

SUMMARY
In many infections, bacteria form surface-associated communities known as biofilms that are substantially more resistant to antibiotics than their planktonic counterparts. Based on the design features of active anti-biofilm peptides, we made a series of related 12-amino acid L-, D- and retro-inverso derivatives. Specific D-enantiomeric peptides were the most potent at inhibiting biofilm development and eradicating pre-formed biofilms of seven species of wild-type and multiply antibiotic resistant Gram-negative pathogens. Moreover, these peptides showed strong synergy with conventional antibiotics, reducing the antibiotic concentrations required for complete biofilm inhibition by up to 64-fold. As shown previously for 1018, these D-amino acid peptides targeted the intracellular stringent response signal (p)ppGpp. The most potent peptides DJK-5 and DJK-6 protected invertebrates from lethal P. aeruginosa infections, and were considerably more active than a previously described L-amino acid peptide 1018. Thus, the protease resistant peptides produced here were more effective both in vitro and in vivo.
Keywords: D-enantiomeric, peptide, biofilm, inhibitor
INTRODUCTION
Bacteria predominantly form biofilms when growing on surfaces or at air-liquid interfaces (Costerton et al., 1999; O’Toole et al., 2000; Kostakiot, et al., 2013). Biofilms are encased in a protective extracellular matrix that contains water, polysaccharides, proteins, extracellular DNA and lipids (López et al., 2010). The transition from a planktonic to a biofilm lifestyle results in increased resistance to exogenous stresses including conventional antimicrobial therapy and host defense mechanisms (de la Fuente-Núñez et al., 2013; O’Toole et al., 2000; Van Acker et al., 2014). Therefore, biofilms are extremely difficult to eradicate with currently available antimicrobial agents. Indeed biofilms play an important role in the pathogenesis of numerous bacterial species because of their ability to persist on medical devices and in the host (Costerton et al., 1999).
There is currently an urgent need to develop new antibacterial agents to treat increasingly prevalent multidrug resistant bacteria (Boucher et al., 2009; Breidenstein et al., 2011). These antibiotic resistant bacteria are capable of forming biofilms that are highly (adaptively) resistant to antibiotics, thus making treatment of these infections even more difficult. Cationic host defense peptides represent a potential alternative to clinically available antibiotics (Fjell et al., 2012; Hancock and Sahl, 2006). These peptides exhibit antimicrobial activity (against free swimming planktonic cells) and/or possess immunomodulatory properties (de la Fuente-Núñez et al., 2014b; Hilchie et al., 2013). In addition it was shown that the human peptide LL-37, despite very poor activity against free swimming (planktonic) cells (MIC>64 μg/ml), is active against Pseudomonas aeruginosa biofilms at a concentration of one sixteenth the MIC (Overhage et al, 2008).
Recently, synthetic cationic peptides with anti-biofilm activity were identified and characterized (Amer et al., 2010; Dean et al., 2011; de la Fuente-Núñez et al., 2012; de la Fuente-Núñez et al., 2014a). Intriguingly, these peptides seem superficially similar to the cationic antimicrobial peptides that are active against planktonic bacteria. These similarities include being short in size (12–50 amino acids long), and containing cationic amino acids (2 to 9 Arg or Lys residues) and a high proportion of hydrophobic residues (~50%). However, these activities can be clearly distinguished. Indeed, peptides with good anti-biofilm but little anti-planktonic cell activity, and vice versa, have been demonstrated (de la Fuente-Núñez et al., 2012). Furthermore these peptides were active against Burkholderia cenocepacia biofilms even though planktonic B. cenocepacia are resistant to antimicrobial peptides. Recently, a broad spectrum anti-biofilm peptide (peptide 1018) was shown to act by binding to and triggering the degradation of the stress-related second messenger nucleotides guanosine penta- and tetra-phosphate [(p)ppGpp] (de la Fuente-Núñez et al., 2014a). These unusual nucleotides play an important role in biofilm development in many bacterial species (Aberg et al., 2006; Chávez de Paz et al., 2012; de la Fuente-Núñez et al., 2014a; Sugisaki et al., 2013).
One limiting feature of natural peptides is that they are extremely susceptible to degradation by bacterial proteases as well as host proteases that are present at sites of infection. Recent work has indicated that a D-amino acid analog of LL-37 was equally active against biofilms in vitro cf. the L-amino acid variant, but showed apparently superior activity in a Galleria model (Dean et al., 2011). Therefore, here we designed and made novel short D-enantiomeric, protease-resistant peptides with broad-spectrum anti-biofilm activity that were shown to be up to 10-fold more potent than previously identified peptides. The lead anti-biofilm peptides DJK-5 and DJK-6 exhibited activity in vivo, as they protected the nematode Caenorhabditis elegans and the insect Galleria mellonella from lethal P. aeruginosa infections. Both peptides synergized with different of classes conventional antibiotics to prevent biofilm formation and eradicate pre-existing biofilms. These peptides also acted by preventing the intracellular accumulation of (p)ppGpp, which plays an important role in biofilm development.
RESULTS
D-enantiomeric peptide screen
In most cases, both L- and D- antimicrobial peptides have been shown to exhibit similar activity against free swimming (planktonic) bacteria (Epand and Vogel, 1999). This has been taken to suggest that there is no receptor-mediated event involved in the antimicrobial activity of these peptides. In contrast, receptor-mediated events could potentially be involved when different activities for L- and D- amino acid peptides of the same amino acid sequence are observed, since e.g. in an α-helix, there would be opposite rotation of the backbone such that side chains would appear in different positions in 3-dimensional space. Here, we tested the impact on anti-biofilm activity of making both retro (D-amino acid) and retro inverso versions (reversed sequence where all amino acids appear in the same position in 3-dimensional space) of a series of peptides related to anti-biofilm peptide 1018, by using the high throughput BioFlux apparatus (Benoit et al., 2010). These peptides were designed based on properties associated with 1018 and/or our most active anti-biofilm peptides from preliminary screens, namely the use of only 9 of the 20 natural amino acids (V,R,L,I,A,W,F,K,Q), including 4 charged residues (most commonly R), 7 or 8 hydrophobic residues, and no more than one Q.
Intriguingly, and in strong contrast to planktonic antimicrobial activity (Epand and Vogel, 1999), there was no obvious relationship between peptide enantiomeric composition and anti-biofilm activity (Table 1). For example, while the retro-inverso version of 1018 retained anti-pseudomonal anti-biofilm activity, it lost activity vs. Klebsiella biofilms. Conversely, RI-1002 was quite active, but the L-version of this peptide was inactive. Overall the D-amino acid versions of the peptides tended to be more active, but there was substantial variability in activity between the D- and RI versions of several peptides. These data thus indicate that there is no simple relationship between enantiomeric composition and activity. Nevertheless, because the D-versions of peptides tended to be more active and had the advantage of being protease resistant, we decided to further evaluate these.
Table 1. Screen to assess the anti-biofilm activity of D-enantiomeric peptides against P. aeruginosa (Pa) and K. pneumonia (Kp) using the BioFlux microfluidics system.
Percentages represent proportion of dead cells in the biofilm population after treatment with 10 μg/ml of the different peptides at the beginning of biofilm growth, as detailed in the Experimental Procedures section. Hyphens (-) denote conditions that were not tested.
| Peptide name | Type of peptide | Sequences (all peptides amidated) | % Biofilm Inhibition @ 10 μg/ml | |
|---|---|---|---|---|
| Pa | Kp | |||
| L1018 | Normal | VRLIVAVRIWRR | 99 | 99 |
| RI1018 | Retro-inverso | RRWIRVAVILRV | 95 | 5 |
| L1012 | Normal | IFWRRIVIVKKF | 41 | 1 |
| RI1012 | Retro-inverso | FKKVIVIRRWFI | 95 | 0 |
| L1002 | Normal | VQRWLIVWRIRK | 7 | 0 |
| RI1002 | Retro-inverso | KRIRWVILWRQV | 72 | 73 |
| LJK1 | Normal | VFLRRIRVIVIR | 6 | 1 |
| DJK1 | D-Enantiomer | VFLRRIRVIVIR | 85 | 87 |
| RIJK1 | Retro-inverso | RIVIVRIRRLFV | 0 | – |
| LJK2 | Normal | VFWRRIRVWVIR | 43 | – |
| DJK2 | D-Enantiomer | VFWRRIRVWVIR | 87 | – |
| RIJK2 | Retro-inverso | RIVWVRIRRWFV | 91 | 91 |
| LJK3 | Normal | VQLRAIRVRVIR | 0 | – |
| RIJK3 | Retro-inverso | RIVRVRIARLQV | 100 | 99 |
| DJK3 | D-Enantiomer | VQLRAIRVRVIR | 45 | – |
| LJK4 | Normal | VQLRRIRVWVIR | 12.7 | 0 |
| RIJK4 | Retro-inverso | RIVWVRIRRLQV | 99.8 | 71 |
| DJK4 | D-Enantiomer | VQLRRIRVWVIR | 99 | 99 |
| LJK5 | Normal | VQWRAIRVRVIR | 0 | – |
| RIJK5 | Retro-inverso | RIVRVRAIRWQV | 0 | – |
| DJK5 | D-Enantiomer | VQWRAIRVRVIR | 99.7 | 99.8 |
| LJK6 | Normal | VQWRRIRVWVIR | 69 | 0 |
| RIJK6 | Retro-inverso | RIVWVRIRRWQV | 74 | 92 |
| DJK6 | D-Enantiomer | VQWRRIRVWVIR | 98.4 | 98 |
Six of the more active D-enantiomeric peptides were screened for their ability relative to inhibit biofilm formation by the bacterial pathogen P. aeruginosa strain PA14 (Table S1). Analogous to previously reported anti-biofilm peptides (e.g. Dean et al., 2011; de la Fuente-Núñez et al., 2012), these peptides exhibited modest antimicrobial activity against planktonic cells (MIC), but relatively strong anti-biofilm activity (MBIC50). These data revealed the importance of even small sequence changes. For example, DJK-2 and DJK-6 exhibited only a single change F2Q but this led to a 10-fold difference in MBIC50 (Table S1). Overall, peptides DJK-5 and DJK-6 were identified as the most active anti-biofilm peptides obtained to date, since they had MBIC50 values vs. P. aeruginosa of 1 μg/ml and 0.5 μg/ml, respectively (Tables 2 and S1).
Table 2. Antimicrobial (MIC), broad spectrum anti-biofilm (MBIC50) activities and synergistic interactions between D-enantiomeric peptides and conventional antibiotics.
MIC refers to the concentration of peptide that resulted in 100% inhibition of planktonic growth. MBIC50 corresponds to the peptide concentration that results in 50% biofilm inhibition. To test for synergy, checkerboard titrations were performed to assess synergistic interactions between D-enantiomeric peptides DJK-5 (A) and DJK-6 (B) and conventional antibiotics to prevent biofilm formation. The result was expressed as the fractional inhibitory concentration (FIC) with the FIC values indicating synergy (FIC<0.5) or near synergy (FIC<0.56) shown in bold. An FIC index of 0.5 is considered to indicate good synergy (representing the equivalent of a four-fold decrease in the MBIC of each compound when used in combination) and an FIC index of 1.0 represents additive activity (a two-fold decrease in the MBIC of each compound in combination). In most cases, peptides when combined with antibiotics reduced the antibiotic MBIC, here depicted as fold decrease in antibiotic concentration at the FIC. CTZ: ceftazidime; CIP: ciprofloxacin; IMI: imipenem; TOB: tobramycin.
| DJK-5 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Strains | MIC (μg/ml) | MBIC50 (μg/ml) | FIC | Fold Decrease in Antibiotic Concentration | ||||||
| CTZ | CIP | IMI | TOB | CTZ | CIP | IMI | TOB | |||
| P. aeruginosa | 16 | 1 | 0.5 | 0.14 | 0.5 | 0.5 | 8X | 16X | 4X | 2X |
| E. coli 0157 | 1.6 | 0.8 | 0.54 | 1 | 1 | 0.56 | 2X | 16X | 64X | 16X |
| A. baumannii | 8 | 4 | 0.75 | 1 | 0.75 | 0.56 | 2X | 1X | 2X | 16X |
| K. pneumoniae | 3.2 | 1.6 | 0.89 | 0.75 | 1 | 0.75 | 16X | 2X | 64X | 4X |
| S. enterica | 3.2 | 0.8 | 0.75 | 0.56 | 1 | 1.03 | 4X | 2X | 2X | 32X |
| DJK-6 | ||||||||||
| P. aeruginosa | 16 | 0.5 | 0.48 | 0.46 | 0.92 | 1.13 | 16X | 4X | 2X | 1X |
| E. coli 0157 | 16 | 8 | 0.35 | 0.5 | 0.67 | 0.5 | 16X | 32X | 2X | 4X |
| A. baumannii | 8 | 2 | 0.5 | 0.53 | 0.46 | 0.75 | 16X | 16X | 4X | 64X |
| K. pneumoniae | 4 | 2 | 1 | 0.75 | 0.75 | 0.63 | 2X | 4X | 4X | 4X |
| S. enterica | 4 | 1 | 1 | 0.56 | 0.63 | 0.75 | 2X | 16X | 4X | 4X |
To confirm these results, we used the more sensitive flow cell method and assessed the activity of peptides DJK-5 and DJK-6 against wild-type P. aeruginosa PA14 biofilms. Peptides were added at 2.5 μg/ml, well below their MICs of 16 μg/ml, to the flow-through medium in one of two ways: (1) Inhibition studies, in which peptides were added at the beginning of biofilm growth and during the subsequent 3 days of the experiment; (2) Eradication studies, whereby peptide was first added after 2 days of biofilm formation when the biofilm structure was already evident. These studies showed that the peptides DJK-5 and DJK-6 were able to prevent biofilm formation in inhibition studies (Fig. 1, center panels), as well as disperse and eradicate bacteria in wild-type P. aeruginosa PA14 mature biofilms (Fig. 1, right panels).
Figure 1. D-enantiomeric peptides completely prevented biofilm formation and eradicated P. aeruginosa biofilms.
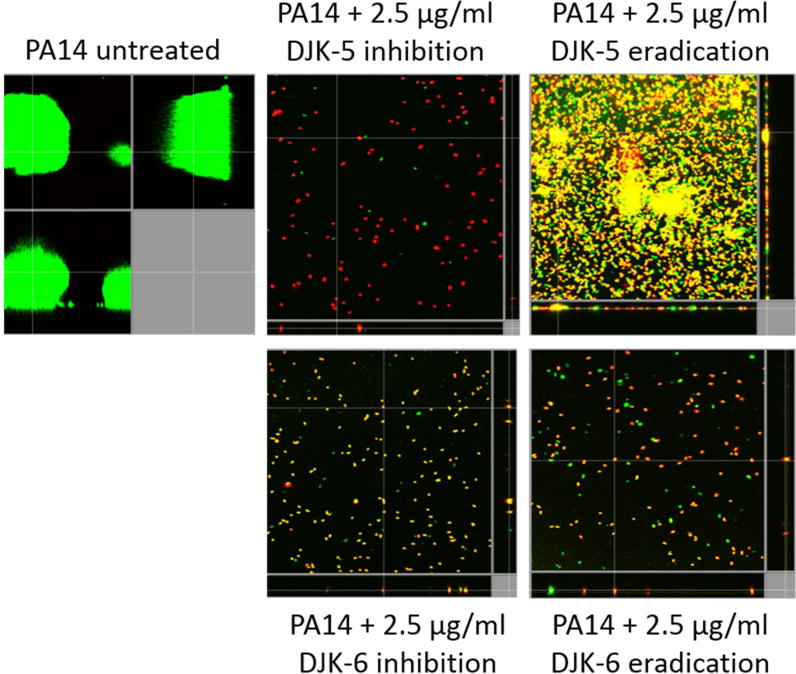
Sub-MIC concentrations (2.5 μg/ml) of peptides DJK-5 and DJK-6 were used. Inhibition of biofilm development was tested by immediately adding peptide into the flow-through medium of the flow cell apparatus and then monitoring biofilm formation for 3 days. Eradication conditions involved waiting two days before addition of either peptide into the flow-through medium. After 3 days, bacteria were stained green with the all bacteria stain Syto-9 and red with the dead-bacteria stain propidium iodide (merge shows as yellow to red) prior to confocal imaging. Each panel shows reconstructions from the top in the large panel and sides in the right and bottom panels (xy, yz and xz dimensions).
D-enantiomeric peptides exhibited broad spectrum anti-biofilm activity
The L-amino acid containing peptide 1018 was previously shown to have broad spectrum activity vs. Gram negative bacteria (de la Fuente-Núñez et al., 2014a). To see if this was also the case for D-enantiomeric peptides, we determined their spectrum of activity. Both peptides prevented biofilm growth in a wide range of bacteria at levels below their MICs for planktonic cells (Table 2). DJK-5 inhibited biofilms at concentrations ranging from 0.8 μg/ml to 4 μg/ml, while DJK-6 was most effective against the wild-type P. aeruginosa strain PA14 (0.5 μg/ml) and showed lower activity against enterohemorrhagic Escherichia coli isolate 0157 (8 μg/ml) (Table 2). As expected, these peptides did not affect the planktonic growth of a clinical isolate of B. cenocepacia, known to be completely resistant to antimicrobial peptides (MIC>256 μg/ml for DJK-5 and MIC>64 μg/ml for DJK-6), but inhibited biofilms of this multidrug resistant strain at only 0.4 μg/ml and 2 μg/ml, respectively (Table S2). The enhanced anti-biofilm activity of these peptides occurred for a broad range (7 different species and 30 strains) of wild-type and multi-drug resistant pathogens, and especially all Gram-negative members of the so called ESKAPE pathogens (Table S2).
D-enantiomeric peptides protected Caenorhabditis elegans and Galleria mellonella from lethal P. aeruginosa infections
To test for the ability of peptides to protect against infections, we utilized two non-vertebrate models of P. aeruginosa infections (Brackman et al., 2011; Cooper et al., 2009; Edwards and Kjellerup, 2012; Stiernagle, 2006). The C. elegans nematode model used here has been shown to consistently develop biofilm infections (Brackman et al., 2011; Edwards and Kjellerup, 2012). Furthermore, Dean et al (2011) argued that protection in the Galleria larvae model reflected anti-biofilm rather than antibiotic activity vs. planktonic bacteria. Consistent with this concept, the tested peptides had very weak MICs of 16–64 μg/ml vs. planktonic P. aeruginosa. The peptides tested included the optimized D-enantiomeric peptides DJK-5, DJK-6, as well as the previously described 1018 (de la Fuente-Núñez et al., 2014a), and its D-analog RI-1018.
Untreated controls infected with P. aeruginosa PAO1 demonstrated 100% death after 48 h in both infection models (Table 3). After 24 hours of infection, each of the peptides significantly (p<0.001) protected C. elegans against lethal P. aeruginosa infections, with DJK-5 and DJK-6 giving nearly complete protection (Table 3). After 48 h of infection, significant protection (p<0.001) was only observed for animals treated with peptides DJK-5 and DJK-6, while mortality was close to 100% (and not significantly different from the peptide untreated control group) for RI-1018 and 1018 (Table 3). The peptides by themselves did not display any toxic activity against C. elegans, since no significant differences in survival were observed after 24 h and 48 h in uninfected C. elegans nematodes treated with peptides compared to untreated animals (Table 3).
Table 3. In vivo anti-biofilm activity of D-enantiomeric peptides.
C. elegans and G. mellonella biofilm survival assays. Percent survival of infected C. elegans and G mellonella (average ± the SD) after treatment with peptides D-enantiomeric peptides RI-1018 (and its L-version 1018), DJK-5 and DJK-6 and P. aeruginosa strain PAO1. The results are expressed as the percent survival after both 24 h and 48 h of infection and peptide treatment. Statistical significance comparing peptide-treated groups to untreated was determined (*, P< 0.001).
| C. elegans survival (%) | ||||
|---|---|---|---|---|
| 24h | 48h post infection | |||
| Peptide | No infection | P. aeruginosa PAO1 | No infection | P. aeruginosa PAO1 |
| None | 100 ± 0 | 61 ± 21 | 95 ± 4 | 1 ± 2 |
| RI1018 | 99 ± 1 | 83 ± 13* | 81 ± 23 | 4 ± 6 |
| 1018 | 97 ± 4 | 91 ± 12* | 88 ± 9 | 1 ± 3 |
| DJK5 | 99 ± 2 | 99 ± 2* | 99 ± 2 | 96 ± 4* |
| DJK6 | 99 ± 2 | 99 ± 2* | 97 ± 4 | 90 ± 5* |
| G. mellonella survival (%) | ||||
| CTRL | 100 ± 0 | 13 ± 11 | 100 ± 0 | 0 ± 0 |
| RI1018 | 90 ± 14 | 50 ± 8* | 80 ± 10 | 18 ± 7* |
| 1018 | 90 ± 14 | 27 ± 11 | 90 ± 14 | 3 ± 5 |
| DJK5 | 100 ± 0 | 90 ± 6* | 100 ± 0 | 42 ± 7* |
| DJK6 | 100 ± 0 | 50 ± 8* | 100 ± 0 | 30 ± 6* |
survival significantly different from untreated control (p<0.001)
Using the G. mellonella infection model, no protective effect was observed after 24 h with peptide 1018, a moderate but significant protective effect was observed for its D-analog RI-1018 as well as DJK-6, and a strong and significant protective effect was conferred by DJK-5 (Table 3). After 48 h, RI-1018 and particularly peptides DJK-5 and DJK-6 resulted in significantly increased survival (18–42% survival; p<0.001), while complete killing was observed in the control group (Table 3). Thus in this model although 1018 and its retro-inverso analog RI-1018, after folding into an α-helix, would have all amino acids positioned in the same place in 3-dimensional space, and had equivalent anti-biofilm activity vs. P. aeruginosa (Table 1), only the latter was protective, indicating an advantage for the protease resistant variant.
Broad-spectrum synergistic interactions between D-enantiomeric peptides and conventional antibiotics to treat biofilms
Previous studies showed that anti-biofilm peptide 1018 demonstrated synergy with conventional antibiotics (Reffuveille et al., 2014). To test whether this was still true for the more active peptides, we adapted the checkerboard methodology (Reffuveille et al., 2014) that is widely used to determine interactions between anti-bacterial compounds. The results obtained when peptides DJK-5 and DJK-6 were combined with four of the most commonly used antibiotics in human medicine, ceftazidime, imipenem, ciprofloxacin or tobramycin, are shown in Table 2. In all cases, we observed either synergy (fractional inhibitory concentration, FIC, of <0.5; indicating that the MBIC of each compound in combination was decreased by at least 4 fold compared to the compounds used alone), near synergy (FIC < 0.56), or additive interactions (FIC=0.5–1) (Table 2). Overall, 42.5% of the combinations showed synergy or near synergy. Interestingly, in 95% of assessments, these peptides led to a substantial decrease in the concentration of antibiotic required for anti-biofilm activity, compared to antibiotic alone, with a 2 to 64-fold drop in antibiotic concentration in combination (Table 2).
These results were confirmed using the flow cell assay at the concentrations of peptide and antibiotic giving the lowest FIC, in checkerboard assays, against each tested bacterial species (shown in Table 2). For example, the lowest FIC value obtained for P. aeruginosa PA14 was 0.14, corresponding to the combination 0.1 μg/ml of DJK-5 with 0.04 μg/ml of ciprofloxacin (Table 2). Flow cell experiments confirmed these results since this combination led to complete biofilm inhibition (Fig. 2A). For all other species tested, complete or nearly complete biofilm prevention was observed at the concentrations giving the lowest FIC for each peptide plus antibiotic combination, with only a few individual cells (some red-stained with the dead-bacteria stain propidium iodide) remaining attached to the surface of the flow cell chambers (Fig. 2A,B).
Figure 2. D-enantiomer peptides DJK-5 (A) and DJK-6 (B) exhibited anti-biofilm activity in flow cells and synergized with conventional antibiotics in preventing biofilm formation by different bacterial species.
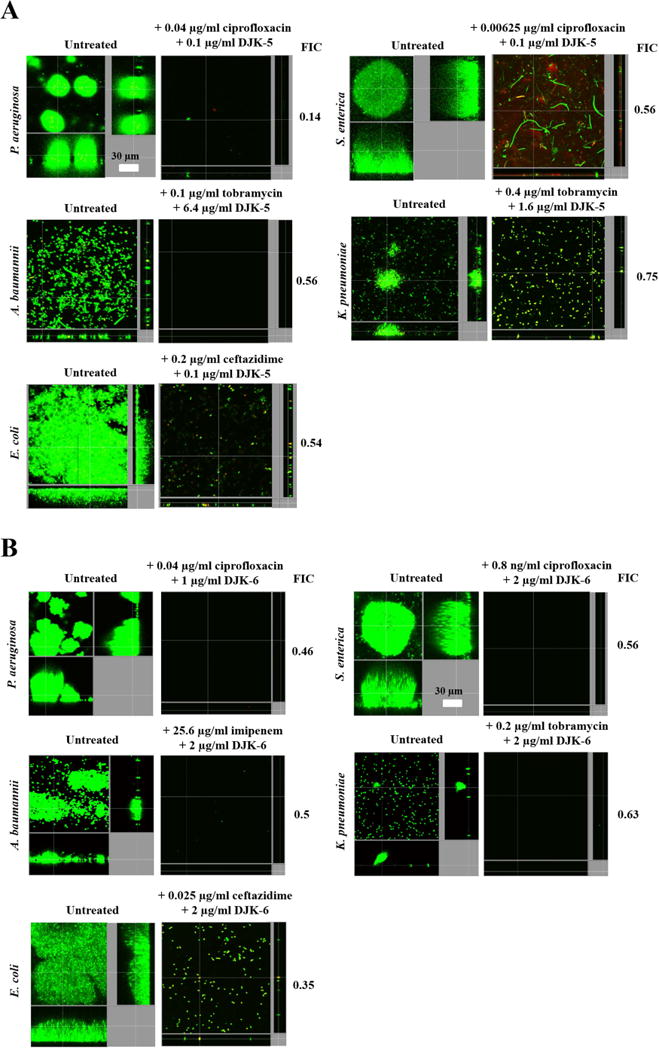
Sub-inhibitory concentrations of peptides DJK-5 and DJK-6 in combination with antibiotics prevented biofilm development of Gram-negative bacteria. Inhibition of biofilm development was tested by immediately (at the beginning of biofilm growth at day 0) adding peptide plus antibiotic into the flow-through medium of the flow cell apparatus and then monitoring biofilm formation for 3 days. After 3 days, bacteria were stained green with the all bacteria stain Syto-9 and red with the dead-bacteria stain propidium iodide (merge shows as yellow to red) prior to confocal imaging. Each panel shows reconstructions from the top in the large panel and sides in the right and bottom panels (xy, yz and xz dimensions). The top FIC combinations of peptide + antibiotic (determined in checkerboard assays) were used.
Similar results were observed using these combinations to eradicate 2-day-old biofilms (Fig. 3). For example, peptide DJK-5, when combined with the antibiotics tobramycin, ceftazidime or ciprofloxacin led to eradication of A. baumannii, S. enterica and Klebsiella pneumoniae mature biofilms, respectively (Fig. 3A). On the other hand, the combination of DJK-5 with ciprofloxacin vs. P. aeruginosa PA14 caused much more limited dispersal but triggered cell death in remaining cells (Fig. 3A). In contrast, the combination of DJK5 and ceftazidime vs. E. coli was not synergistic in eradication studies (Fig. 3A). Combinations of DJK-6 with any of the antibiotics tested led to disruption of pre-formed biofilms in all cases (Fig. 3B), with at most only a few cells remaining attached to the surface of the flow cell chambers.
Figure 3. Synergistic interactions of D-enantiomer peptides DJK-5 (A) and DJK-6 (B) with different classes of antibiotics in treating mature biofilms.
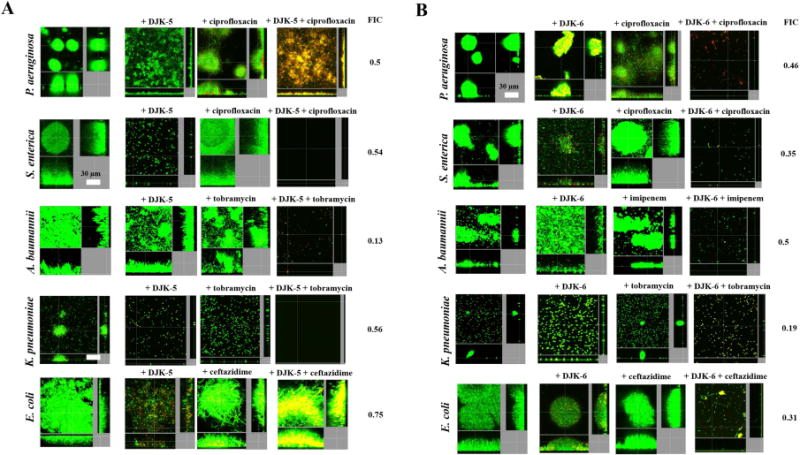
Bacteria were grown in flow cells and treated at day 2 of biofilm formation with peptide, antibiotic, or the combination of both. The top FIC combinations of peptide + antibiotic (determined in checkerboard assays) were used (as in Figure 2). In some cases, at the concentrations selected, the activity of the peptides led to complete eradication of the flow cell biofilms. Thus, we decreased the levels of peptide used, which lowered the FIC values (see on the right hand side of panels) compared to the checkerboard assay results shown in Table 2. Specifically, in (A) 0.8 (μg/ml of DJK-5 (instead of 6.4 (μg/ml) was used in combination with tobramycin vs A. baumannii. In (B), 0.5 (μg/ml of DJK-6 were used instead of 2 (μg/ml combined with imipenem vs. A. baumannii, 1 (μg/ml of DJK-6 (as opposed to 2 (μg/ml) was used in conjunction with ciprofloxacin vs S. enterica, and 0.5 (μg/ml of DJK-6, instead of 2 (μg/ml, was used in combination with tobramycin vs K. pneumoniae. After 3 days, bacteria were stained green with the all bacteria stain Syto-9 and red with the dead-bacteria stain propidium iodide (merge shows as yellow to red) prior to confocal imaging. Each panel shows reconstructions from the top in the large panel and sides in the right and bottom panels (xy, yz and xz dimensions).
Mechanism of action
Recently, the anti-biofilm peptide 1018 was shown to bind to and promote degradation of the signal for biofilm formation and maintenance, (p)ppGpp (de la Fuente-Núñez et al., 2014a). Here, we performed selected experiments to demonstrate that the more potent D-enantiomeric peptides, DJK-5 and DJK-6, operated through the same mechanism. Thus, overproduction of the potential target (p)ppGpp by treatment of P. aeruginosa with 80 μM of serine hydroxamate (SHX) (Tosa and Pizer, 1971; Raskin et al., 2007), led to reduced susceptibility of P. aeruginosa biofilms to peptide action (Fig. S1). To examine the fate of (p)ppGpp upon peptide treatment, we treated planktonic cells with 500 μM serine hydroxamate to enable them to accumulate (p)ppGpp (Nguyen et al., 2011). Direct measurement of the cellular levels of (p)ppGpp by thin layer chromatography (TLC) revealed that treatment with 1 μg/ml of peptides DJK-5 and DJK-6 resulted in the complete loss of (p)ppGpp from P. aeruginosa cells (Fig. 4A). Treatment with 0.5 μg/ml of RI-1018 also led to the absence of (p)ppGpp accumulation, whereas the enantiomeric L-form equivalent 1018 required 5 μg/ml to achieve similar activity (Fig. 4B). In addition, peptide DJK-6 appeared to be more effective at enhancing degradation of pre-accumulated (p)ppGpp compared to its retro-inverso version RI-JK6, and peptide RI-1018 (Fig. 4C). Treatment with RI-JK6 and DJK-6 degraded most of the pppGpp but not the ppGpp nucleotide pool within 10 minutes (Fig. 4C). After 20 minutes, however, both RI-JK6 and DJK-6 led to almost complete disappearance of ppGpp and pppGpp (Fig. 4C).
Figure 4. D-enantiomeric peptides prevented (p)ppGpp accumulation and led to disappearance of (p)ppGpp in vivo.
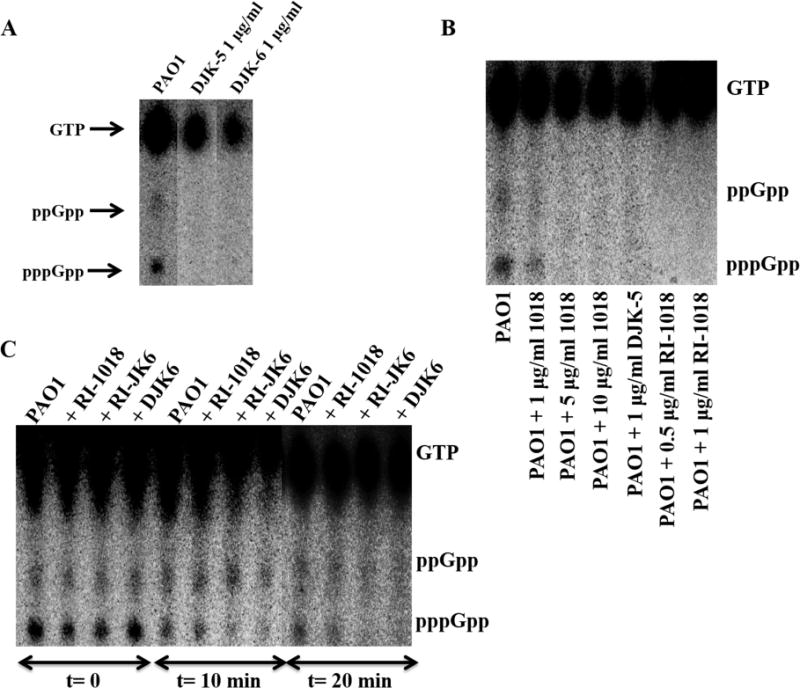
(A) Anti-biofilm peptides DJK-5 and DJK-6 at 1 (μg/ml led to the absence of (p)ppGpp accumulation as revealed by TLC separation of guanine nucleotides extracted from intact cells as described in Experimental Procedures. (B) D-enantiomeric peptides RI-1018 and DJK-5 led to complete disappearance of (p)ppGpp more potently than L-peptide 1018. (C) D-enantiomeric peptide DJK-6 exhibited increased ability to trigger the degradation of pre-formed (p)ppGpp compared to RI-1018 and RI-JK6. Twenty (μg/ml of each of the three peptides were used.
DISCUSSION
Multidrug resistant Gram-negative pathogens are becoming increasingly prevalent, including members of the ESKAPE pathogens, E. coli/Enterobacter, Pseudomonas, Klebsiella and Acinetobacter, for which no fundamentally-new drugs are under development in the antibiotic pipeline (Boucher et al., 2009; Payne et al., 2007). An additional concern is adaptive resistance, whereby the growth state of the organism leads to non-mutational high-level resistance to most currently available antibiotics (de la Fuente-Núñez et al., 2013). For example, biofilm growth leads to multidrug adaptive resistance (up to 10 to 1000 increased resistance compared to planktonic bacteria) and is associated with at least 65% of all human clinical infections (Kostakiot et al., 2013). Moreover, there are currently no specific treatments for biofilm-related infections.
Bacterial resistance strategies to antimicrobial peptides have been previously described that include enzymatic degradation of L-enantiomeric peptides, while host proteases can also degrade such peptides during therapy (Fjell et al., 2012). Here, we overcame these limitations, by designing D-enantiomeric peptides, which cannot be recognized by bacterial or host proteases that abound during infections and can cleave peptides composed entirely of L-amino acids (Sieprawska-Lupa et al., 2004). We then characterized the anti-biofilm activities of D-enantiomeric peptides against a range of Gram-negative bacterial species, including multidrug resistant strains (Table S2). Our data demonstrated that the best peptides share many of the features of L-amino acid peptides, but appeared to be superior to previously-described peptides, particularly in invertebrate animal protection models. For example the broad-spectrum peptide 1018 showed weak to no activity in two non-vertebrate models. Conversely, the retro-inverso-analog RI-1018 was more active in both models. However, the most effective peptides were the optimized D-enantiomeric peptides DJK-5 and DJK-6, which protected C. elegans nematodes and G. mellonella larvae against lethal Pseudomonas infections. Importantly, the C. elegans model used here is an established biofilm infection model (Brackman et al., 2011; Edwards and Kjellerup, 2012), and together with the weak activity of these peptides vs. planktonic cells is consistent with the D-enantiomeric peptides having anti-biofilm activity in vivo. This is also true for the Galleria model that was suggested by Dean et al (2011) to demonstrate the anti-biofilm activity of D-LL-37 in vivo, although the level of protection by that peptide was substantially lower than that presented here. Thus, it can be concluded that D-enantiomeric peptides offer real advantages with regards to activity in animal models where proteases abound.
Intriguingly, despite examining the activity of more than 100 L-amino acid containing peptides to date, 1018 appears to be the most active. Nevertheless the two most active D-peptides described here, DJK-5 and DJK-6, were by and large considerably more active. This likely reflects their resistance to proteases encountered in the process of action on bacteria, and possibly also the increased ability of D-peptides to stimulate degradation or prevent accumulation of (p)ppGpp (Fig. 4B). Unlike some antimicrobial peptides that appear to be able to act on membranes or membrane-associated processes, anti-biofilm peptides that target (p)ppGpp must be able to translocate into cells and would thus be especially susceptible to intracellular or membrane-bound proteases. In this regard it is important to note that amphipathic cationic peptides, like those described here, have the characteristics of cell penetrating peptides and can freely translocate across membranes (Fjell et al., 2012). The overall activity of these D-enantiomeric peptides is thus likely to reflect their relative ability to be taken up into cells (i.e. ability to cross both the outer and cytoplasmic membrane), as well as their relative affinity for their target (p)ppGpp, which we have assessed here for some peptides (Fig. 4). Given the lack of a suitable translocation assay into cells for any cationic peptide, to assess the combined effects of translocation and affinity we have measured cellular (p)ppGpp nucleotide pools in TLC assays in the absence and presence of different concentrations of peptides (Fig. 4). The structure-activity relationships are likely very complex, as also observed for antimicrobial peptides where dozens of physico-chemical properties influenced activity, including inductive properties that reflected 3-D structure (Cherkasov et al, 2009). For example, although RI-JK1 and RI-JK5 differed from RI-JK6 by only 2 and 3 amino acids, respectively, the former were inactive while the latter was very active (Table 1). Similarly, a single amino acid substitution Q2F in DJK-2 cf. DJK-6 led to a 10-fold difference in MBIC50 (Table S1).
There were no apparent major differences in the properties of D-enantiomer peptides assessed in vitro when compared to L-enantiomers. Thus, anti-biofilm activities were often superior to activity vs. planktonic cells, synergy was often observed with antibiotics, and the previously described target was still evidently the same (de la Fuente-Núñez et al., 2014a). Previous reports showed that, at the concentrations used in synergy studies, peptide treatment causes biofilm dispersion (de la Fuente-Núñez et al., 2014a; Reffuveille et al., 2014), and that the antibiotic susceptibility of these dispersed cells was similar to that of planktonic cells used in MIC assays (Reffuveille et al., 2014). Therefore, we propose that synergy reflects, at least in part, peptide-mediated bacterial dispersal from biofilms increasing their susceptibility to antibiotics.
The data favored a mechanism similar to that observed for peptide 1018 (de la Fuente-Núñez et al., 2014a), whereby peptides DJK-5 and DJK-6 inhibited biofilm formation and suppressed mature biofilms by entering cells and subsequently targeting and causing the degradation of the intracellular nucleotides (p)ppGpp, which are important for the formation and maintenance of biofilms. Furthermore, we have shown here that the D-peptide RI-1018 was more potent at stimulating degradation and/or preventing accumulation of (p)ppGpp in P. aeruginosa compared to its L-form peptide 1018 (Fig. 4B). D-peptides were also capable of promoting degradation of pre-formed (p)ppGpp, as treatment with RI-JK6 and DJK-6 for 10 minutes substantially eliminated pppGpp, and after 20 minutes led to almost the complete disappearance of both ppGpp and pppGpp nucleotide pools (Fig. 4C). This represents increased activity compared to results obtained with L-1018, which did not substantially decrease (p)ppGpp pool after 10 minutes, and was only able to lead to complete degradation of pre-formed (p)ppGpp after 30 minutes of treatment (de la Fuente-Núñez et al., 2014a). Further, we found that within the first 10 minutes of treatment, the D-peptides, particularly RI-JK6 and DJK-6, led to degradation of pppGpp but not ppGpp, which took twice as long to disappear (Fig. 4C). Thus either the peptide interacts more strongly with pppGpp to promote degradation (which is possible since it is more highly negatively charged than ppGpp) or first promotes the transition from pppGpp to ppGpp. Interestingly, although another second messenger guanidine nucleotide cyclic-di-GMP influences the switch between planktonic and biofilm lifestyles (Hengge, 2009; Römling et al., 2013) we have no direct evidence that it is involved in the events described here and it is noteworthy that there was no obvious change in GTP pools that can affect cyclic-di-GMP concentrations. Nor was there any obvious influence of a specific growth condition, since collectively we observed the biofilm inhibitory effects of peptides in both nutrient and minimal medium.
Overall, in addition to protease resistance, which appears to be an asset, the D-enantiomeric peptides retain two very potent activities for countering drug resistance. First, they kill bacteria growing as biofilms, which are known to be associated with more than two thirds of all infections in humans, and demonstrate high adaptive resistance to multiple classes of antibiotics. Second, the peptides showed synergy or additive effects with highly utilized conventional antibiotics, rendering biofilms more susceptible to these agents. Thus, the combination of D-enantiomeric peptides with antibiotics enhanced the activity of antibiotics to target bacterial biofilms, both at the initial stages of growth and in their mature state. The in vivo protective activity of these peptides against otherwise lethal P. aeruginosa infections, demonstrates the stand-alone potential of these peptides. Future studies will focus on the synergistic interactions of peptides, in combination with antibiotics, in different animal models.
SIGNIFICANCE
There are relatively few novel compounds or strategies under development or entering the clinic to treat multidrug-resistant Gram-negative bacterial pathogens, especially when they become even more resistant growing as biofilms (the growth state of bacteria in two thirds of infections). Investigations of the anti-biofilm activities of a series of related 12-mer peptides comprised of either L-amino acids or D-amino acids, indicated that the latter generally had better in vitro activities. The best D-enantiomeric peptides had broad-spectrum activity in vitro, and were able to confer protection in two non-vertebrate models against lethal infections caused by P. aeruginosa, thus demonstrating their potential in vivo. As observed for other L-enantiomeric peptides, the observed anti-biofilm activities were often superior to activity vs. planktonic cells, and synergy was often observed with antibiotics. In addition, the peptides targeted the stringent response nucleotides (p)ppGpp, which play an important role in biofilm formation. Thus the D-enantiomeric anti-biofilm peptides described here have the potential to be used in novel adjuvant therapies that might be effective in combination with antibiotics against biofilms formed by antibiotic-resistant bacteria.
EXPERIMENTAL PROCEDURES
Bacterial strains
Strains utilized included wild-type strains of Pseudomonas aeruginosa PA01 and PA14, Burkholderia cenocepacia isolate 4813 (isolate from a CF patient attending Vancouver Children’s Hospital), Escherichia coli 0157, Klebsiella pneumoniae ATTC 13883 (a colistin-heteroresistant reference strain from American Type Culture Collection, Rockville, MD), multidrug resistant Acinetobacter baumannii SENTRY C8 (a polymyxin B resistant blood clinical isolate from the U.S.A. obtained through the SENTRY surveillance system), and Salmonella enterica serovar Typhimurium isolate 14028S were used. A complete list of the strains tested in this study is provided in Supplementary Table 2. The growth conditions of these strains were generally as described previously (de la Fuente-Núñez et al., 2014a).
Peptide Synthesis
All D-enantiomeric peptides were synthesized by CPC Scientific using solid-phase 9-fluorenylmethoxy carbonyl (Fmoc) chemistry and purified to ~95% using reverse-phase high-performance liquid chromatography. Correct peptide mass was confirmed by mass spectrometry.
BioFlux microfluidic studies
BioFlux studies were performed as previously described (Benoit et al., 2010) using a Klebsiella pneumoniae strain LM21 gfp (Balestrino et al., 2005) and a Pseudomonas aeruginosa gfp strain. For use in biofilm experiments, LB cultures were grown to an OD620 of ~ 0.5 and seed into BioFlux™ 48-well flow-channel plates (Fluxion P/N 950-0010) for ~5 seconds and incubated with no flow for ~45 minutes to allow bacterial attachment. One ml of diluted synthetic peptide suspension was added to the inlet wells at the beginning of biofilm growth. Shear flow was applied at 5 dynes/cm2 overnight. Biofilm development was periodically checked via brightfield microscopy and at the end of the study residual cells detected by GFP fluorescence with dead cells being detected by using the fluorescent dead-cell stain propidium iodide using a Nikon Eclipse Ti inverted epifluorescence scope and associated digital camera for biofilm visualization and micrograph collection. Quantitative green (total bacteria) and red (dead bacteria) fluorescence intensity data was extracted from micrographs using Montage Offline (Fluxion 940-0004).
Minimal Inhibitory Concentration (MIC, MBIC50) assays
The broth microdilution method with minor modifications for cationic peptides (Wiegand et al., 2008) was used for measuring the MIC of all D-enantiomeric peptides used. Minimal biofilm inhibitory concentrations leading to 50% decrease in adherent (biofilm) growth (MBIC50) were obtained using 96-well plate assays and crystal violet staining of adherent biofilms as previously described (de la Fuente-Núñez et al., 2012).
Biofilm growth conditions in checkerboard assays
The medium used was generally LB, except for S. enterica serovar Typhimurium isolate 14028S that was grown in BM2 minimal medium (62 mM potassium phosphate buffer, pH 7.0, 7 mM [(NH4)2SO4, 2 mM MgSO4, 10 μM FeSO4] containing 0.5% casarnino acids and 0.4% (wt/vol) glucose as a carbon source, and K. pneumoniae that was grown in Todd Hewitt broth medium containing 0.4% yeast extract. Bacteria were grown for 24 h in all cases, except for K. pneumoniae that was allowed to grow for 48 h. In checkerboard assays, the MBIC values represented the concentration (or combinations of concentrations when using peptides in combination with antibiotic) at which 100% biofilm inhibition was observed. The result was expressed as the fractional inhibitory concentration (FIC) index, calculated as follows: FIC = [A]/MBICA + [B]/MBICB, where MBICA and MBICB are the MBICs of peptides A and B alone and [A] and [B] are the MBICs of A and B when in combination.
Biofilm cultivation in flow cell chambers and microscopy
Experiments were done as described previously (de la Fuente-Núñez et al., 2014a). Biofilms were grown in BM2 glucose minimal medium for 72 h, in the absence or presence of the desired concentration of peptides DJK-5, DJK-6 and/or the different antibiotics tested, at 37°C in flow chambers with channel dimensions of 1 by 4 by 40 mm. Biofilm cells were stained using the LIVE/DEAD BacLight Bacterial Viability kit (Molecular Probes, Eugene, OR) prior to microscopy experiments. Microscopy was done using a confocal laser scanning microscope (Olympus, Fluoview FV1000) and three-dimensional reconstructions were generated using the Imaris software package (Bitplane AG).
(p)ppGpp measurement by thin layer chromatography
Measurement of (p)ppGpp was performed by thin layer chromatography (TLC) of cells grown overnight in modified MOPS minimal medium containing 0.4% glucose, 2 mM phosphate (KH2PO4), and 0.2% CAA and treated with 500 μM serine hydroxamate (SHX) to induce (p)ppGpp synthesis, in the presence or absence of peptides DJK-5 and DJK-6. Cells were labelled with 10 μCi/ml 32P for 3 h, prior to analysis by TLC. After chromatography, nucleotide spots were visualized by autoradiography and quantified with a Molecularlmager FX Phosphorlmager and Quantity One software (Bio-Rad).
Strains and culture conditions for in vivo experiments
P. aeruginosa PAOl was cultured in Mueller-Hinton broth (MH; Oxoid, Basingstoke, England) at 37°C. E. coli OP50 was grown in TSB (Oxoid) at 37°C. C. elegans N2 (glp-4; sek-1) was propagated under standard conditions, synchronized by hypochlorite bleaching, and cultured on nematode growth medium using E. coli OP50 as a food source (Cooper et al, 2009; Stiernagle, 2006). Adult G. mellonella larvae (De Poorter, Gent, Belgium) were stored in wood chips at 15°C in darkness prior to use. Larvae weighing between 200 and 300 mg were used for all experiments.
Caenorhabditis elegans survival assay
The C. elegans survival assay was carried out as previously described (Brackman et al., 2011). In brief, synchronized worms (L4 stage) were suspended in a medium containing 95% M9 buffer, 5% brain heart infusion broth (Oxoid), and 10 μg of cholesterol (Sigma-Aldrich) per ml. One half ml of this suspension of nematodes was transferred to the wells of a 24-well microtiter plate. An overnight bacterial culture was centrifuged, resuspended in the assay medium, and standardized to 108 CFU/ml. Next, 250 μl of this standardized suspension were added to each well, while 250 μl of sterile medium was added to the positive control. Peptides were added to the test wells at a final concentration of 20 μg/ml. The assay plates were incubated at 25°C for up to 2 days. The fraction of dead worms was determined by counting the number of dead worms and the total number of worms in each well, using a dissecting microscope. Peptides were tested at least four times in each assay, and each assay was repeated at least three times (n ≥ 12). At least 100 C. elegans nematodes were used for each condition in each assay (n ≥ 300 nematodes/condition).
G. mellonella survival assay
The G mellonella survival assay was carried out as previously described (Brackman et al., 2011). In brief, prior to injection in G mellonella, bacterial cells were washed with PBS and then diluted to IO4 CFU per 10 μl. A Hamilton syringe was used to inject 10 μl in the G mellonella last left proleg. The peptides (20 μg/10 μl were administered by injecting 10 μl into a different proleg within 1 h after injecting the bacteria. Two control groups were used: the first group included uninfected larvae injected with PBS to monitor killing due to physical trauma; the second group included uninfected larvae receiving no treatment at all. Results from experiments in which one or more larvae in either control group died were discarded and the experiments were repeated. To evaluate the toxicity of the peptides, uninfected larvae were injected with peptides. Larvae were placed in the dark at 37°C and were scored as dead or alive 24 h and 48 h post-infection. Larvae were considered dead when they displayed no movement in response to shaking or touch. At least 20 larvae were injected for each treatment. For each treatment, data from at least six independent experiments were combined (n ≥ 120 G mellonella larvae/condition).
Supplementary Material
Highlights.
Identified novel D-enantiomeric peptides with potent anti-biofilm activity
These peptides conferred protection in two different invertebrate infection models
Peptides demonstrated synergistic interactions with conventional antibiotics
D-enantiomeric peptides acted by preventing the accumulation of (p)ppGpp
Acknowledgments
We thank Carmen Gibbs Allen and Neel Doshi for their expertise using the BioFlux device. We would also like to acknowledge George A. Mackie for his expertise and technical advice with the TLC work. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R21AI098701, by a grant from the Canadian Institutes for Health Research MOP-74493 by the Fund for Scientific Research – Flanders (FWO-Vlaanderen), by the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen, SBO programme) and the Interuniversity Attraction Poles Programme initiated by the Belgian Science Policy Office. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. R.E.W.H. holds a Canada Research Chair in Health and Genomics. C.D.L.F.-N. received a scholarship from the Fundación “la Caixa” and Fundación Canadà (Spain). Additionally, C.D.L.F.-N. and RE.W.H. are co-inventors of a provisional patent application on the use of cationic anti-biofilm and innate defense regulator (IDR) peptides (U.S. Patent Application No. 61/870,655).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberg A, Shingler V, Balsalobre C. (p)ppGpp regulates type 1 fimbriation of Escherichia coli by modulating the expression of the site-specific recombinase FimB. Mol Microbiol. 2006;60:1520–33. doi: 10.1111/j.1365-2958.2006.05191.x. [DOI] [PubMed] [Google Scholar]
- Amer LS, Bishop BM, van Hoek ML. Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem Biophys Res Commun. 2010;396:246–51. doi: 10.1016/j.bbrc.2010.04.073. [DOI] [PubMed] [Google Scholar]
- Balestrino D, Haagensen JA, Rich C, Forestier C. Characterization of type 2 quorum sensing in Klebsiella pneumoniae and relationship with biofilm formation. J Bacteriol. 2005;187:2870–80. doi: 10.1128/JB.187.8.2870-2880.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow PG, Svoboda P, Mackellar A, Nash AA, York IA, Pohl J, Davidson DJ, Donis RO. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS One. 2011;6:e25333. doi: 10.1371/journal.pone.0025333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit MR, Conant CG, Ionescu-Zanetti C, Schwartz M, Matin A. New device for high-throughput viability screening of flow biofilms. Appl Environ Microbiol. 2010;76:4136–42. doi: 10.1128/AEM.03065-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Brackman G, Cos P, Maes L, Nelis HJ, Coenye T. Quorum sensing inhibitors increase the susceptibility of bacterial biofilms to antibiotics in vitro and in vivo. Antimicrob Agents Chemother. 2011;55:2655–61. doi: 10.1128/AAC.00045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenstein EBM, de la Fuente-Núñez C, Hancock REW. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011;19:419–26. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Chávez de Paz LE, Lemos JA, Wickström C, Sedgley CM. Role of (p)ppGpp in biofilm formation by Enterococcus faecalis. Appl Environ Microbiol. 2012;78:1627–30. doi: 10.1128/AEM.07036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock REW. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic resistant Superbugs. ACS Chemical Biol. 2009;4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]
- Cooper VS, Carlson WA, LiPuma JJ. Susceptibility of Caenorhabditis elegans to Burkholderia infection depends on prior diet and secreted bacterial attractants. PLoS One. 2009;4:e7961. doi: 10.1371/journal.pone.0007961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–22. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Dean SN, Bishop BM, van Hoek ML. Susceptibility of Pseudomonas aeruginosa biofilm to alpha-helical peptides: D-enantiomer of LL-37. Front Microbiol. 2011;2:128. doi: 10.3389/fmicb.2011.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C, Korolik V, Bains M, Nguyen U, Breidenstein EBM, et al. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic Peptide. Antimicrob Agents Chemother. 2012;56:2696–704. doi: 10.1128/AAC.00064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C, Reffuveille F, Fernández L, Hancock REW. Bacterial biofilm development as a multicellular adaptation: antibiotic resistance and new therapeutic strategies. Curr Opin Microbiol. 2013;16:580–9. doi: 10.1016/j.mib.2013.06.013. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Núñez C, Reffuveille F, Haney EF, Straus SK, Hancock REW. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014a;10:e1004152. doi: 10.1371/journal.ppat.1004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C, Mansour SC, Wang Z, Jiang L, Breidenstein EB, Elliott M, Reffuveille F, Speert DP, Reckseidler-Zenteno SL, Shen Y, et al. Anti-biofilm and immunomodulatory activities of peptides that inhibit biofilms formed by pathogens isolated from cystic fibrosis patients. Antibiotics. 2014b;3:509–526. doi: 10.3390/antibiotics3040509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DM, Nijnik A, Mayer ML, Hancock REW. Potential of immunomodulatory host defense peptides as novel anti-infectives. Trends Biotechnol. 2009;27:582–90. doi: 10.1016/j.tibtech.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Kjellerup BV. Exploring the applications of invertebrate host-pathogen models for in vivo biofilm infections. FEMS Immunol Med Microbiol. 2012;65:205–14. doi: 10.1111/j.1574-695X.2012.00975.x. [DOI] [PubMed] [Google Scholar]
- Epand RM, Vogel HJ. Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- Fjell CD, Hiss JA, Hancock REW, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2011;11:37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- Hancock REW, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–7. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–73. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- Hilchie AL, Wuerth K, Hancock REW. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat Chem Biol. 2013;9:761–8. doi: 10.1038/nchembio.1393. [DOI] [PubMed] [Google Scholar]
- Hilpert K, Volkmer-Engert R, Walter T, Hancock REW. High-throughput generation of small antibacterial peptides with improved activity. Nat Biotechnol. 2005;23:1008–12. doi: 10.1038/nbt1113. [DOI] [PubMed] [Google Scholar]
- Kostakiot M, Hadjifrangiskou M, Hultgren SJ. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med. 2013;3:a010306. doi: 10.1101/cshperspect.a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 2011;334:982–6. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Annu Rev Microbiol. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- Overhage J, Campisano A, Bains M, Torfs EC, Rehm BH, Hancock REW. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun. 2008;76:4176–82. doi: 10.1128/IAI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Potrykus K, Cashel M. (p)ppGpp: still magical? Annu Rev Microbiol. 2008;62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- Raskin DM, Judson N, Mekalanos JJ. Regulation of the stringent response is the essential function of the conserved bacterial G protein CgtA in Vibrio cholerae. Proc Natl Acad Sci USA. 2007;104:4636–41. doi: 10.1073/pnas.0611650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reffuveille F, de la Fuente-Núñez C, Mansour S, Hancock REW. A broad-spectrum anti-biofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob Agents Chemother. 2014;58:5363–71. doi: 10.1128/AAC.03163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, Dhand A, Pogliano J, Yeaman MR, Hensler ME, Bayer AS, Nizet V. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med (Berl) 2014;92:139–49. doi: 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprawska-Lupa M, Mydel P, Krawczyk K, Wójcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 2004;48:4673–9. doi: 10.1128/AAC.48.12.4673-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. Maintenance of C. elegans. Wormbook. 2006;11:1–11. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugisaki K, Hanawa T, Yonezawa H, Osaki T, Fukutomi T, Kawakami H, Yamamoto T, Kamiya S. Role of (p)ppGpp in biofilm formation and expression of filamentous structures in Bordetella pertussis. Microbiology. 2013;159:1379–89. doi: 10.1099/mic.0.066597-0. [DOI] [PubMed] [Google Scholar]
- Taylor CM, Beresford M, Epton HA, Sigee DC, Shama G, Andrew PW, Roberts IS. Listeria monocytogenes relA and hpt mutants are impaired in surface-attached growth and virulence. J Bacteriol. 2002;184:621–8. doi: 10.1128/JB.184.3.621-628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosa T, Pizer LI. Biochemical bases for the antimetabolite action of L-serine hydroxamate. J Bacteriol. 1971;106:972–82. doi: 10.1128/jb.106.3.972-982.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Acker H, Van Dijck P, Coenye T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 2014;22:326–33. doi: 10.1016/j.tim.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Wiegand I, Hilpert K, Hancock REW. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 2008;3:163–75. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.