonds,22 a mercapto group, triazole or tetrazole ring,21 oxadiazole-amide,34 or 2-aminobenzothiazole34 scaffold. A dipeptide containing arginine and aspartic acid (RD) displayed higher binding affinity for MtSK than shikimate. Additionally, pyrazolone analogs31 displayed inhibition against MtSK.
Conclusion
The discovery of MtSK inhibitors offers potential for development of new antitubercular drugs that are selective for M. tuberculosis. Multiple SK inhibitors have been determined through the use of in silico virtual screening in which the docking score and interactions can be determined. These SK inhibitors bind to the same active site as shikimate through similar interactions. The development of an UF-LC/MS binding assay and an LC/MS functional assay has initiated in vitro studies; however, further in vivo assays and clinical studies will need to be conducted before an SK inhibitor is put on the market as an antitubercular agent.
Acknowledgments
JS is grateful to the Secretaría Nacional de Ciencia y Tecnología (SENACYT) in collaboration with the Instituto para la Formación de Recursos Humanos (IFARHU) of the Panamanian government for Ph.D. scholarship.
Footnotes
ACADEMIC EDITOR: Yitzhak Tor, Editor in Chief
FUNDING: The work was supported by Auburn University Intramural Grants Program (AU-IGP) through the Office of the Vice President for Research (OVPR). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
Author Contributions
Wrote the first draft of the manuscript and made corrections: SG. Did the literature search for the manuscript and provided critical comments: JS. Jointly developed the structure and arguments for the paper: SG, AIC. Made critical revisions and approved the final version: DCG, AIC. All authors reviewed and approved the final manuscript: SG, JS, DCG, AIC.
REFERENCES
- 1.World Health Organization (WHO) Global tuberculosis report 2013. WHO/HTM/TB/2013.11. Geneva, Switzerland: 2014. Available at: http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- 2.Thomas K. F.D.A Approves New Tuberculosis Drug. New York: The New York Times; 2012. [Google Scholar]
- 3.Mattelli A, Carvalho AC, Dooley KE, Kritski A. TMC207: the first compound of a new class of potent anti-tuberculosis drugs. Future Microbiol. 2010;5:849–58. doi: 10.2217/fmb.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bentley R. The shikimate pathway – a metabolic tree with many branches. Biochem Mol Bio. 1990;25:307–84. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- 5.Parish T, Stoker NG. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology. 2002;148:3069–77. doi: 10.1099/00221287-148-10-3069. [DOI] [PubMed] [Google Scholar]
- 6.Gan J, Gu Y, Li Y, Yan H, Ji X. Crystal structure of Mycobacterium tuberculosis shikimate kinase in complex with shikimic acid and an ATP analogue. Biochemistry. 2006;45:8539–45. doi: 10.1021/bi0606290. [DOI] [PubMed] [Google Scholar]
- 7.Pereira JH, de Oliveira JS, Canduri F, et al. Structure of shikimate kinase from Mycobacterium tuberculosis reveals the binding of shikimic acid. Acta Crystallogr D Biol Crystallogr. 2004;60:2310–9. doi: 10.1107/S090744490402517X. [DOI] [PubMed] [Google Scholar]
- 8.Dhaliwal B, Nichols CE, Ren J, et al. Crystallographic studies of shikimate binding and induced conformational changes in Mycobacterium tuberculosis shikimate kinase. FEBS Lett. 2004;574:49–54. doi: 10.1016/j.febslet.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Krell T, Maclean J, Boam DJ, et al. Biochemical and X-ray crystallographic studies on shikimate kinase: the important structural role of the P-loop lysine. Protein Sci. 2001;10:1137–49. doi: 10.1110/ps.52501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu Y, Reshetnikova L, Li Y, et al. Crystal structure of shikimate kinase from Mycobacterium tuberculosis reveals the dynamic role of the LID domain in catalysis. J Mol Biol. 2002;319:779–89. doi: 10.1016/S0022-2836(02)00339-X. [DOI] [PubMed] [Google Scholar]
- 11.Hartmann MD, Bourenkov GP, Oberschall A, Strizhov N, Bartunik HD. Mechanism of phosphoryl transfer catalyzed by shikimate kinase from Mycobac-terium tuberculosis. J Mol Biol. 2006;364:411–23. doi: 10.1016/j.jmb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Faim LM, Kias MVB, Vasconcelos IG, et al. Crystal structure of shikimate kinase from Mycobacterium tuberculosis in complex with AMP-PNP. Deposited 11/11/2008. Protein Data Bank. doi: 10.2210/pdb3baf/pdb. [DOI] [Google Scholar]
- 13.Dias MV, Faim LM, Vasconcelos IB, et al. Effects of the magnesium and chloride ions of shikimate on the structure of shikimate kinase from Mycobacterium tuberculosis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:1–6. doi: 10.1107/S1744309106046823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otero JM, Garcia-Doval C, Llamas-Saiz AL, et al. Mycobacterium tuberculosis shikimate kinase inhibitors: design and simulation studies of the catalytic turnover. J Am Chem Soc. 2013;135:12366–76. doi: 10.1021/ja405853p. [DOI] [PubMed] [Google Scholar]
- 15.Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–21. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 16.Vianna CP, de Azevedo WF., Jr Identification of new potential Mycobacterium tuberculosis shikimate kinase inhibitors through molecular docking simulations. J Mol Model. 2012;18:755–64. doi: 10.1007/s00894-011-1113-5. [DOI] [PubMed] [Google Scholar]
- 17.Bender A, Glen RC. A discussion of measures of enrichment in virtual screening: comparing the information content of descriptors with increasing levels of sophistication. J Chem Inf Model. 2005;45:1369–75. doi: 10.1021/ci0500177. [DOI] [PubMed] [Google Scholar]
- 18.Miteva MA, Violas S, Montes M, Gomez D, Tuffery P, Villoutreix BO. FAF-drugs: free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006;34(Web Server issue):W738–44. doi: 10.1093/nar/gkl065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 20.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Prot Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 21.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 22.Coracini JD, de Azevedo WF., Jr Shikimate kinase, a protein target for drug design. Curr Med Chem. 2014;21:592–604. doi: 10.2174/09298673113206660299. [DOI] [PubMed] [Google Scholar]
- 23.Kumar M, Verma S, Sharma S, Srinivasan A, Singh TP, Punit K. Structure-based In silico design of a high-affinity dipeptide inhibitor for novel protein drug target shikimate kinase of Mycobacterium tuberculosis. Chem Biol Drug Des. 2010;76:277–84. doi: 10.1111/j.1747-0285.2010.01005.x. [DOI] [PubMed] [Google Scholar]
- 24.Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model. 2003;21:289–307. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- 25.Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP. eHiTS: a new fast, exhaustive flexible ligand docking system. J Mol Graph Model. 2007;26:198–212. doi: 10.1016/j.jmgm.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Segura-Cabrera A, Rodríguez-Pérez MA. Structure-based prediction of Mycobacterium tuberculosis shikimate kinase inhibitors by high-throughput virtual screening. Bioorg Med Chem Lett. 2008;18:3152–7. doi: 10.1016/j.bmcl.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–6. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 28.Sinha S, Rajasulochana P, Ramesh Babu PB, Krishnamoorthy P. Comparative modeling of shikimate kinase (M Tb) and molecular docking studies of its known inhibitors. Res J Pharm Biol Chem Sci. 2013;4:715–20. [Google Scholar]
- 29.Blanco B, Prado V, Lence E, et al. Mycobacterium tuberculosis shikimate kinase inhibitors: design and simulation studies of the catalytic turnover. J Am Chem Soc. 2013;135:12366–76. doi: 10.1021/ja405853p. [DOI] [PubMed] [Google Scholar]
- 30.Cambridge Crystallographic Data Centre (CCDC) CCDC: Cambridge, UK. Gold - Protein ligand binding interactions (binding site prediction/docking) 2014. Available at: http://www.ccdc.cam.ac.uk/products/life_sciencies/gold/
- 31.Mulabagal V, Calderón A. Development of an ultrafiltration-liquid chromatography/mass spectrometry (UF-LC/MS) based ligand-binding assay and an LC/MS based functional assay for Mycobacterium tuberculosis shikimate kinase. Anal Chem. 2010;82:3616–21. doi: 10.1021/ac902849g. [DOI] [PubMed] [Google Scholar]
- 32.Tanramluk D, Schreyer A, Pitt WR, Blundell TL. On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine. Chem Biol Drug Des. 2009;74:16–24. doi: 10.1111/j.1747-0285.2009.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bandodkar BS, Schmitt S. Pyrazolone derivatives for the treatment of tuberculosis. WO/2007/020426 A1. Patent. 2007
- 34.Simithy J, Reeve N, Hobrath JV, Reynolds RC, Calderón AI. Identification of shikimate kinase inhibitors among anti-Mycobacterium tuberculosis compounds by LC-MS. Tuberculosis. 2014;94:152–58. doi: 10.1016/j.tube.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Hsu KC, Cheng WC, Chem YF, et al. Core site-moiety maps reveal inhibitors and binding mechanisms of orthologous proteins by screening compound libraries. PLoS One. 2012;7:e32142. doi: 10.1371/journal.pone.0032142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han C, Zhang J, Chen L, Chen K, Shen X, Jiang H. Discovery of Helicobacter pylori shikimate kinase inhibitors: bioassay and molecular modeling. Bioorg Med Chem. 2007;15:656–62. doi: 10.1016/j.bmc.2006.10.058. [DOI] [PubMed] [Google Scholar]
- 37.Hsu KC, Cheng WC, Chen YF, Wang WC, Yang JM. Pathway-based screening for multitarget inhibitors of diverse proteins in metabolic pathways. PLoS Comput Biol. 2013;9:31003127. doi: 10.1371/journal.pcbi.1003127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang JM, Chen CC. GEMDOCK. Proteins. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]