

Summary
The microbial communities inhabiting the root interior of healthy plants, as well as the rhizosphere, which consists of soil particles firmly attached to roots, engage in symbiotic associations with their host. To investigate the structural and functional diversification among these communities, we employed a combination of 16S rRNA gene profiling and shotgun metagenome analysis of the microbiota associated with wild and domesticated accessions of barley (Hordeum vulgare). Bacterial families Comamonadaceae, Flavobacteriaceae, and Rhizobiaceae dominate the barley root-enriched microbiota. Host genotype has a small, but significant, effect on the diversity of root-associated bacterial communities, possibly representing a footprint of barley domestication. Traits related to pathogenesis, secretion, phage interactions, and nutrient mobilization are enriched in the barley root-associated microbiota. Strikingly, protein families assigned to these same traits showed evidence of positive selection. Our results indicate that the combined action of microbe-microbe and host-microbe interactions drives microbiota differentiation at the root-soil interface.
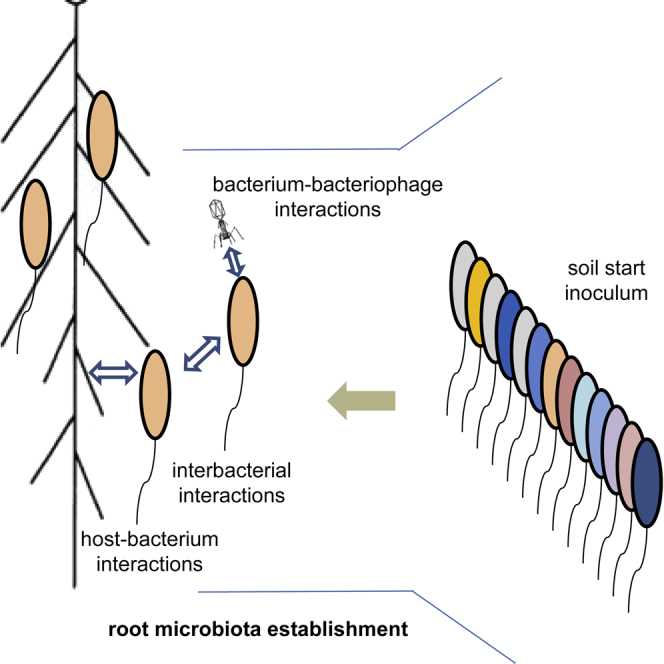
Graphical Abstract
Highlights
-
•
A small number of bacterial families dominate the root-enriched barley microbiota
-
•
The host genotype determines the profile of a subset of community members
-
•
Functions relevant for host interactions are enriched in root-associated taxa
-
•
Genes mediating host, bacteria, and phage interactions show signs of positive selection
Microbial communities inhabiting the root interior and surrounding soil contribute to plant growth. Bulgarelli et al. examine the microbiota that populates the roots of barley (Hordeum vulgare) and present evidence that integrated actions of microbe-microbe and host-microbe interactions drive root microbiota establishment through physiological processes occurring at the root-soil interface.
Introduction
Land plants host rich and diverse microbial communities in the thin layer of soil adhering to the roots, i.e., the rhizosphere, and within the root tissues, designated rhizosphere and root microbiota, respectively (Bulgarelli et al., 2013). Roots secrete a plethora of photosynthesis-derived organic compounds to the rhizosphere (Dakora and Phillips, 2002). This process, known as rhizodeposition, has been proposed as the major mechanism that enables plants to sustain their microbiota (Jones et al., 2009). In turn, members of the rhizosphere and root microbiota provide beneficial services to their host, such as indirect pathogen protection and enhanced mineral acquisition from surrounding soil for plant growth (Bulgarelli et al., 2013; Lugtenberg and Kamilova, 2009). Thus, the dissection of the molecular mechanisms underlying plant-microbe community associations at the root-soil interface will be a crucial step toward the rational exploitation of the microbiota for agricultural purposes. Recent studies performed using the model plant Arabidopsis thaliana revealed that the soil type and, to a minor extent, the host genotype shape root microbiota profiles (Bulgarelli et al., 2012; Lundberg et al., 2012). The structure of the microbial communities thriving at the root-soil interface appears to be resilient to host evolutionary changes, as indicated by a largely conserved composition of the root bacterial microbiota in A. thaliana and related species that spans 35 Ma of divergence within the family Brassicaceae (Schlaeppi et al., 2014). However, it is unclear whether microbiota divergence is greater in host species belonging to other plant families and whether the process of domestication, which gave rise to modern cultivated plants (Abbo et al., 2014) and which cannot be studied in A. thaliana, has left a human footprint of selection on crop-associated microbiota.
Barley (Hordeum vulgare) is the fourth-most cultivated cereal worldwide (Newton et al., 2011) and one of the earliest cereals consumed by humans, with evidence of presence of wild barley (Hordeum vulgare ssp. spontaneum) in human diets dating back to 17,000 BC (Kislev et al., 1992). Barley was one of the first plants subjected to domestication, which culminated ∼10,000 years ago when the cultivation of domesticated barley (Hordeum vulgare ssp. vulgare) began in the Fertile Crescent. Anthropic pressure on barley evolution continued through diversification, which progressively differentiated early domesticated plants into several genetically distinct accessions whose area of cultivation radiated from the Middle East to the rest of the globe (Comadran et al., 2012). Nowadays, wild and cultivated barley accessions still coexist, providing an excellent experimental framework to investigate the structure and the evolution of the microbiota associated with a cultivated plant.
Here, we used an amplicon pyrosequencing survey of the bacterial 16S rRNA gene and combined it with state-of-the-art metagenomics and computational biology approaches to investigate the structure and functions of the bacterial microbiota thriving at the barley root-soil interface. We found evidence for positive selection being exerted on a significant proportion of the proteins encoded by root-associated microbes, with a bias for cellular components mediating microbe-plant and microbe-microbe interactions.
Results
The Structure of the Barley Bacterial Microbiota
We have grown barley accessions in soil substrates collected from a research field located in Golm, near Berlin (Bulgarelli et al., 2012), under controlled environmental conditions (Experimental Procedures). We subjected total DNA preparations from 6 bulk soil, 18 rhizosphere, and 18 root samples to selective amplification of the prokaryotic 16S rRNA gene with PCR primers encompassing the hypervariable regions V5-V6-V7 (Schlaeppi et al., 2014), and we generated 691,822 pyrosequencing reads. After in silico depletion of error-containing sequences, and chimeras as well as sequencing reads assigned to plant mitochondria, we identified 1,374 prokaryotic operational taxonomic units (OTUs) at 97% sequence similarity (Database S1; Experimental Procedures).
Taxonomic classification of the OTU-representative sequences to phylum level highlighted that Actinobacteria, Bacteroidetes, and Proteobacteria largely dominate the barley rhizosphere and root communities, where 88% and 96% of the pyrosequencing reads, respectively, were assigned to these three phyla. Of note, other members of the soil biota, such as Firmicutes and Chloroflexi, were virtually excluded from the plant-associated assemblages (Figure 1). The enrichment of members of the phylum Bacteroidetes significantly discriminated rhizosphere and root samples from bulk soil samples irrespective of the accession tested (moderated t test, false discovery rate-adjusted [FDR], p value < 0.05; Figure 1) At family level, Comamonadaceae, Flavobacteriaceae, and Rhizobiaceae designated a conserved barley microbiota whose enrichment differentiated the rhizosphere and root communities from bulk soil irrespective of the accessions tested (moderated t test, FDR, p < 0.05; Figure 1). Of note, the enrichment of a fourth family, Oxalobacteraceae, also significantly discriminated between root samples and unplanted soil in wild, landrace, and modern accessions (moderated t test, FDR < 0.05; Figure 1). Taken together, these results highlight a shift in community composition at the barley root-soil interface, which progressively differentiated the rhizosphere and root bacterial assemblages from the surrounding soil biota.
Figure 1.

The Barley Rhizosphere and Root Microbiota Are Gated Communities
Average relative abundance (RA ± SEM) of the five most abundant (A) phyla and (B) families in soil, rhizosphere, and root samples as revealed by the 16S rRNA gene ribotyping. For each sample type, the number of replicates is n = 6. Stars indicate significant enrichment (FDR, p < 0.05) in the rhizosphere and root samples compared to bulk soil. Vertical lines denote a simultaneous enrichment of the given taxa in all three barley accessions. Only taxa with a RA > 0.5% in at least one sample were included in the analysis.
To gain insights into the richness of the barley microbiota we compared the total number of observed OTUs, Chao1, and the Shannon diversity indices of the communities retrieved from bulk soil and plant-associated microhabitats. All the indices revealed a significant reduction of the bacterial richness and diversity in the root samples (TukeyHSD, p < 0.05; Figure S1), while the rhizosphere microbiota displayed an intermediate composition between soil and root samples (Figure S1).
To elucidate whether the composition of the bacterial communities correlated or was independent of the sample type and the host genotype, we used the OTU count data to construct dissimilarity matrices with the UniFrac (Lozupone et al., 2011) and Bray-Curtis metrics. We applied a previously used relative abundances threshold (0.5%; Bulgarelli et al., 2012) to focus our analysis on PCR-reproducible OTUs. Permutational multivariate ANOVA based on distance matrices (ADONIS) revealed a marked contribution of the microhabitat (Bray-Curtis R2 = 0.11584; R2 Unweighted Unifrac R2 = 0.08851, p < 0.05) as well as phylogenetic-dependent contributions of the host genotype to the composition of the barley microbiota (Weighted Unifrac R2 = 0.24427; R2 Unweighted Unifrac R2 = 0.15262, p < 0.05). We used a canonical analysis of principal coordinates (CAP; Anderson and Willis, 2003) to better quantify the influence of these factors on the beta diversity. CAP analysis constrained by the environmental variables of interest revealed that the microhabitat explained 22% of the variance (p < 0.005; 95% confidence interval = 17%, 30%). Consistently, we observed a clear separation between plant-associated microhabitats and bulk soil samples followed by segregation of the rhizosphere and root samples (Figure 2A).
Figure 2.

Constrained Principal Coordinate Analysis on the Soil and Barley Bacterial Microbiota
(A) Variation between samples in Bray-Curtis distances constrained by microhabitat (22% of the overall variance; p < 5.00E−2) and (B) by accession (5.7% of the overall variance; p < 5.00E−2). In both panels, triangles correspond to rhizosphere and circles to root samples. The percentage of variation explained by each axis refers to the fraction of the total variance of the data explained by the constrained factor. In (B) soil samples were not included.
The host genotype alone could explain 5.7% of the overall variance of the data, and the constrained ordination showed a clear clustering of the samples corresponding to the wild, landrace, and modern accessions (Figure 2B). This proportion of the variation, albeit small, was found significant by permutation-based ANOVA (p < 0.005; Figure 2). Further exploration of these analyses revealed that the OTUs with the largest contribution to both constrained ordinations had a distinct taxonomic membership, mostly belonging to the phyla Proteobacteria and Bacteroidetes, and could explain most of the observed variation among microhabitats and genotypes (Figure S2A). Bootstrapping analysis of the constrained ordination (Experimental Procedures) indicated that the significance of the observed genotype effect could not be attributed to any individual OTUs. Only after randomly permuting the abundances of the 83 OTUs with the largest contribution (72.23% and 65.67% of the root and rhizosphere communities, respectively), the statistical significance was lost (Figure S2C). Consistently, CAP analyses generated using weighted UniFrac distance matrix, sensitive to OTU phylogenetic affiliations and OTU relative abundances, further supported the observed differentiation of the barley microbiota (Figure S2B). However, transformations based on unweighted UniFrac distance, which is sensitive to unique taxa, but not to OTU relative abundances, showed a drastic reduction of the variance explained by the microhabitat and failed to identify a significant host-genotype-dependent effect on the barley microbiota (Figure S2B). Together, these results further support the hypothesis that the barley rhizosphere and root are two microhabitats colonized by communities with taxonomically distinct profiles, which emerge from the soil biota through progressive differentiation.
To identify bacteria responsible for the diversification between the two root-associated microhabitats we employed a linear model analysis (Supplemental Experimental Procedures) to determine bacterial OTUs significantly enriched in root and rhizosphere compared to unplanted soil. With this approach we identified three distinct bacterial sub-communities thriving at the root-soil interface (Figure 3; Database S1). One sub-community, designated Root_OTUs, was defined by bacteria significantly enriched in the root samples and discriminating this sample type from bulk soil. Root_OTUs accounted for the largest fraction of the bacteria enriched in the barley microbiota in the wild and modern accessions (Database S1). A second sub-community was defined by bacteria enriched in both the rhizosphere and root samples and discriminating these samples from the bulk soil. This second sub-community, designated RR_OTUs, represented the largest fraction of the barley microbiota retrieved from the landrace accession (Database S1). Finally, a third sub-community defined by the bacteria discriminating the rhizosphere samples from bulk soil was identified. This sub-community, designated Rhizo_OTUs, represented the minor fraction of the barley microbiota irrespective of the accession tested (Database S1). Consistent with the constrained ordinations, taxonomic affiliations of the OTU-representative sequences assigned to RR_OTUs and Root_OTUs were largely represented by Bacteroidetes and Proteobacteria members (Database S1). We previously demonstrated that the root microbiota of the model plant Arabidopsis thaliana is dominated by members of Actinobacteria, Bacteroidetes, and Proteobacteria (Bulgarelli et al., 2012). We took advantage of the similar experimental platform used for the barley and Arabidopsis surveys, including the same soil type, to compare the bacterial microbiota retrieved from these monocotyledonous and dicotyledonous hosts. First, we re-processed the A. thaliana data set using exactly the same analysis pipeline we employed in the present study. Taxonomic classification using the representative sequences of the OTUs enriched in the root microbiota of barley and A. thaliana (Figure 4) revealed a similar taxonomic composition, with few bacterial taxa belonging to a limited number of bacterial families from different phyla, including members of Comamonadaceae, Flavobacteriaceae, Oxalobacteraceae, Rhizobiaceae, and Xanthomonadaceae. Notably, this analysis also revealed clear differences between the two host species. In particular, the enrichment in root samples of the families Pseudomonadaceae, Streptomycetaceae, and Thermomonosporaceae differentiated the Arabidopsis root-associated communities from barley. Conversely, the enrichment of members of the Microbacteriaceae family appears to be a distinctive feature of the barley root microbiota in the tested conditions. Excluding these qualitative differences, we found a very high correlation between the two sub-communities (0.90 Pearson correlation coefficient, p = 0.005).
Figure 3.

OTU Enrichment at the Barley Root/Soil Interface
Ternary plots of all OTUs detected in the data set with RA > 0.5% in at least one sample in (A) Hordeum vulgare ssp. spontaneum, (B) H. vulgare ssp. vulgare Landrace, and (C) H. vulgare ssp. vulgare Modern. Each circle represents one OTU. The size of each circle represents its relative abundance (weighted average). The position of each circle is determined by the contribution of the indicated compartments to the total relative abundance. Dark blue circles mark OTUs significantly enriched in the root microhabitat (Root_OTUs, FDR, p < 0.05), magenta circles mark OTUs significantly enriched in the rhizosphere microhabitat (Rhizo_OTUs, FDR, p < 0.05), and cyan circles mark OTUs significantly enriched in both microhabitats (RR OTUs, FDR, p < 0.05).
Figure 4.

Taxonomic Representation of the Barley and Arabidopsis Root-Enriched Bacterial Taxa
The tree represents a subset of the NCBI taxonomy containing all OTUs found to be enriched in the barley and Arabidopsis root samples with respect to soil. The branches of the tree do not reflect evolutionary distances. The position of the dots corresponds to the taxonomic placement of each OTU-representative sequence in the taxonomy. The size of the dots illustrates the aggregated relative abundance of all OTUs assigned to a given taxon (log scale). OTUs enriched in Arabidopsis roots are depicted in red, whereas Barley root OTUs are shown in blue. Note that the relative abundance of each subset of root-enriched taxa with respect to its respective root community varies (Barley root OTUs, 45.44%; Arabidopsis root enriched OTUs, 59.02%).
The Barley Rhizosphere Microbiome
To gain further insights into the significance of the marked barley rhizosphere effect detected by the 16S rRNA gene survey, we reasoned that, unlike roots, where DNA is mostly plant derived, DNA isolated from the rhizosphere should mainly originate from microbes, and we used the same rhizosphere DNA preparations for independent Illumina shotgun sequencing. We obtained two metagenome samples per host genotype, each corresponding to a different soil batch (Table S2) and generated an average of 75 million 100-bp paired-end reads per sample, adding up to a total of 44.90 Gb of sequence data. We then assembled the filtered reads of each sample independently using SOAPdenovo (Heger and Holm, 2000; Experimental Procedures). Despite the heterogeneity of the data, an average of 69.85% of the reads per sample were assembled into contigs (Table S2).
The partially assembled metagenome sequences (including unassembled singleton reads) were taxonomically classified with taxator-tk (Dröge et al., 2014), a tool for the taxonomic assignment of shotgun metagenomes (Experimental Procedures). Relative abundances were calculated by mapping the reads back to the assembled contigs and determining the number of reads assigned to each taxon. In total, 27.35% of all reads were assigned at least to the domain level. Of those, 94.04% and 0.054% corresponded to Bacteria and Archaea, respectively, and 5.90% to Eukaryotes (Database S1).
Comparison of SSU rRNA Genes and Metagenome Taxonomic Abundance Estimates
The availability of barley rhizosphere 16S rRNA gene amplicon and shotgun metagenome data provided an opportunity to compare both data sets. Toward this end, we classified the OTU-representative sequences onto the NCBI reference database (Sayers et al., 2009). This allowed us to cross-reference the relative abundances of each taxonomic bin from the rhizosphere metagenome with each OTU from the 16S rRNA gene analysis using the NCBI taxonomy and to directly compare the results of the two approaches (Figure 5). The analysis of the metagenome samples revealed the presence of Archaea (0.058% relative abundance) in the rhizosphere microhabitat, as well as members of bacterial phyla whose presence we did not detect in our 16S rRNA gene analysis, such as the Cyanobacteria (0.024% relative abundance). Our results also indicated an overrepresentation for Beta- and Gammaproteobacteria in the 16S rRNA gene taxonomic profiling, representing 10.12% and 9.64% of the whole community, respectively, compared with 7.73% and 5.50% as found in the metagenome samples. These quantitative differences can be at least partially attributed to the fact that Beta- and Gammaproteobacteria possess multiple ribosomal RNA operon copies (Case et al., 2007). The observed differences in detected taxa can furthermore be explained by known biases of 16S rRNA gene primers, in particular, the 799F primer was designed to avoid contamination from chloroplast 16S sequences, a side effect of which is a strong bias against Cyanobacteria (Chelius and Triplett, 2001).
Figure 5.

Comparison of 16S rRNA Amplicon and Metagenome Abundances
The tree represents the NCBI taxonomy for all taxonomically classified OTUs from the rhizosphere samples of the 16S rRNA survey as well as all metagenome bins, resolved down to the order rank. The branches of the tree do not reflect evolutionary distances. The position of the dots in the tree corresponds to the taxonomic placement of the representative sequences in the NCBI taxonomy. The size of the dots illustrates the average relative abundances per sample of each taxa (log scale). Blue dots represent abundances as found in the shotgun metagenome classification, red dots correspond to abundances from the 16S rRNA amplicon data, and green depicts an overlap.
We further assessed the variability in abundance estimates for bacterial taxa which could be detected in both analyses (excluding Cyanobacteria) and found several discrepancies, despite the overall high correlation (0.86 Pearson coefficient; p < 1.75E−12). The largest differences were found in taxonomic groups for which 16S rRNA gene pyrotagging was reported to be either biased or lacking in resolution, due to either copy number variation or primer biases, especially for soil bacteria belonging to Chloroflexi, Deltaproteobacteria, and Bacteroidetes (Hong et al., 2009; Klindworth et al., 2013).
The taxonomic classification of fragments of 16S rRNA genes found in the metagenome shotgun reads allowed us to calculate the relative abundances of bacterial taxa not affected by primer biases. We found a high correlation between the results obtained for the two different 16S rRNA gene data sets (Figure 5; 0.89 Pearson correlation coefficient; p < 21.55E−14), indicating that the negative impact of the 799F primer bias on the beta-diversity estimates for the barley rhizosphere is only marginal, further validating the results reported above.
We also retrieved and analyzed 18S rRNA sequences following the same approach, which allowed us to compare eukaryotic and bacterial abundances in a quantifiable way. We found an increase in the relative abundance of eukaryotes (11.06%) when comparing 16S and 18S sequences relative to the estimate obtained from taxonomically classifying the metagenome sequences (5.90%), which could be partially explained by the high number and variability of rRNA operon copy number in eukaryotes (Amaral-Zettler et al., 2009). Furthermore, we were able to characterize the relative abundances of the major taxonomic groups found in the rhizosphere (Figure S3), revealing that fungi constitute the most abundant eukaryotic phylum in the barley rhizosphere (33.31% of all Eukaryotes).
Enrichment of Biological Functions in Root- and Rhizosphere-Associated Bacterial Taxa
The 16S rRNA gene survey revealed a clear dichotomy between the taxonomic composition of soil and root bacterial communities, a differentiation which, in barley, starts in the rhizosphere. Furthermore, a large fraction of bacterial taxa enriched in roots (Root_OTUs) was also enriched in the rhizosphere relative to unplanted soil (designated RR_OTUs). To determine if this differentiation process is linked to specific biological functions, we identified and annotated protein coding sequences (Experimental Procedures) and tested whether particular biological traits were significantly enriched in family-level taxonomic bins corresponding to RR_OTUs (containing 29.51% of all annotated protein coding sequences) with respect to soil-associated bins, i.e., bins corresponding to OTUs which were not enriched in the root or in the rhizosphere (57.86% of the annotated sequences). Genes found in contigs that could not be taxonomically assigned, as well as those assigned to Cyanobacteria (12.81% of the total), were not included in this analysis.
We identified 12 functional categories which were significantly enriched in root and rhizosphere bacterial taxa (Table 1). These correspond to traits likely important for the survival or adaptation in the root-associated microhabitats, such as adhesion, stress response, and secretion. Importantly, categories relating to host-pathogen interactions (type III secretion system T3SS, regulation of virulence, invasion, and intracellular resistance) as well as microbe-microbe interactions (type VI secretion system; T6SS) and microbe-phage interactions (transposable elements, bacteriophage integration) were also significantly enriched. Interestingly, root- and rhizosphere-associated taxa were also significantly enriched in protein families related to iron mobilization (siderophore production) and sugar transport (sugar phosphotransferase systems).
Table 1.
Biological Functions in Root- and Rhizosphere-Associated Bacterial Taxa
| Functional Category | p Valuea |
|---|---|
| Protein secretion system type III | 0.0013 |
| Adhesion | 0.0014 |
| Regulation of virulence | 0.0024 |
| Siderophores | 0.0024 |
| Secretion | 0.0072 |
| Transposable elements | 0.0177 |
| Periplasmic stress | 0.0188 |
| Sugar phosphotransferase systems | 0.0251 |
| Bacteriophage integration excision lysogeny | 0.0346 |
| Invasion and intracellular resistance | 0.0346 |
| Protein secretion system type VI | 0.0379 |
| Detoxification | 0.0379 |
Functional categories significantly enriched in taxonomic bins corresponding to RR_OTUs found in the barley rhizosphere metagenome.
Calculated using a Mann-Whitney test, controlling for false discovery rate (FDR).
To further assess the ecological significance of these functional enrichments, we performed a comparison with functional representation in sequenced isolates. We retrieved and analyzed 1,233 genomes from the NCBI database (Experimental Procedures; Supplemental Information) belonging to the soil- and root-associated bacterial taxa found in the barley rhizosphere and performed the same enrichment tests. We found only one functional category to be significantly enriched in the root-associated taxa with respect to the soil background taxa, namely, the T3SS (p = 0.044).
Positive Selection in the Barley Rhizosphere
To gain further insights on the molecular mechanisms driving the functional diversification of the barley rhizosphere microbiota, the gene families identified in the assembled barley metagenome were annotated based on matches to TIGRFAM (Haft et al., 2013) hidden Markov models (HMMs; Experimental Procedures), and we calculated, for each TIGRFAM, the ratio between the number of nonsynonymous (Dn) and synonymous (Ds) changes, a proxy for evolutionary pressure. Our analyses showed that 9% of the gene families had on average significantly higher Dn values and lower Ds values than the mean value calculated over all annotated sequences (one-sided Fisher test, FDR < 0.05), suggesting that they have been under positive (diversifying) selection. Interestingly, a closer investigation of these gene families revealed that positive selection signatures markedly characterize diverse proteins involved in pathogen-host interactions, including bacterial secretion, as well as proteins essential for phage defense (Figures 6A and S5). Strikingly, these proteins encode for a subset of the functions enriched in RR_OTUs and Root_OTUs (Table 1). Furthermore, we determined that 10.66% (115) of protein families encoded by the barley metagenome displayed a Dn/Ds ratio significantly greater than the metagenome mean Dn/Ds value in at least one of the barley genotypes tested (Table S3).
Figure 6.

Proteins under Selection in the Barley Rhizosphere Microbiome
(A) Top-ranking protein families under positive selection with significantly increased Dn/Ds statistic. The distribution at the top shows the density function over all protein families smoothed with a Gaussian kernel function. The green bar indicates the average ∼Dn/Ds over all the samples, the blue bar the average ∼Dn/Ds for all TIGFRAMS annotated with the term “patho” and/or “secretion.” The boxplot shows the distribution of the ∼Dn/Ds across all samples for the top 50 ranked TIGRFAM families under positive selection, with families sorted by their median ∼Dn/Ds in descending order. TIGRFAMs annotated with “repeat” or with a mean repetitive value of more than 50% were discarded.
(B) Sequence clusters of residues under positive selection in selected protein families. Top: dots indicate ∼Dn/Ds for a given position in the protein sequence, and their color corresponds to the proportion of gaps in the multiple sequence alignment (MSA). Gray-shaded areas indicate significant clusters of residues under positive selection. Gray-shaded horizontal lines indicate repetitive elements. Bottom: Jensen-Shannon divergence as a function of the positions in the MSA.
Of note, we identified significant signs of positive selection for a component of the T3SS, which is found in most Gram-negative bacteria and is used to suppress plant immune responses (Cornelis and Van Gijsegem, 2000; Table S6). Our findings are in line with previous studies, which reported evidence of positive selection for T3SS components in the bacterial phytopathogens Pseudomonas syringae (Guttman et al., 2006) and Xanthomonas campestris (Weber and Koebnik, 2006). Furthermore, we detected positive selection for components of the T6SS, a contact-dependent transport system mediating microbe-microbe interactions (Table S4; Russell et al., 2014). In particular, we found the forkhead-associated (FHA) domain to be under strong positive selection. This domain is a phosphopeptide recognition domain embedded in diverse bacterial regulatory proteins, which control various cellular processes including pathogenic and symbiotic interactions (Durocher and Jackson, 2002).
Microbial Elicitors and Effectors of Plant Immunity under Positive Selection
One branch of the plant immune system recognizes and is activated by a variety of evolutionary conserved microbial epitopes, designated microbe-associated molecular patterns (MAMPs) (Boller and Felix, 2009). The co-evolutionary arms race between the plant host and microbial pathogens leads to reciprocal selective pressure for the interacting proteins to change. To avoid activation of plant defenses, phytopathogens have evolved different mechanisms such as the diversifying evolution of elicitor epitopes by mutation or reassortment, and the injection of strain-specific pathogen effector proteins into host cells to intercept intracellular immune signaling (Shames and Finlay, 2012).
To identify putative elicitors of plant immune responses at the root-soil interface, we searched for genes that contained clusters of residues under positive selection using a sliding window approach (Figure 6B; Experimental Procedures). A total of 56 putative elicitors of plant immune responses were previously identified in the genomes of six plant pathogenic and a soil-dwelling bacterium using a similar approach (McCann et al., 2012). Remarkably, we found a semantic overlap of nine protein families under selection in the barley rhizosphere microbiome (Table S5). For example, the GGDEF domain, a previously reported putative bacterial elicitor, essential for motility and biofilm formation (Simm et al., 2004), was under positive selection in the rhizosphere of the wild accession (p = 0.027). Of the protein families that had a Dn/Ds ratio significantly higher than the mean, 85.3% had such clusters, whereas they were found in only 34.9% of all detected protein families (p < 2.2 E−16, one-sided Fisher’s exact test). On average, we found 0.66 ± 1.54 (SD) clusters for each protein family, which spanned 4.0% ± 7.9% (SD) of their amino acid sequence among all families. For the protein families already shown to exhibit significant signatures of positive selection, an average of 6.7 ± 9.0 (SD) clusters were detected.
Furthermore, we identified by de novo prediction 16 putative polymorphic type III secreted effector proteins (T3SEs), of which 30% were under positive selection (Experimental Procedures; Table S6). In addition, 31.5% of these candidate effector proteins contained an average of 5.2 ± 9.8 (SD) clusters of residues under positive selection. This shows that, in the barley rhizosphere microbiota, highly polymorphic bacterial protein families, some of which are known to function in the suppression of plant immune responses, have similar footprints of positive selection as the evolutionary conserved MAMPs (McCann et al., 2012).
Positive Selection Acting on Phages and CRISPR Systems
Interestingly, in our Dn/Ds analysis we found that endoribonuclease gene cas2 was under strong positive selection. This gene is associated with the clustered, regularly interspaced short palindromic repeat (CRISPR) system, a defense mechanism composed of an array of repeats with dyad symmetry separated by spacer sequences, which, together with a set of CRISPR-associated (CAS) genes, provides protection against phages in Bacteria and Archaea (Westra et al., 2014). In particular, Cas2 participates in the acquisition of new spacers (Barrangou et al., 2007), indicating that the ability to develop resistance to new phages might be an important trait for the bacterial community of the barley rhizosphere (Figure 6B). The enrichment of functional categories related to interactions with bacterial phages in RR_OTUs (Table 1) further supports this notion. In addition, we found that the coding sequences of bacteriophage tail and head morphogenesis genes were under positive selection. The phage tail serves as a channel for the delivery of the phage DNA from the phage head into the cytoplasm of the bacteria. Thus, interactions between bacteria and their phages might have contributed to the positive selection on both the CRISPR-cas adaptive immune system of bacteria and on a subset of the bacteriophage proteins observed in the barley rhizosphere.
Discussion
Here, we characterized the rhizosphere and the root microbiota of soil-grown wild, traditional, and modern accessions of barley using a pyrosequencing survey of the 16S rRNA gene. This revealed that the enrichment of members of the families Comamonadaceae, Flavobacteriaceae, and Rhizobiaceae and the virtual exclusion of members of the phyla Firmicutes and Chloroflexi differentiate rhizosphere and root assemblages from the surrounding soil biota. This microbiota diversification begins in the rhizosphere, where a marked initial community shift occurs, and continues in the root tissues by additional differentiation, leading to the establishment of a community inside roots, which is more distinct from the surrounding soil biota.
A comparison to the root and rhizosphere microbial assemblages retrieved from the distantly related dicotyledonous plants Arabidopsis thaliana and A. thaliana relatives (Bulgarelli et al., 2012; Lundberg et al., 2012; Schlaeppi et al., 2014) revealed both striking differences as well as common features. First, we detected in each of the three tested barley genotypes a marked “rhizosphere effect,” i.e., a structural and phylogenetic diversification of this microhabitat from the surrounding soil biota (Figure 3), which we failed to detect in previous studies of A. thaliana and A. thaliana relatives (Bulgarelli et al., 2012; Schlaeppi et al., 2014). Second, taxonomic classification using the representative sequences of the OTUs enriched in the root microbiota of monocotyledonous barley and dicotyledonous A. thaliana, grown in the same soil type, revealed a similar enrichment pattern, although some clear differences were identified (Figure 4). On the basis of our study, the enrichment of members of the families Pseudomonadaceae, Streptomycetaceae, and Thermomonosporacea in root samples of Arabidopsis is not seen in barley. Consistently, recent cultivation-independent surveys of the rhizosphere of field-grown maize (Peiffer et al., 2013) and wheat (Turner et al., 2013), two grasses like barley, also revealed almost no enrichment of the aforementioned two actinobacterial taxa. By contrast, enrichment of members of the Microbacteriaceae family appears to be a distinct feature of the barley root microbiota. This suggests the existence of host lineage-specific molecular cues contributing to the differentiation of the root-associated microbiota from the surrounding soil type-dependent bacterial start inoculum. However, the overall conserved microbiota composition in the roots of the monocot barley and the dicot Arabidopsis, which diverged ∼200 Ma, could be indicative of an ancient plant trait that preceded the emergence of flowering plants. Alternatively, but not mutually exclusive, the conserved microbiota composition might indicate that microbe-microbe interactions serve as a dominant structuring force of the root microbiota in flowering plants.
Our results revealed also a host-genotype-dependent stratification of both the barley root and rhizosphere microbiota (Figure 2B). The host influence on the microbiota profiles is limited, since ∼5.7% of the variance can be explained by the factor host genotype and is entirely quantitative. Notably, the host genotype effect is manifested by variations in the abundance of many OTUs from diverse phyla, rather than by single OTUs. Re-analysis of root microbiota abundance data from three A. thaliana ecotypes (Schlaeppi et al., 2014), generated with the same 16S rRNA gene primers and using the same computational approach, failed to detect a significant ecotype-dependent effect. By contrast, our results from barley are congruent with a recent investigation of the rhizosphere microbiota of 27 field-grown modern maize inbreds (Peiffer et al., 2013). This study reported a similar proportion of variation attributed to the host genotype (5.0%–7.7% using unweighted or weighted UniFrac distances, respectively) and also a lack of individual bacterial taxa predictive for a given host genotype. Bouffaud and co-workers reported a stratification of the maize rhizosphere microbiota reflecting the major genetic groups emerged during maize diversification, rather than their genetic distance (Bouffaud et al., 2012). These results concur with our findings of accession-dependent microbiota differentiation (Figure 2B) owing to the fact that the tested wild, landrace, and modern accessions represent three distinct phases of the domestication and diversification history of barley (Meyer et al., 2012).
The availability of barley rhizosphere microbiome sequences prompted us to compare the taxonomic classification generated by shotgun DNA sequencing without PCR amplification with the 16S rRNA gene amplicon profiles. This allowed us to determine the presence of microorganisms whose presence cannot be estimated using the 16S rRNA gene primers we have adopted, such as Protists, Fungi, and Archaea. Furthermore, the use of assembly as an intermediate step to improve taxonomic classification of reads and abundance estimates is likely to introduce biases which are not fully understood. In order to assess this effect we retrieved marker genes from the unassembled metagenome reads to be analyzed and used as a control. Correlation tests between the abundance estimates for bacterial taxa obtained with the two methods (0.86 Pearson correlation coefficient; p < 1.75E−12) indicated that known 16S primer biases, differential ribosomal operon copy number, as well as assembly biases have a minor, but notable, impact on the analysis of beta-diversity, further underlining the importance of using complementary methods for the study of microbial diversity.
Strikingly, we found that Bacteria dominate the annotated barley rhizosphere, whereas the relative abundance of Eukaryotes accounted for only a small fraction. A recent study employing metatranscriptomics to estimate microbial abundances reported a 5-fold higher abundance of Eukaryotes in the oat and pea rhizosphere (16.6% and 20.7%, respectively) compared to that of wheat (3.3%) (Turner et al., 2013). However, since both metatranscriptome and metagenome abundance estimates are based on taxonomic classification using a reference-based method, database-related biases likely play a role in this apparent skew in the community in favor of bacterial taxa. Analysis of 18S rRNA sequences found in the shotgun reads revealed an increased relative abundance of Eukaryotes compared to the results obtained for the metagenome data (11.06% and 5.9%, respectively). However, given the large variation in rRNA operon copy number in eukaryotic genomes, abundance estimates based on 18S read counts are likely to be inflated. We conclude that further studies, combining alternative markers such as the 18S rRNA gene or internal transcribed spacers (ITSs), targeting broader microbial communities (e.g., Fungi and Oomycetes), are needed to better estimate the phylogenetic composition of the microbiota thriving at the root-soil interface.
Combining our findings from the 16S rRNA gene survey, i.e., that some bacterial taxa are significantly enriched in root and rhizosphere samples with respect to soil (RR_OTUs), together with the functional analyses of the rhizosphere metagenome, we were able to map functions to root- and soil-associated taxa. Functional categories significantly enriched in root and rhizosphere (Table 1) corresponded to important traits for the survival and adaptation in these microhabitats, as well as traits related to microbe-microbe interactions and microbe-phage interactions. Importantly, several functions appeared to be relevant for interactions with the host (pathogenic as well as mutualistic), such as the T3SS, regulation of virulence, siderophore production, sugar transport, secretion, invasion, and intracellular resistance, further supporting the hypothesis that the presence of the host plant triggers a functional diversification in the rhizosphere. This is congruent with the observations that plants, through the release of photosynthesis-derived organic compounds into soil (Dakora and Phillips, 2002), can modify the physical, chemical, and biological properties of the rhizosphere to enhance the acquisition of important resources such as water and minerals (McCully, 1999). The growth of barley, like other graminaceous monocotyledons, relies on the secretion and subsequent reuptake of iron-chelating phytosiderophores for the acquisition of scarcely mobile iron ions from soil (Jeong and Guerinot, 2009). Therefore, the observed enrichment of bacterium-derived siderophores in the barley-associated microbial communities indicates that the combined action of microbiota- and host-derived siderophores maximizes the mobilization and bioavailability of the soil-borne iron micronutrient in the rhizosphere.
Out of the 12 categories found to be significantly enriched in the root-associated metagenome bins, only the T3SS was also detected as enriched when we analyzed sequenced isolates. This suggests that the T3SS is a relevant feature of root-associated bacterial taxa in general, whereas the remaining enriched functions detected only by analysis of the metagenome data (Table 1) could correspond to environment-specific features.
Analyzing the coding sequences found in the metagenome data, we observed strong positive selection in proteins that are known to directly interact with the plant host, such as the bacterial T3SS and other outer surface proteins, which might be related to plant-pathogen interactions and secretion (Figure 6). These signs of positive selection are evidence of plant-microbe co-evolution in the rhizosphere and suggest that host-microbe and microbe-microbe interactions exist in these natural community systems that are reminiscent of the arms race co-evolution model established for binary plant-pathogen interactions. Thus, our findings predict that the innate immune system of plants contributes to the selection of bacterial community structure as early as at the root-soil interface. Interestingly, it has been recently noted that balanced polymorphism of resistance genes in A. thaliana is maintained in the population through complex community-wide interactions encompassing many pathogen species (Karasov et al., 2014). The substantial number of protein families and the overall scale of positive selection which we identified indicate that metagenomic data are a sensitive tool for studying microevolution within natural environments. However, caution must be exercised when interpreting signatures of positive selection in this context, where the interplay between numerous species, including pathogens, mutualists, and commensals, creates a much more complex system than described by current models of co-evolution.
Previous comparative genomic studies of bacterial CAS genes surprisingly indicated no signs of positive selection, which was attributed to the additional roles of these genes in transcriptional regulation (Takeuchi et al., 2012). A high SNP density, indicative of positive selection, was also found for the CAS proteins csy1 and cse2 in metagenome samples of human gut microbiomes (Schloissnig et al., 2013). The strong signs of positive selection that cas2, one of the three essential proteins of the CRISPR system, exhibited in the barley rhizosphere, along with the positive selection identified for a subset of phage proteins, indicates that natural community systems might allow a more sensitive detection of such effects compared to comparative studies of a relatively small number of isolates. The role of the cas2 gene in the acquisition of resistance to new phages might be of particular importance in a metabolically active and proliferating bacterial community, such as the rhizosphere microbiota (Ofek et al., 2014), which represents an ideal substrate for bacteriophage infections. Alternatively, the cas2 gene product could be an elicitor of MAMP-triggered immunity in the host, which preferentially targets indispensable, evolutionary conserved, and broadly distributed microbial epitopes, such as flagellin or EF-Tu (McCann et al., 2012). Thus, the positive selection on CAS genes might simultaneously reflect the pressure exerted by bacteriophages and the host on members of the root-associated microbiota.
The observed overlap of bacterial traits under diversifying selection in the rhizosphere and those found to be significantly enriched in RR_OTUs provides direct and independent evidence for the contribution of host-microbe interactions in the selection of the root-associated bacterial microbiota from the surrounding soil biota (e.g., T3SS, virulence regulation and pathogenicity, siderophore production, sugar uptake). Our findings imply that the host innate immune system as well as the supply and demand of functions of root metabolism are relevant host factors for bacterial recruitment. In addition, both the analysis of the metagenome data (e.g., enrichment of T6SS) and the existence of a largely conserved phylogenetic pattern in the root-enriched bacterial taxa in barley and A. thaliana (Figure 4) imply that microbe-microbe interactions are also a driving force in the taxonomic differentiation of the root-associated bacterial assemblages. Thus, collectively, our results point toward a model in which the integrated action of microbe-microbe and host-microbe interactions drives root microbiota establishment through specific physiological processes from the surrounding soil biota.
Experimental Procedures
Experimental Design
Surface-sterilized seeds of barley genotypes Morex, Rum, and HID369 were sown onto pots filled with experimental soil collected at the Max Planck Institute of Molecular Plant Physiology, Potsdam, in September 2010 and September 2011. For each accession we organized three biological replicates and repeated the entire experiment using two different samplings of soil substrate (Table S1). At early stem elongation we excavated the plants from the soil and detached the root systems from the stems. We employed a combination of washing and ultrasound treatments to simultaneously separate the rhizosphere fraction from the roots and enrich for root endophytes. In parallel, bulk soil controls, i.e., pots filled with the same soil and exposed to the same environmental conditions as the plant-containing pots, were processed.
16S Data Analysis
16S rRNA gene sequences were subjected to demultiplexing, quality filtering, dereplication, abundance sorting, OTU clustering, and chimera identification using UPARSE pipeline (Edgar, 2013). Briefly, after removal of barcode and primer sequences, reads were truncated to a length of 290 bp, and only reads with a quality score Q > 15 and no ambiguous bases were retained for the analysis. Chimeras were identified using the “gold” reference database (http://drive5.com/uchime/gold.fa), and OTUs were defined at 97% sequence identity. OTU-representative sequences were taxonomically classified using the RDP classifier (Wang et al., 2007) trained on the Greengenes reference database. The resulting OTU table was used to determine taxonomic relative abundances and subsequent statistical analyses of alpha- and beta-diversity (see Supplemental Experimental Procedures).
Metagenome Data Analysis
Paired-end Illumina reads were subjected to trimming, filtering, and quality control using a combination of custom scripts and the CLC Workbench v5.5.1 and assembled using SOAPdenovo (Heger and Holm, 2000). A small fraction of the partially assembled metagenome samples (on average 3.02% of the reads) was mapped to the annotated barley genomic sequences, and the corresponding contigs or singleton reads were removed (Table S2; Supplemental Experimental Procedures). We used taxator-tk (Dröge et al., 2014) to taxonomically classify the partially assembled metagenome sequences (including unassembled singleton reads) using the NCBI database as a reference. Coding sequences were predicted using MetaGeneMark (Zhu et al., 2010) and annotated using matches to HMM (HMMER v3.0) profiles to the TIGRFAM (Haft et al., 2013) and PFAM (Punta et al., 2012) databases as well as a k-mer-based matching using the SEED (Edwards et al., 2012) API and server scripts. To test for a significant enrichment of functional categories in the root-associated bins relative to the remaining bins, we assumed a correspondence at the family level between metagenome bins and root- and rhizosphere-enriched OTUs (RR_OTUs) of these families found in the amplicon survey. To search for signatures of positive selection we first employed HMMER to obtain multiple sequence alignments (MSAs) of orthologous sequences found in the metagenome samples. From each MSA, we calculated neighbor-joining trees and used them to infer Ds and Dn changes. Clusters of residues with significant signs of positive selection were calculated using a sliding window approach. A detailed description of the methods and tools used for the analysis of the metagenome is available in the Supplemental Experimental Procedures.
Author Contributions
D.B. and P.S.-L. conceived of and designed the experiments. D.B. performed the experiments. D.B. and R.G.-O. analyzed the pyrosequencing data. R.G.-O., P.C.M., J.D., A.W., Y.P., and A.C.M. conceived of and performed the metagenomics analysis. D.B., R.G.-O., P.C.M., A.C.M., and P.S.-L. wrote the paper.
Acknowledgments
We thank Isa Will and Maren Winnacker for their excellent technical assistance during the preparation of metagenomic DNA samples, Dr. Bruno Huettel and Diana Kuehn (Max Planck Genome Centre Cologne) for the preparation and sequencing of the 454 and Illumina libraries, and Dr. Kurt Stueber for the bioinformatic support. We thank Nina Dombrowski, Dr. Stijn Spaepen, Dr. Girish Srinivas, Dr. Stéphane Hacquard, Dr. Marc Erhardt, and Dr. Daniel Falush for their valuable comments on the manuscript. This work was supported by funds to P.S.-L. from the Max Planck Society, a European Research Council advanced grant (ROOTMICROBIOTA), and the “Cluster of Excellence on Plant Sciences” program funded by the Deutsche Forschungsgemeinschaft. D.B. was supported by a Royal Society of Edinburgh/Scottish Government Personal Research Fellowship co-funded by Marie Curie Actions.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Contributor Information
Alice C. McHardy, Email: alice.mchardy@helmholtz-hzi.de.
Paul Schulze-Lefert, Email: schlef@mpipz.mpg.de.
Accession Numbers
The sequences generated in the barley pyrosequencing survey and the raw and assembled metagenomics reads reported in this study are deposited in the European Nucleotide Archive (ENA) under the accession number PRJEB5860. Individual metagenomes are also retrievable on the MG-RAST server under the IDs 4529836.3, 4530504.3, 4524858.3, 4524596.3, 4524591.3, and 4524575.3. The scripts used to analyze the data and generate the figures of this study are available at http://www.mpipz.mpg.de/R_scripts.
Supplemental Information
References
- Abbo S., Pinhasi van-Oss R., Gopher A., Saranga Y., Ofner I., Peleg Z. Plant domestication versus crop evolution: a conceptual framework for cereals and grain legumes. Trends Plant Sci. 2014;19:351–360. doi: 10.1016/j.tplants.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Amaral-Zettler L.A., McCliment E.A., Ducklow H.W., Huse S.M. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS ONE. 2009;4:e6372. doi: 10.1371/journal.pone.0006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M.J., Willis T.J. Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology. 2003;84:511–525. [Google Scholar]
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Boller T., Felix G. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 2009;60:379–406. doi: 10.1146/annurev.arplant.57.032905.105346. [DOI] [PubMed] [Google Scholar]
- Bouffaud M.L., Kyselková M., Gouesnard B., Grundmann G., Muller D., Moënne-Loccoz Y. Is diversification history of maize influencing selection of soil bacteria by roots? Mol. Ecol. 2012;21:195–206. doi: 10.1111/j.1365-294X.2011.05359.x. [DOI] [PubMed] [Google Scholar]
- Bulgarelli D., Rott M., Schlaeppi K., Ver Loren van Themaat E., Ahmadinejad N., Assenza F., Rauf P., Huettel B., Reinhardt R., Schmelzer E. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- Bulgarelli D., Schlaeppi K., Spaepen S., Ver Loren van Themaat E., Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013;64:807–838. doi: 10.1146/annurev-arplant-050312-120106. [DOI] [PubMed] [Google Scholar]
- Case R.J., Boucher Y., Dahllöf I., Holmström C., Doolittle W.F., Kjelleberg S. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl. Environ. Microbiol. 2007;73:278–288. doi: 10.1128/AEM.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelius M.K., Triplett E.W. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 2001;41:252–263. doi: 10.1007/s002480000087. [DOI] [PubMed] [Google Scholar]
- Comadran J., Kilian B., Russell J., Ramsay L., Stein N., Ganal M., Shaw P., Bayer M., Thomas W., Marshall D. Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat. Genet. 2012;44:1388–1392. doi: 10.1038/ng.2447. [DOI] [PubMed] [Google Scholar]
- Cornelis G.R., Van Gijsegem F. Assembly and function of type III secretory systems. Annu. Rev. Microbiol. 2000;54:735–774. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- Dakora F.D., Phillips D.A. Root exudates as mediators of mineral acquisition in low nutrient environments. Plant Soil. 2002;245:35–47. [Google Scholar]
- Dröge J., Gregor I., McHardy A.C. Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu745. Published online November 10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D., Jackson S.P. The FHA domain. FEBS Lett. 2002;513:58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Edwards R.A., Olson R., Disz T., Pusch G.D., Vonstein V., Stevens R., Overbeek R. Real time metagenomics: using k-mers to annotate metagenomes. Bioinformatics. 2012;28:3316–3317. doi: 10.1093/bioinformatics/bts599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman D.S., Gropp S.J., Morgan R.L., Wang P.W. Diversifying selection drives the evolution of the type III secretion system pilus of Pseudomonas syringae. Mol. Biol. Evol. 2006;23:2342–2354. doi: 10.1093/molbev/msl103. [DOI] [PubMed] [Google Scholar]
- Haft D.H., Selengut J.D., Richter R.A., Harkins D., Basu M.K., Beck E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2013;41(Database issue):D387–D395. doi: 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A., Holm L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins. 2000;41:224–237. doi: 10.1002/1097-0134(20001101)41:2<224::aid-prot70>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Hong S., Bunge J., Leslin C., Jeon S., Epstein S.S. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009;3:1365–1373. doi: 10.1038/ismej.2009.89. [DOI] [PubMed] [Google Scholar]
- Jeong J., Guerinot M.L. Homing in on iron homeostasis in plants. Trends Plant Sci. 2009;14:280–285. doi: 10.1016/j.tplants.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Jones D.L., Nguyen C., Finlay R.D. Carbon flow in the rhizosphere: carbon trading at the soil-root interface. Plant Soil. 2009;321:5–33. [Google Scholar]
- Karasov T.L., Kniskern J.M., Gao L., DeYoung B.J., Ding J., Dubiella U., Lastra R.O., Nallu S., Roux F., Innes R.W. The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature. 2014;512:436–440. doi: 10.1038/nature13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislev M.E., Nadel D., Carmi I. Grain and fruit diet 19.000 years old at Ohalo II, Sea of Galilee, Israel. Rev. Palaeobot. Palynol. 1992;73:161–166. [Google Scholar]
- Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., Glöckner F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., Lladser M.E., Knights D., Stombaugh J., Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugtenberg B., Kamilova F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009;63:541–556. doi: 10.1146/annurev.micro.62.081307.162918. [DOI] [PubMed] [Google Scholar]
- Lundberg D.S., Lebeis S.L., Paredes S.H., Yourstone S., Gehring J., Malfatti S., Tremblay J., Engelbrektson A., Kunin V., del Rio T.G. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann H.C., Nahal H., Thakur S., Guttman D.S. Identification of innate immunity elicitors using molecular signatures of natural selection. Proc. Natl. Acad. Sci. USA. 2012;109:4215–4220. doi: 10.1073/pnas.1113893109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully M.E. ROOTS IN SOIL: unearthing the complexities of roots and their rhizospheres. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999;50:695–718. doi: 10.1146/annurev.arplant.50.1.695. [DOI] [PubMed] [Google Scholar]
- Meyer R.S., DuVal A.E., Jensen H.R. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 2012;196:29–48. doi: 10.1111/j.1469-8137.2012.04253.x. [DOI] [PubMed] [Google Scholar]
- Newton A.C., Flavell A.J., George T.S., Leat P., Mullholland B., Ramsay L., Revoredo-Giha C., Russell J., Steffenson B.J., Swanston J.S. Crops that feed the world 4. Barley: a resilient crop? Strengths and weaknesses in the context of food security. Food Security. 2011;3:141–178. [Google Scholar]
- Ofek M., Voronov-Goldman M., Hadar Y., Minz D. Host signature effect on plant root-associated microbiomes revealed through analyses of resident vs. active communities. Environ. Microbiol. 2014;16:2157–2167. doi: 10.1111/1462-2920.12228. [DOI] [PubMed] [Google Scholar]
- Peiffer J.A., Spor A., Koren O., Jin Z., Tringe S.G., Dangl J.L., Buckler E.S., Ley R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA. 2013;110:6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M., Coggill P.C., Eberhardt R.Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell A.B., Peterson S.B., Mougous J.D. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol. 2014;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers E.W., Barrett T., Benson D.A., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Edgar R., Federhen S. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009;37(Database issue):D5–D15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeppi K., Dombrowski N., Oter R.G., Ver Loren van Themaat E., Schulze-Lefert P. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. USA. 2014;111:585–592. doi: 10.1073/pnas.1321597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloissnig S., Arumugam M., Sunagawa S., Mitreva M., Tap J., Zhu A., Waller A., Mende D.R., Kultima J.R., Martin J. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames S.R., Finlay B.B. Bacterial effector interplay: a new way to view effector function. Trends Microbiol. 2012;20:214–219. doi: 10.1016/j.tim.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Simm R., Morr M., Kader A., Nimtz M., Römling U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 2004;53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi N., Wolf Y.I., Makarova K.S., Koonin E.V. Nature and intensity of selection pressure on CRISPR-associated genes. J. Bacteriol. 2012;194:1216–1225. doi: 10.1128/JB.06521-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner T.R., Ramakrishnan K., Walshaw J., Heavens D., Alston M., Swarbreck D., Osbourn A., Grant A., Poole P.S. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 2013;7:2248–2258. doi: 10.1038/ismej.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber E., Koebnik R. Positive selection of the Hrp pilin HrpE of the plant pathogen Xanthomonas. J. Bacteriol. 2006;188:1405–1410. doi: 10.1128/JB.188.4.1405-1410.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra E.R., Buckling A., Fineran P.C. CRISPR-Cas systems: beyond adaptive immunity. Nat. Rev. Microbiol. 2014;12:317–326. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- Zhu W., Lomsadze A., Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.