

Abstract
In response to gamma-irradiation (IR) induced DNA damage, activation of cell cycle checkpoints results in cell cycle arrest, allowing time for DNA repair prior to cell cycle reentry. Human cells contain G1 and G2 cell cycle checkpoints. While G1 checkpoint is defective in most cancer cells, commonly due to mutations and/or alterations in the key regulators of G1 checkpoint (e.g. p53, Cyclin D), G2 checkpoint is rarely impaired in cancer cells, which is important for cancer cell survival. G2 checkpoint activation involves activation of ataxia telangiectasia mutated (ATM)/ATM- and rad3-related (ATR) signalings, which leads to inhibition of Cdc2 kinase and subsequent G2/M cell cycle arrest. Previous studies from our laboratory show that G2 checkpoint activation following IR exposure of MCF-7 breast cancer cells is dependent on the activation of extracellular signal-regulated protein kinase 1 and 2 (ERK1/2) signaling. Since HER receptor tyrosine kinases (RTKs), which play important roles in cell proliferation and survival, have been shown to activate ERK1/2 signaling in response to various stimuli, we investigated the role of HER RTKs in IR-induced G2/M checkpoint response in breast cancer cells.
Results of the present studies indicate that IR exposure resulted in a striking increase in phosphorylation of HER1, HER2, HER3 and HER4 in MCF-7 cells, indicative of activation of these proteins. Furthermore, specific inhibition of HER2 using an inhibitor, short hairpin RNA and dominant negative mutant HER2 abolished IR-induced activation of ATM/ATR signaling, phosphorylation of Cdc2-Y15 and subsequent induction of G2/M arrest. Moreover, the inhibition of HER2 also abrogated IR-induced ERK1/2 phosphorylation. In contrast, inhibition of HER1 using specific inhibitors or decreasing expression of HER3 or HER4 using shRNAs did not block the induction of G2/M arrest following IR. These results suggest an important role of HER2 in the activation of G2/M checkpoint response following IR.
Keywords: Irradiation, G2/M checkpoint activation, HER receptor tyrosine kinases, ATM/ATR
Introduction
Cells rely on G1 and G2 cell cycle checkpoints to maintain their genomic integrity.1 While most cancer cells are defective in G1 checkpoint, commonly due to the mutation/alteration of key regulators of G1 checkpoint,2 the G2 checkpoint is rarely impaired in cancer cells.1
In response to ionizing irradiation (IR), cell cycle checkpoints are rapidly activated, resulting in either cell cycle arrest, which allows time for repairing the damage, or apoptosis induction, which eliminates the deregulated cells.3 G2 checkpoint is tightly controlled by the Cdc2/Cyclin B complex, whose activity is required for G2/M transition of the cell cycle.4 Previous studies identify Cdc2-Y15 as a vital site involved in G2 checkpoint control in response to IR. Cdc2-Y15 is phosphorylated during radiation-induced G2/M arrest and introduction in fission yeast of a mutant Cdc2-Y15F abolished DNA-damage induced G2/M arrest.5-7 Cdc2-Y15 is phosphorylated by Wee1 and Myt1 kinases8,9 and dephosphorylated by Cdc25 dual-specificity phosphatases.10
In response to DNA-damage, ATM and ATR kinases are rapidly activated, which, in turn, induces the phosphorylation/activation of their respective downstream targets, Chk1 and Chk2 kinases. Activation of Chk1 and Chk2 results in phosphorylation of Cdc25, leading to the subcellular sequestration, degradation and/or inhibition of the Cdc25 that normally activate Cdc2/Cyclin B at the G2/M boundary.11
Cell cycle transition from G2 to mitotic-phase requires histone H3-Ser10 phosphorylation, which is associated with chromosome condensation prior to cell division.12 Since both G2 and mitotic cells contain 4N-DNA content and are not distinguishable from each other by DNA content analysis, H3-Ser10 phosphorylation is commonly used as a specific marker for mitotic cells within the 4N-DNA content cell population.13 Furthermore, the initial H3-Ser10 phosphorylation occurs in the late G2 phase but only on the pericentromeric chromatin. As cells progress through mitosis, the phosphorylation spreads along chromosomes and is completed at the end of prophase.14,15 Thus, there is a gradual increase in H3-Ser10 phosphorylation from the beginning to the end of mitosis. In log-phase cells, H3-Ser10 phosphorylation in mitotic cells is detected in a wide range by flow cytometry analysis.16,17 Upon induction of G2/M arrest, H3-Ser10 phosphorylation is inhibited due to the blockage of G2/M transition of the cell cycle.4,16,17
ERK1/2 signaling plays a critical role in cell proliferation and survival, and has been implicated in the development of cancer therapy resistance.18-21 Studies from our laboratory and others have shown that ERK1/2 signaling is often activated in breast cancer cells by IR and chemotherapy drugs,17,22-24 and that this is associated with a G2/M cell cycle arrest.17,24-26 Recent studies from our laboratory demonstrated that ERK1/2 inhibition alters the response of breast cancer cells to IR and chemotherapy drugs, resulting in an attenuation of G2/M arrest and a concomitant induction of apoptosis.24,26,27 These results indicate a necessary role of ERK1/2 signaling in the response of breast cancer cells to IR and chemotherapy drug treatment.
HER [also called ERBB or EGFR] family of receptor tyrosine kinases (RTKs) consists of HER1, HER2, HER3 and HER4, which localize on the membrane.18 HER RTKs share a similar protein structure that contains an extracellular region (ligand binding and dimerization domains), a transmembrane region and an intracellular region (protein tyrosine kinase domain and phosphorylation regulatory tail).28 Among HERs, HER2 has no known ligand and HER3 expresses very low kinase activity.28 Binding of ligands to the ligand binding domain of HER1, HER3 and HER4 results in homo- or hetero-dimerization of the receptors followed by trans-phosphorylation of several tyrosines within the regulatory tail at the c-termini of the receptor.28 The phosphorylated-tyrosines serve as docking sites for downstream adaptors and signal transducers, activating the HER receptor signaling network.29 HER RTKs are essential for normal cell physiology including proliferation and survival.20 Furthermore, HER1 and HER2 are frequently upregulated or mutated in a broad spectrum of cancer types and approximately 25% of human breast cancers overexpress HER2.29-31 Moreover, HER1 and HER2 are reported to be necessary for the activation of ERK1/2 and AKT signaling in response to various stimuli.20
HER1 activation following IR has been reported previously.32-34 However, the effects of IR on the other HER RTKs are not known. Furthermore, inhibition of HER RTKs has been shown to increase radiosensitivity of cancer cells. While inhibition of HER1/2/3/4 by HER pan-inhibitor CI-1033 significantly enhances radiosensitivity of human colon carcinoma cells both in vitro and in vivo,35 HER2 inhibition by Herceptin and HER1 inhibition by gefitinib respectively sensitizes breast cancer cells and EGFR amplified glioma cells to radiation.36,37 However, the impact of HER RTKs on IR-induced cell cycle checkpoint response was not examined in these previous studies. In the current study, we investigated the effect of IR on HER receptors and the role of HER receptors in the activation of G2/M checkpoint response following IR in human breast cancer cells. Results in this report indicate that IR activates HER1/2/3/4 in human breast cancer cells and that HER2 activation is specifically required for G2 checkpoint activation following IR.
Results
IR induces phosphorylation of HER RTKs in breast cancer cells
To investigate the role of HER RTKs in the response of breast cancer cells to IR, we analyzed HER expression in SkBr3, ZR-75-1 and MCF-7 cells. MCF-7 and ZR-75-1 are lines derived from the Luminal A subtype of breast cancer cells and SkBr3 is a line derived from the HER2 overexpressing subtype of breast cancer cells (Table 1).38 As shown in Figure 1a, the expressions of HER1/2/3 were detected in all three breast cancer cell lines, whereas HER4 expression was not detected in SkBr3 cells.
Table 1.
The characteristics of the indicated breast cancer cell lines.38
| Cell line | ER | PR | HER2 | EGFR | Subtype |
|---|---|---|---|---|---|
| MCF-7 | + | + | 0-1+ | 1+ | Luminal A |
| ZR-75-1 | + | + | 2+ | 1+ | Luminal A |
| SKBr3 | - | - | 3+ | 2+ | HER2 |
Figure 1.

IR induces activation of EGFR/HER family members in breast cancer cells. (a) Expression of HER1, 2, 3 and 4 in the indicated cells were assessed by Western blot analysis. The HER1, 2, 3 and 4 proteins were detected at molecular weights of 170-, 185-, 180- and 180-KD respectively. (b) MCF-7 cells were exposed to 10-Gy IR and incubated for the time indicated. The cells were analyzed for phosphorylation and level of HER1, 2, 3 and 4 by immunoblotting, as described in Materials and Methods (p-HER1, 2, 3, 4 and HER1, 2, 3, 4). The level of Actin was assessed for protein loading control. (c) SkBr3 cells were exposed to 10-Gy IR, incubated for the times indicated and analyzed for phosphorylation and level of HER1, 2 and 3 by immunoblotting.
We next examined the effect of IR on HER-phosphorylation in MCF-7 cells. As shown in Figure 1b, immunoblotting respectively detected 1.5-, 9- and 2-fold increases in the phosphorylation of HER1, HER2 and HER3 in MCF-7 cells within 15 min post IR. At 1 h post IR, HER2 phosphorylation further increased, while HER1 and HER3 phosphorylation decreased to a level even lower than that of the unirradiated cells (Figure 1b). IR also induced HER4 phosphorylation in MCF-7 cells at 1 h post IR (Figure 1b). There was no change in protein levels of HER1, HER3 and HER4 following IR. However, an increase of HER2 protein was detected in the irradiated cells at 1 h post IR (Figure 1b).
We also examined the HER-phosphorylation in SkBr3 cells following IR. As shown in Figure 1c, while IR had little effect on the HER1 phosphorylation in SkBr3 cells, IR induced phosphorylation of both HER2 and HER3 in SkBr3 cells. Furthermore, the increase of HER2 phosphorylation was observed within 15 min following IR and the increase of HER3 phosphorylation was detected at 1 h post IR. Collectively, these results indicate an activation of HER RTKs in breast cancer cells following IR.
IR induces G2/M arrest in breast cancer cells
We examined the effect of IR on cell cycle response of breast cancer cells. Log-phase cells were exposed to IR and analyzed for DNA content by fluorescence-activated cell sorting (FACS) at 24 h following IR. As shown in Figure 2a, IR exposure induced a dose-dependent increase in the amount of 4N-DNA content cells, indicative of G2/M cell cycle arrest, in MCF-7 cells.4 Furthermore, as shown in Figure 2a (lower right panel), while the induction of G2/M arrest following IR was time-dependent with a maximum detected at 6 h post IR, the amounts of G1-and S-phase cells were decreased flowing IR. Consistent with the results obtained from MCF-7 cells, IR also induced a dose-dependent accumulation in G2/M phase cells in ZR-75-1 and SkBr3 cells (Figure 2b).
Figure 2.
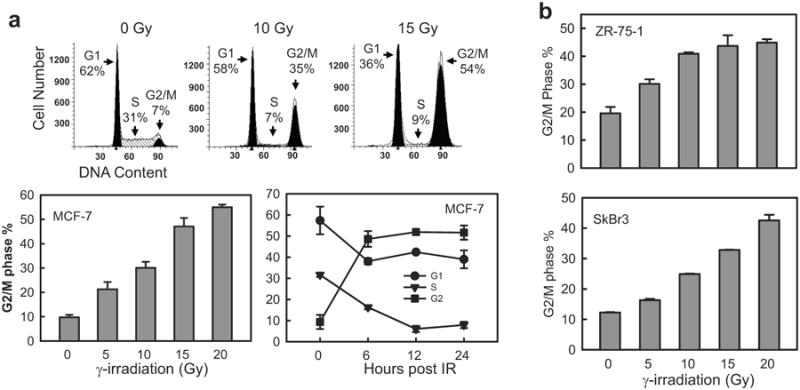
IR induces G2/M arrest in breast cancer cells. (a) Log-phase MCF-7 cells were exposed to IR at the indicated doses. The cells were incubated for 24 h and analyzed for cell cycle by FACS. Upper panels: The histograms shown are representative FACS analyses. Amounts of cells in G1, S and G2/M phase of the cell cycle are indicated. Lower left panel: MCF-7 cells were exposed to IR at the dose indicated, incubated for 24 h and analyzed for cell cycle. Results depict the percentage of cells in G2/M phase and represent the mean±s.d. of two sets of experiment in duplicates. Lower right panel: MCF-7 cells were exposed to 15-Gy IR and incubated for the time indicated. Results depict the percentage of cells in G1, S and G2/M phase of the cell cycle and represent the mean±s.d. of three sets of experiments in duplicates. (b) ZR-75-1 and SkBr3 cells were exposed to IR at the indicated doses, incubated for 24 h and analyzed for cell cycle by FACS. Results depict the percentage of cells in G2/M phase and represent the mean±s.d. of two sets of experiment in duplicates. (c) MCF-7 cells were exposed to 10-Gy IR, incubated for the indicated times and analyzed for the phosphorylation of Chk1-S317 and Chk2-T68 by immunoblotting. As controls, amounts of Chk1, Chk2 and Actin in the cell lysates were assessed by immunoblotting. (d) MCF-7 cells were exposed to 10-Gy IR, incubated for the time indicated and analyzed by flow cytometry for mitotic cells, which contain both 4N-DNA content and Histone H3-Ser10 phosphorylation. Upper panel: The histograms shown are representative flow cytometry analyses for mitotic cells in samples treated with/without IR and incubated for 3 h. The location of mitotic cells in each sample is indicated (M). Lower panel: The bar graph depicts the percentage of mitotic cells and is shown as mean±s.d. of two sets of experiments in duplicates.
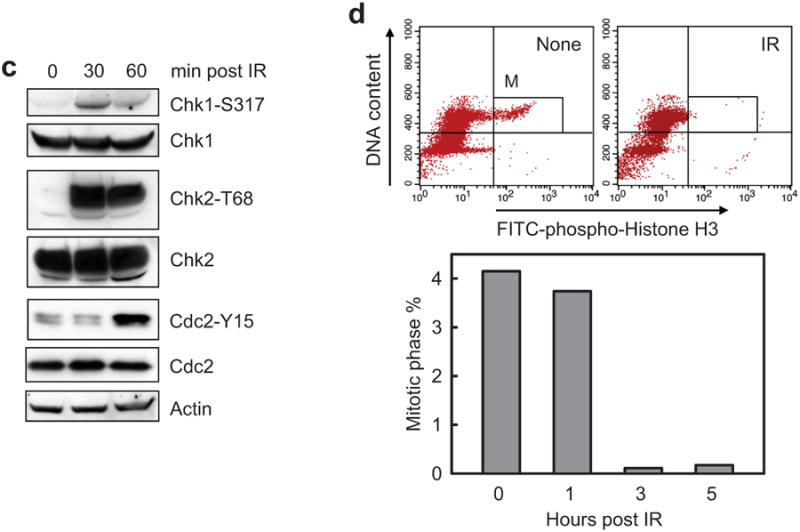
To confirm the activation of G2 checkpoint in these cells, irradiated MCF-7 cells were analyzed for markers of G2 checkpoint. As shown in Figure 2c, immunoblotting detected an increase in phosphorylation of Chk1-S317, Chk2-T68 and Cdc2-Y15, indicative of activation of the G2 checkpoint signaling,39 in irradiated MCF-7 cells. Since G2 checkpoint activation inhibits the G2 to M transition of the cell cycle,4 we examined the proportion of mitotic cells after IR using histone-H3 phosphorylation as a marker of mitotic cells.40 As shown in Figure 2d, IR resulted in a marked reduction in the percentage of mitotic cells. At 3 h post IR, a 96% decrease in mitotic cells was noted in the irradiated cells relative to unirradiated cells (Figure 2d).
Inhibition of HER RTKs abolishes the induction of G2/M arrest following IR in breast cancer cells
Using CI1033, a pan-inhibitor of the HER family,41 we examined the role of HER RTKs in IR-induced G2/M cell cycle arrest. For these studies, MCF-7 cells were incubated for 1 h with increasing doses of CI1033 prior to IR. As shown in Figure 3a, incubation of MCF-7 cells with 20 μM CI1033 resulted in near complete inhibition in IR-induced phosphorylation of HER1/2/3/4 (at 15 min following IR for HER1/2/3, and at 1 h post IR for HER4). Incubation with CI1033 had no effect on the levels of these proteins. As shown in Figure 3b, incubation with 20 μM CI1033 abrogated the induction of G2/M arrest following IR in MCF-7, ZR-75-1 and SkBr3 cells. In contrast, incubation with CI1033 without IR had little, if any, effect on the percentage of cells in G2/M phase relative to control cells (Figure 3b, solid bars). Furthermore, pre-incubation of cells with 2 μM CI1033, a dose that did not inhibit the IR-induced HER-phosphorylation, had no significant effect on the IR-induced G2/M arrest in these cells (Figure 3b).
Figure 3.


Inhibition of HER receptors by CI1033 abrogates IR-induced G2/M arrest in breast cancer cells. (a) MCF-7 cells were incubated for 1 h with CI1033 at the doses indicated, treated with/without 10-Gy IR and then incubated for 15 min (analysis of HER1, 2 and 3) or 1 h (analysis of HER4) at 37°C. The cells were analyzed for phosphorylation and level of HER1, 2, 3 and 4 by immunoblotting using specific antibodies. As protein loading controls, Actin levels in the cell lysates were assessed by immunoblotting. (b) MCF-7, ZR-75-1 and SkBr3 cells were incubated with CI1033 for 1 h at the indicated doses, exposed to IR at 10-Gy (SkBr3 and ZR-75-1) or 15-Gy (MCF-7). The cells were incubated for 24 h and analyzed for cell cycle by FACS. Results depict the percentage of cells in G2/M phase and represent the mean±s.d. of two sets of experiments in duplicates.
Inhibition of HER1 does not block IR-induced G2/M arrest in breast cancer cells
Using AG1478, a HER1 specific inhibitor,42 we examined the effect of HER1 on IR-induced HER1/2/3/4 phosphorylation. As shown in Figure 4a, the presence of 10 μM AG1478 effectively abrogated IR-induced HER1 phosphorylation in MCF-7 cells, whereas it did not block IR-induced phosphorylation of HER2 and HER3. Incubation with 10 μM AG1478 also abolished the increase of HER4 phosphorylation following IR in MCF-7 cells. Incubation with AG1478 had little effect on the protein levels of HER RTKs (Figure 4a).
Figure 4.


Inhibition of EGFR/HER1 by AG1478 does not block the induction of G2/M arrest in breast cancer cells following IR. (a) MCF-7 cells were incubated with AG1478 at the indicated concentrations for 1 h and exposed to 10-Gy IR. Following incubation for 15 min (HER1, 2 and 3) or 1 h (HER4) post IR, the cells were assessed for the phosphorylation and level of HER1, 2, 3 and 4 by immunoblotting. As controls, Actin levels in cell lysates were assessed by immunoblotting. (b) MCF-7 and SkBr3 cells were respectively exposed to 15-Gy and 10-Gy IR in the presence of increasing doses of AG1478, incubated for 24 h and analyzed for DNA content by FACS. Results depict the percentage of cells in G2/M phase and represent the mean±s.d. of two sets of experiment in duplicates. (c) Upper panel: MCF-7 cells were incubated with Erlotinib at the indicated concentrations for 1 h and exposed to 15-Gy IR. Following incubation for 15 min post IR, the cells were analyzed for the phosphorylation and level of HER1 by immunoblotting. Lower panel: In the presence of increasing doses of Erlotinib, MCF-7 cells were exposed to 15-Gy IR, incubated for 24 h and analyzed for DNA content by FACS. Results depict the percentage of cells in G2/M phase and represent the mean±s.d. of two sets of experiments in duplicates.
We next examined the effect of HER1 inhibition on the induction of G2/M arrest following IR. MCF-7 and SkBr3 cells were exposed to 10-Gy IR in the presence of increasing doses of AG1478, incubated for 24 h and assessed for the cells in G2/M phase by FACS. As shown in Figure 4b, the presence of AG1478 did not block the induction of G2/M arrest following IR in both MCF-7 and SkBr3 cells.
To verify the effect of AG1478 on IR-induced G2/M arrest, we examined Erlotinib, a clinically used HER1 inhibitor for cancer therapy.43 As shown in Figure 4c (upper panel), incubation with Erlotinib at ≥5 μM efficiently inhibited IR-induced HER1 phosphorylation in MCF-7 cells. However, incubation with Erlotinib up to 10 μM had no effect on the induction of G2/M arrest following IR in MCF-7 cells. These results indicate that HER1 activation is not required for the IR-induced G2/M arrest in MCF-7 cells.
HER2 inhibition by CP724714 abrogates IR-induced G2 checkpoint activation
We next examined the effect of HER2 on IR-induced G2/M checkpoint response using HER2 specific inhibitor CP724714. Previous studies indicate that CP724714 is a potent inhibitor of HER2 auto-phosphorylation with no detectable effect on EGF-induced HER1 phosphorylation.44 As shown in Figure 5a (upper panel), incubation with 50 μM CP724714 effectively abolished IR-induced HER2 phosphorylation in MCF-7 cells. This effect is dose-dependent, as CP724714 at 10 μM only inhibited 40% of IR-induced HER2 phosphorylation in MCF-7 cells (Figure 5a, upper panel). We also tested the effect of CP724714 on HER2 phosphorylation in SkBr3 cells, which overexpress HER2 (Figure 1a). As shown in Figure 5a (lower panel), incubation with CP724714 also resulted in a dose-dependent inhibition of HER2 auto-phosphorylation in log-phase SkBr3 cells. Incubation with CP724714 at 10 μM and 30 μM inhibited HER2 phosphorylation by 21% and 92%, respectively, in SkBr3 cells. Since HER2 interacts with and regulates the other HER family members, we examined the effect of CP724714 on the phosphorylation of other HER members. As shown in Figure 5b, incubation of MCF-7 cells with 50 μM CP724714 also abrogated IR-induced HER3/4 phosphorylation but not the IR-induced HER1 phosphorylation.
Figure 5.


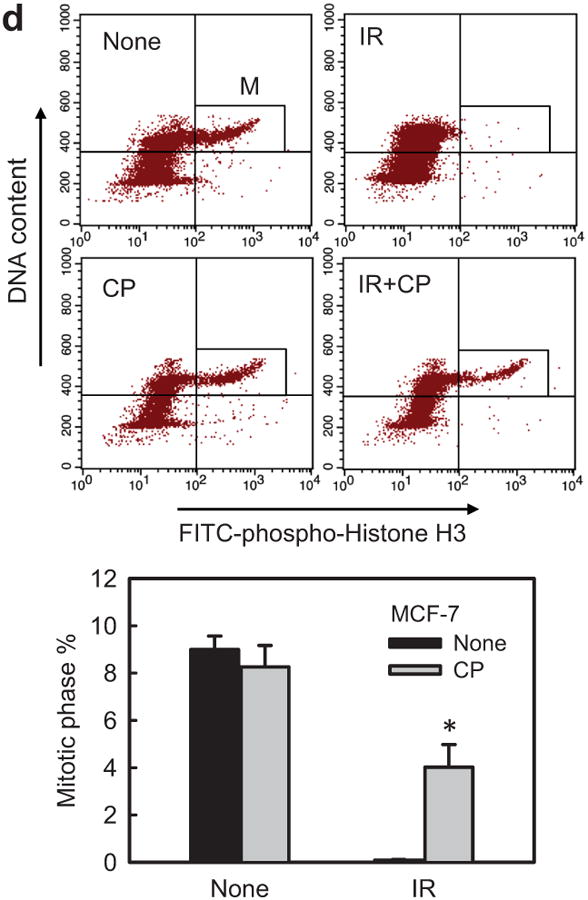
Incubation with CP724714 HER2 selective inhibitor abrogates the induction of G2/M arrest following IR. (a) Upper panel: MCF-7 cells were incubated with CP724714 at the doses indicated for 1 h, exposed to 10-Gy IR and incubated for 15 min. The cells were analyzed for levels of HER2 phosphorylation, HER2 protein and Actin by immunoblotting. Lower panel: SkBr3 cells were incubated with CP724714 at the indicated doses for 1 h and analyzed for HER2 phosphorylation and total protein by immunoblotting. (b) MCF-7 cells were incubated in the presence/absence of 50 μM CP-724714 for 1 h, exposed to 10-Gy IR and incubated for 15 min (HER1 and HER3) or 1 h (HER4). The resulting cells were analyzed for phosphorylation and protein level of HER1, 3 and 4. The levels of GAPDH in the lysates were assessed to confirm the equal protein loadings. (c) Upper and lower left panels: breast cancer cells were incubated with increasing doses of CP-724714 for 1 h and exposed to IR at 15-Gy (MCF-7) or 10-Gy (SkBr3 and ZR-75-1). The cells were incubated for 24 h post IR and analyzed for cell cycle by FACS. Result depicts the percentage of cells in G2/M phase and is shown as mean±s.d. of two separate experiments in duplicates. Lower right panel: MCF-7 cells were incubated in the presence/absence of CP724714 for 1 h and exposed to increasing doses of IR. The cells were incubated for 8 h post IR and analyzed for cell cycle. Result depicts the percentage of cells in G2/M phase and is shown as mean±s.d. of two separate experiments in duplicates. (d) MCF-7 cells were incubated with/without 50 μM CP-724714 for 1 h, exposed to 10-Gy IR, incubated for 3 h and analyzed for mitotic cells. Upper panel: the histograms shown are representative flow cytometry analyses for mitotic cells in the samples treated with/without IR in the presence or absence of CP-724714. The location of mitotic cells is indicated (M). Lower panel: the bar graph depicts the percentage of mitotic cells and is shown as mean±s.d. of triplicate samples. *, p=<0.001 (n=3), significant difference from cells exposed to IR in the absence of CP-724714.
Subsequently, we tested the effect of CP724714 on IR-induced G2/M arrest in MCF-7, ZR-75-1 and SkBr3 cells. As shown in Figure 5c, the induction of G2/M arrest after IR was effectively abrogated by incubation of cells with 50 or 100 μM CP724714 with little effect noted in cells incubated with 10 μM CP724714, a dose that had little inhibitory effect on HER2 phosphorylation in both MCF-7 and SkBr3 cells (Figure 5a). Our previous published data demonstrates that the IR-induced increase in the amount of G2/M phase cells can be detected as early as 8 h post IR.45 We therefore tested the effect of CP724714 on IR-induced G2/M arrest at 8 h following IR. As shown in Figure 5c (lower right panel), incubation with 50 μM CP724714 also respectively abrogated the induction of G2/M arrest in MCF-7 cells exposed to 6- and 12-Gy IR.
Phosphorylation of histone-H3 by Cdc2/Cyclin B is required for cells entering into mitosis.40 We therefore tested the effect of CP724714 on the amount of mitotic cells following IR. As shown in Figure 5d, >90% decrease in the proportion of mitotic cells was detected in irradiated MCF-7 cells relative to control non-irradiated cells (black bars). In contrast, incubation with CP724714 blocked the effect of IR, resulting in a significant increase in the proportion of mitotic cells in irradiated cells compared to the control irradiated cells (Figure 5d, IR). Incubation with CP724714 alone resulted in a slight decrease in the amount of mitotic cells compared to the control untreated cells (Figure 5d, None), but the effect was not statistically significant.
Since ATM/ATR signaling play important roles in the activation of G2 checkpoint response,11 we examined the effect of CP724714 on the activation of ATM and ATR signaling following IR. As shown in Figures 6a and 6b (upper panels), incubation with CP724714 prior to IR prominently inhibited the activation of ATM and ATR following IR in MCF-7 cells. Furthermore, incubation with CP724714 also markedly diminished the IR-induced activation of Chk2 (downstream target of ATM) and Chk1 (downstream target of ATR) in MCF-7 as well as in ZR-75-1 cells (Figure 6a and 6b). We also noticed that CP724714 had more effect on the IR-induced Chk2 activation in MCF-7 cells compared to ZR-75-1 cells. While CP724714 completely inhibited the IR-induced Chk2 activation in MCF-7 cells, it reduced IR-induced Chk2 activation by 47% in ZR-75-1 cells. The cause for this difference in the effect of CP724714 is unclear. It may be attributed to cell-type specificity.
Figure 6.


Incubation with CP724714 HER2 selective inhibitor abolishes IR-induced G2/M checkpoint activation. (a) After pre-incubation for 1 h at 37°C in the presence or absence of 50 μM CP724714, MCF-7 and ZR-75-1 cells were exposed to 10-Gy IR or left non-irradiated and then incubated for an additional 1 h at 37°C. Upper panel: ATM was immunoprecipitated from MCF-7 cell lysates using Ab-3 anti-ATM antibody and assayed for kinase activity, in the presence of [gamma-32P]ATP, using p53 recombinant protein as substrate. ATM kinase activity is expressed as the amount of phosphorylated-p53 recombinant protein detected by autoradiography (ATM activity). As controls, the level of ATM, detected as a 370-KD protein, was assessed by immunoblotting in the immunoprecipitates and in the cell lysates, respectively (ATM IP-WB and ATM). Chk2 was immunoprecipitated from cell lysate and assayed for kinase activity using Cdc25C recombinant protein as substrate. Chk2 activity is expressed as the amount of phosphorylated-Cdc25C recombinant protein detected by autoradiography (Chk2 activity). As controls, Chk2 protein (62-KD) in the immunoprecipitates as well as in the cell lysates was measured by immunoblotting (Chk2 IP-WB and Chk2). Lower panel: Chk2 was immunoprecipitated from ZR-75-1 cell lysate and assayed for kinase activity using Cdc25C recombinant protein as substrate (Chk2 activity). As a control, Chk2 protein in the immunoprecipitates was measured by immunoblotting (Chk2 IP-WB). (b) Upper panel: ATR was immunoprecipitated from MCF-7 cell lysates using N-19 anti-ATR antibody and assayed for kinase activity using p53 recombinant protein as substrate (ATR activity). ATR kinase activity is expressed as the amount of phosphorylated-p53 recombinant protein detected by autoradiography (ATR activity). As controls, levels of ATR, detected as a 250-KD protein, in the immunoprecipitates and cell lysates (ATR IP-WB and ATR) were determined by immunoblotting. Chk1 was immunoprecipitated from MCF-7 cell lysates using G-4 anti-Chk1 antibody and assayed for kinase activity using Cdc25C recombinant protein as substrate. Chk1 activity is expressed as the amount of phosphorylated-Cdc25C recombinant protein detected by autoradiography (Chk1 activity). As controls, Chk1 protein (56-KD) in the immunoprecipitates and in the cell lysates was respectively measured by immunoblotting (Chk1 IP-WB and Chk1). Lower panel: Chk1 was immunoprecipitated from ZR-75-1 cell lysates and assayed for kinase activity using Cdc25C recombinant protein as substrate (Chk1 activity). As controls, Chk1 protein in the immunoprecipitates was determined by immunoblotting (Chk1 IP-WB). (c) Cdc2, a 34-KD protein, was immunoprecipitated from the cell lysates and analyzed for Cdc2-Y15 phosphorylation and Cdc2 protein by immunoblotting (Cdc2-Y15 and Cdc2 IP-WB). As controls, levels of Cdc2 and Actin in the cell lysates were assessed by immunoblotting.
The activation of ATM/ATR signaling leads to the induction of Cdc2-Y15 phosphorylation, which inhibits Cdc2 activity.4 We then tested the effect of CP724714 on Cdc2-Y15 phosphorylation in irradiated MCF-7 cells. As shown in Figure 6c, IR-induced Cdc2-Y15 phosphorylation in MCF-7 cells was abolished by the presence of CP724714 prior to IR. Incubation with CP724714 alone had no effect on Cdc2-Y15 phosphorylation in MCF-7 cells.
Collectively, these results suggest possible roles for HER2, HER3 and/or HER4 in the activation of G2 checkpoint response in breast cancer cells.
Decreased HER2 expression by short-hairpin RNA (shRNA) diminishes IR-induced G2/M cell cycle arrest
Using specific shRNAs, we directly examined the role of HER2, HER3 and HER4 in IR-induced G2/M arrest in MCF-7 cells.
As shown in Figure 7a (upper panel), HER2-shRNA expressing clones exhibited a marked decrease in HER2 protein levels compared to Control-shRNA transduced cells. Furthermore, DNA-content analysis revealed that HER2-shRNA expressing cells displayed a significant diminution of IR-induced G2/M arrest compared to control cells. In contrast to the 4.4-fold increase in the amount of G2/M DNA-content cells in the irradiated control cells relative to non-irradiated control cells, there is only a 1.6-fold increase in the amount of G2/M DNA-content cells in the irradiated HER2-shRNA expressing cells compared to non-irradiated HER2-shRNA expressing cells (Figure 7a, middle panel). No difference in IR-induced G2/M arrest was observed between control-shRNA expressing cells and non-transduced cells (data not shown).
Figure 7.



Decrease of HER2 protein expression by HER2-shRNA attenuates the induction of G2/M arrest in MCF-7 cells following IR. MCF-7 cells were infected with retroviral vector expressing shRNA targeting HER2, HER3, HER4 or, as a control, non-targeting shRNA. The infected cells were selected for stable shRNA expressing clone as described in Materials and Methods. (a) Upper panel: HER2-shRNA expressing clones were analyzed for HER2 protein level by immunoblotting. GAPDH in the cell lysates were measured by immunoblotting as a protein loading control. Middle panel: the HER2-shRNA expressing cells were exposed to IR at the indicated doses, incubated for 24 h and analyzed for DNA content. Result depicts the percentage of cells in G2/M phase and is shown as mean±s.d. of two separate experiments in duplicates. *, p=0.013 (n=4); **, p=<0.001 (n=4), significant difference between control and HER2-shRNA expressing cells. Lower panel: HER2-shRNA expressing and control MCF-7 cells were exposed to 10-Gy IR, incubated for 2 h and analyzed for mitotic cells. Bar graph depicts the percentage of mitotic cells and is shown as mean±s.d. of triplicate samples. *, p=<0.001 (n=3), significant difference from cells expressing control-shRNA exposed to IR. (b) Upper panel: HER3-shRNA expressing clones were analyzed for levels of HER3 protein and GAPDH by immunoblotting. Lower panel: the HER3-shRNA expressing and control cells were exposed to IR at the indicated doses, incubated for 24 h and analyzed for cell cycle. Result depicts the percentage of cells in G2/M phase and is shown as mean±s.d. of three separate experiments in duplicates. (c) Upper panel: HER4-shRNA expressing clones were analyzed for levels of HER4 protein and GAPDH by immunoblotting. Lower panel: the HER4-shRNA expressing cells were exposed to IR at the indicated doses, incubated for 24 h and analyzed for cell cycle. Result depicts the percentage of cells in G2/M phase and is shown as mean±s.d. of three separate experiments in duplicates. (d) Indicated cells were treated with/without 10-Gy IR and incubated for 1 h. Chk1 and Chk2 were immunoprecipitated from cell lysate and assayed for kinase activity using Cdc25C recombinant protein as substrate (Chk2 activity).
We also assessed the effect of HER2-shRNA expression on mitotic cells following IR. As shown in Figure 7a (lower panel), HER2-shRNA expression resulted in a significant increase in the proportion of mitotic cells in irradiated cells compared to control irradiated cells.
We then examined the effect of HER3- and HER4-shRNA on IR-induced G2/M arrest. As shown in Figure 7b and 7c, specific shRNA expression significantly decreased the protein expression of HER3 and HER4 in MCF-7 cells compared to the control-shRNA transduced cells. However, decrease in either HER3 or HER4 expression by shRNA did not block the induction of G2/M arrest in MCF-7 cells after IR.
The activation of Chk1 and Chk2 following IR was assessed using kinase assay in the cells expressing HER2-, HER3- or HER4-shRNA. As shown in Figure 7d, the IR-induced Chk1 and Chk2 activations were markedly diminished in the HER2-shRNA expressing cells compared to control-shRNA transduced cells. In contrast, decreased HER3 or HER4 expression by shRNA did not block the activation of Chk1 and Chk2 following IR in MCF-7 cells. These results suggest a requirement for HER2-mediated signaling in the IR-induced Chk1/2 activation.
Together, these results provide direct evidence supporting a vital role for HER2 in the IR-induced G2 checkpoint response.
Ectopic expression of HER2 dominant negative mutant abolishes the G2 checkpoint activation following IR
We also explored the effects of HER2 dominant negative mutant (HER2-mut)46 on IR-induced G2 checkpoint response. The HER2-mut construct contains the extracellular and transmembrane portion of HER2 protein but lacks the intracellular portion of HER2, which contains the tyrosine kinase domain and phosphoregulatory tail of HER2. Previous studies demonstrate that the HER2-mut lacks kinase activity and functions as a dominant negative mutant.46 Results in Figure 8a show that expression of the HER2-mut in MCF-7 cells abrogated the induction of G2/M arrest following IR. Since the HER2-mut still contains the dimerization domain and can bind to other HER RTKs, we examined the effect of HER2-mut expression on the phosphorylation and level of other HER receptors. Results in Figure 8b showed that HER2-mut expression in MCF-7 cells abolished IR-induced HER1 and HER2 phosphorylation, but did not block the increase of HER3 phosphorylation following IR. Of interest was the finding that the steady-state level of HER1 protein was slightly decreased in HER2-mut expressing cells compared to controls cells, while the levels of HER2, HER3 and HER4 protein were noticeably increased in HER2-mut expressing cells relative to control cells. Furthermore, the HER2-mut expression by itself resulted in a striking increase in HER4 phosphorylation (Figure 8b). These results suggest that mut-HER2 expression has an impact on the activities and/or levels of each of the HER family members.
Figure 8.



Ectopic expression of dominant negative mutant (mut) HER2 abrogates IR-induced G2/M checkpoint activation in MCF-7 cells. (a) MCF-7 cells stably transfected with a vector expressing myc-tagged HER2-mut or a relevant control empty vector were exposed to 10-Gy IR or left un-irradiated. Upper panel: The transfected cells were analyzed for HER2-mut expression by immunoblotting using anti-myc antibody. The mut-HER2 is detected as an ∼80-KD protein by Western blotting. Lower panel: the cells were exposed to 10-Gy IR, incubated for 24 h at 37°C and analyzed for cell cycle. Result depicts the percentage of cells in G2/M phase and is shown as mean±s.d. of triplicate samples, which represents two separate experiments. (b) HER2-mut expressing and control cells were exposed to 10-Gy IR and incubated for 15 min (HER1 and HER3) or 1 h (HER4). The cells were analyzed for phosphorylation and protein level of HER1, 2, 3 and 4. The levels of GAPDH in the lysates were assessed to confirm the equal protein loadings. (c) mut-HER2 expressing and control cells were exposed to 10-Gy IR, incubated for 1 h and analyzed for ATM and Chk2 activities as described above. As controls, the levels of ATM and Chk2 in the immunopreciptate and in cell lysates were analyzed by immunoblotting. GAPDH in cell lysates were probed as a protein loading control. (d) The cell samples above were analyzed for ATR and Chk1 kinase activities. As controls, the levels of ATR and Chk1 in the immunopreciptate and in cell lysates were analyzed by immunoblotting. GAPDH in cell lysates were analyzed as a protein loading control. (e) Cdc2 was immunoprecipitated from the cell samples described above in (c) and analyzed for levels of Cdc2-Y15 phosphorylation and Cdc2 protein by immunoblotting (Cdc2-Y15 and Cdc2 IP-WB). As controls, levels of Cdc2 and Actin in the cell lysates were assessed by immunoblotting.
We next tested the effect of HER2-mut on the activation of ATM and ATR signalings following IR. As shown in Figure 8c, IR-induced activation of ATM and Chk2 activities was effectively abrogated by the expression of HER2-mut in MCF-7 cells. The expression of HER2-mut also abolished the activation of ATR and Chk1 in irradiated MCF-7 cells (Figure 8d). We also unexpectedly observed an increase in the steady-state level of ATM, ATR and Chk1 protein in the HER2-mut expressing cells compared to control cells (Figure 8d, ATM, ATR and Chk1). However, these increases apparently are not associated with ATM, ATR and Chk1 activities. The mechanism causing this effect of HER2-mut is unclear and requires future studies.
Since Cdc2-Y15 phosphorylation is the target of G2 checkpoint signaling, we also examined the effect of mut-HER2 on IR-induced Cdc2-Y15 phosphorylation. As shown in Figure 8e, immunoblot analysis revealed no increase in Cdc2-Y15 phosphorylation in mut-HER2 expressing cells following IR.
Collectively, these results indicate that expression of HER2-mut in MCF-7 cells inhibited IR-induced activation of HER1 and HER2 and abrogated the G2 checkpoint activation following IR.
Effect of HER signaling on IR-induced ERK1/2 activation
Previous studies from our laboratory demonstrated that IR exposure of breast cancer cells activates ERK1/2 signaling and that this is required for G2 checkpoint activation following IR.17 We therefore examined the effect of HER RTKs on IR-induced ERK1/2 activation.
We first tested the effect of CI1033 HER pan-inhibitor on IR-induced ERK1/2 activation. MCF-7 and ZR-75-1 cells were incubated for 1 h in the presence or absence of 20 μM CI1033 and exposed to 10-Gy IR. As shown in Figure 9a, incubation with CI1033, which inhibited the IR-induced phosphorylation of all HER RTKs (Figure 3a), abolished IR-induced ERK1/2 phosphorylation in both MCF-7 and ZR-75-1 cells.
Figure 9.

Effect of HER2 inhibition on IR-induced ERK1/2 activation. (a) MCF-7 and ZR-75-1 cells were incubated in the presence or absence of 20 μM CI1033 for 1 h, exposed to 10-Gy IR and incubated for 15 min. The cells were analyzed for levels of ERK1/2 phosphorylation (p-ERK1/2) and ERK1/2 protein (ERK1/2). The ERK1/2 are detected as 42-KD/44-KD proteins by Western blotting. (b) MCF-7 cells were incubated with 50 μM CP724714 for 1 h, exposed to 10-Gy IR, incubated for 15 min and analyzed for ERK1/2 phosphorylation and ERK1/2 protein. (c) MCF-7 cells expressing mut-HER2 and control cells were exposed to 10-Gy IR, incubated for 15 min and analyzed for ERK1/2 phosphorylation and ERK1/2 protein. (d) MCF-7 cells expressing HER2-shRNA (clone HER2-2-4) and control cells were exposed to 10-Gy IR, incubated for 15 min and analyzed for ERK1/2 phosphorylation and ERK1/2 protein. (e) MCF-7 cells expressing HER3-shRNA (clone HER3-P-3), HER4-shRNA (clone HER4-P-4) and control cells were exposed to 10-Gy IR, incubated for 15 min and analyzed for ERK1/2 phosphorylation and ERK1/2 protein.
We next tested the effect of HER2 specific inhibitor CP724714 on IR-induced ERK1/2 activation. As shown in Figure 9b, incubation with 50 μM CP724714, which inhibited the IR-induced phosphorylation of HER2/3/4 (Figure 5b), abrogated the IR-induced ERK1/2 phosphorylation in MCF-7 cells.
We also examined the effect of HER2-mut on IR-induced ERK1/2 activation. Results in Figure 9c showed that the expression of HER2-mut, which inhibited the IR-induced HER1/2 phosphorylation (Figure 6), abolished ERK1/2 activation in MCF-7 cells following IR.
Lastly, we tested the effect of HER2-shRNA expression on ERK1/2 activation following IR. As shown in Figure 9d, expression of HER2-shRNA, which decreased HER2 protein in MCF-7 cells (Figure 7a), diminished the ERK1/2 activation following IR.
To verify the effect of HER2 inhibition on IR-induced ERK1/2 activation, we assessed the ERK1/2 phosphorylation following IR in cells expressing HER3- or HER4-shRNA. As shown in Figure 9e, decreasing either HER3 or HER4 expression by shRNA had no effect on the IR-induced ERK1/2 activation in MCF-7 cells.
Collectively, these results suggest a requirement for HER2 in the IR-induced ERK1/2 activation in breast cancer cells.
Discussion
HER receptors play critical roles in cell proliferation and survival.28 While their effect on radiosensitivity has been studied before,35-37 their impact on cell cycle checkpoint in response to IR remains largely undefined. The present studies investigated the role of HER RTKs in activation of G2 checkpoint following IR exposure of breast cancer cells.
Previous studies reported a HER1 activation in response to IR, whereas the effects of IR on HER2/3/4 were not examined in these studies.34,51 Results of the current studies reveal that, in various patterns, IR not only activates HER1 but also activates HER2, 3 and 4 in breast cancer cells. The mechanism causing the activation of HER RTKs following IR is unclear. However, previous studies demonstrate that receptor protein tyrosine phosphatases (PTPs), which suppress HER RTKs, can be efficiently inhibited by reactive oxygen/nitrogen species (ROS/RNS) through oxidation.52 Previous studies also show that IR can induce ROS/RNS production via a mitochondria-dependent mechanism.53 Thus, the ROS/RNS induced by IR could lead to the inhibition of PTPs, resulting in the activation of HER RTKs. We will investigate this possible mechanism in future studies.
We also noticed that the IR-induced HER phosphorylations are in various patterns. While IR-induced HER2/4 phosphorylation sustains at 1 h post IR in MCF-7 cells, the IR-induced HER1/3 phosphorylation is diminished at 1 h post IR in the MCF-7 cells (see Figure 1b). The mechanism causing the differences is unclear. However, previous studies reveal a negative feedback regulation between HER1/3 and their respective downstream signalings. For instance: Inhibition of BRAF (V600E) (a downstream target of EGFR signaling) using specific inhibitor PLX4032 results in a feedback activation of EGFR in colon cancer cells.54 Another study shows that the inhibition of MEK signaling (downstream signaling of HER receptors) using AZD6244 causes activation of both HER1 and HER3 phosphorylation in several cancer cell lines via feedback regulatory mechanisms.55 Therefore, the diminution of HER1/3 phosphorylation observed in MCF-7 cells at 1 h post IR may also be caused by negative feedback regulations from the downstream signalings of HER1/3. We also observed that IR induces HER1 phosphorylation in MCF-7 but not SkBr3 cells (see Figure 1). This result suggests a possible involvement of HER4 in IR-induced HER1 activation, as HER4 is expressed in MCF-7 but not SkBr3 cells. We will investigate these possible mechanisms in future studies.
The HER2-mut used in the present study contains the dimerization domain but lacks the kinase domain of HER2.46 Thus, mut-HER2 can still dimerize with its partners but is incapable of activating downstream signalings. In the MCF-7 cells expressing HER2-mut, we surprisingly observed an upregulation in the steady state levels of HER2, HER3 and HER4 and a concomitant decrease in HER1 level (Figure 8b). The mechanism causing these effects of HER2-mut is unclear. It is possible that the protein expressions of HER2/3/4 are negatively regulated by the downstream signalings of HER2 via feedback loops, whereas the maintenance of HER1 protein expression simply requires the kinase activity of HER2. Furthermore, it is noticeable that HER2 and HER3 phosphorylation are not associated with the changes in levels of these proteins (Figure 8b), suggesting a requirement for HER2 kinase activity in the phosphorylation of HER2 and HER3.
The current studies assessed the effect of HER RTKs on IR-induced G2 checkpoint activation in various types of breast cancer cells. Results of these studies indicate that IR-induced G2/M arrest is abrogated by the presence of the HER pan-inhibitor CI1033, the HER2 inhibitor CP724714, the HER2-mut or the HER2-shRNA. In contrast, inhibition of HER1 using specific inhibitor AG1478 or Erlotinib (Figure 4), or decrease of HER3 or HER4 expression by shRNA (Figure 7) does not block the induction of G2/M arrest following IR in breast cancer cells. We also compared the MCF-7 cells overexpressing wild-type HER2 (HER2-wt) with the MCF-7 cells expressing HER2-mut for the induction of G2/M arrest following IR. Results in Supplementary Figure show that IR-induced G2/M arrest is detected in the cells overexpressing HER2-wt but not in the cells expressing HER2-mut. Therefore, the results in this report suggest a requirement for HER2 in the induction of G2/M arrest following IR.
Several studies including our own have reported an essential role for ERK1/2 signaling in the activation of G2/M checkpoint response following DNA damage, which involves the activation of ATR and Chk1 kinases.17,22,23 Results presented in this report indicate that the HER2 inhibition by specific inhibitor, HER2-mut or HER2-shRNA abolishes the activation of ATM and ATR signaling after IR, as well as the IR-induced ERK1/2 phosphorylation (Figure 9). These observations suggest HER2 as a vital upstream regulator of the IR-induced ERK1/2 activation and subsequent G2 checkpoint response in breast cancer cells.
Materials and Methods
Cell culture and treatment
Cell culture and treatment are described in Supplemental Material.
Antibodies and recombinant proteins
Antibodies and recombinant proteins are described in Supplemental Material.
Immunoblotting, immunoprecipitation and kinase assay
Immunoblotting, immunoprecipitation and kinase assay are described in Supplemental Material.
Cell cycle analysis
Cell cycle analysis is described in Supplemental Material.
Analysis for mitotic cells
Mitotic cells were analyzed as described in Supplemental Material.
shRNA retroviral vectors and viral infection
Retroviral vectors expressing shRNAs were obtained from OriGene Technologies (Rockville, MD). The sequences of shRNAs and methods for retrovirus infection are described in Supplemental Material.
Expressing vectors and transfection
Expressing vectors and cell transfection are described in Supplemental Material.
Supplementary Material
Acknowledgments
We thank Dr. Helen Piwnica-Worms for the GST-Cdc25C construct, Victoria Smith and Dr. Charles Kuzynski for assistance with FACS analysis and Dr. Janina Baranowska-Kortylewicz for assistance with the Mark I 68A Cesium-137 Irradiator. This work was supported by Nebraska DHHS-LB506 grant 2010-40 to Y.Y., NCI Training Grant (NCI T32 CA009476) to R.K. and NCI Cancer Center Support Grant (P30CA036727) to K.C..
Footnotes
Conflict of Interst: The authors declare that they have no conflicts of interest.
References
- 1.Kuntz K, O'Connell MJ. The G2 DNA damage checkpoint: Could this ancient regulator be the Achilles heel of cancer? Cancer Biology & Therapy. 2009;8:1433–1439. doi: 10.4161/cbt.8.15.9081. [DOI] [PubMed] [Google Scholar]
- 2.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Research. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 3.Motoyama N, Naka K. DNA damage tumor suppressor genes and genomic instability. Current Opinion in Genetics & Development. 2004;14:11. doi: 10.1016/j.gde.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Smits VA, Medema RH. Checking out the G(2)/M transition. Biochim Biophys Acta. 2001;1519:1–12. doi: 10.1016/s0167-4781(01)00204-4. [DOI] [PubMed] [Google Scholar]
- 5.Rhind N, Furnari B, Russell P. Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev. 1997;11:504–511. doi: 10.1101/gad.11.4.504. [DOI] [PubMed] [Google Scholar]
- 6.Kharbanda S, Saleem A, Datta R, Yuan ZM, Weichselbaum R, Kufe D. Ionizing radiation induces rapid tyrosine phosphorylation of p34cdc2. Cancer Res. 1994;54:1412–1414. [PubMed] [Google Scholar]
- 7.O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. Embo J. 1997;16:545–554. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell. 1991;64:1111–1122. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- 9.Parker LL, Atherton-Fessler S, Piwnica-Worms H. p107wee1 is a dual-specificity kinase that phosphorylates p34cdc2 on tyrosine 15. Proc Natl Acad Sci U S A. 1992;89:2917–2921. doi: 10.1073/pnas.89.7.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bulavin DV, Higashimoto Y, Demidenko ZN, Meek S, Graves P, Phillips C, et al. Dual phosphorylation controls Cdc25 phosphatases and mitotic entry. Nat Cell Biol. 2003;5:545–551. doi: 10.1038/ncb994. [DOI] [PubMed] [Google Scholar]
- 11.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 12.Prigent C, Dimitrov S. Phosphorylation of serine 10 in histone H3, what for? J Cell Sci. 2003;116:3677–3685. doi: 10.1242/jcs.00735. [DOI] [PubMed] [Google Scholar]
- 13.Xu B, Kastan MB. Analyzing cell cycle checkpoints after ionizing radiation. Methods Mol Biol. 2004;281:283–292. doi: 10.1385/1-59259-811-0:283. [DOI] [PubMed] [Google Scholar]
- 14.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 15.Sauve DM, Anderson HJ, Ray JM, James WM, Roberge M. Phosphorylation-induced rearrangement of the histone H3 NH2-terminal domain during mitotic chromosome condensation. J Cell Biol. 1999;145:225–235. doi: 10.1083/jcb.145.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22:1049–1059. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Y, Black CP, Cowan KH. Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene. 2007;26:4689–4698. doi: 10.1038/sj.onc.1210268. [DOI] [PubMed] [Google Scholar]
- 18.Navolanic PM, Steelman LS, McCubrey JA. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy (Review) Int J Oncol. 2003;22:237–252. [PubMed] [Google Scholar]
- 19.Hersey P, Zhuang L, Zhang XD. Current Strategies in Overcoming Resistance of Cancer Cells to Apoptosis Melanoma as a Model. In: Kwang WJ, editor. International Review of Cytology. Vol. 251. Academic Press; 2006. pp. 131–158. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi H, Chang SS, Hsu JL, Hung MC. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene. 2013 doi: 10.1038/onc.2013.74. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–369. [PubMed] [Google Scholar]
- 22.Abbott DW, Holt JT. Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J Biol Chem. 1999;274:2732–2742. doi: 10.1074/jbc.274.5.2732. [DOI] [PubMed] [Google Scholar]
- 23.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, et al. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 24.Kolb RH, Greer PM, Cao PT, Cowan KH, Yan Y. ERK1/2 signaling plays an important role in topoisomerase II poison-induced G2/M checkpoint activation. Plos One. 2012;7:e50281. doi: 10.1371/journal.pone.0050281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene. 2005;24:3285–3296. doi: 10.1038/sj.onc.1208492. [DOI] [PubMed] [Google Scholar]
- 26.Yan Y, Greer PM, Cao PT, Kolb RH, Cowan KH. RAC1 GTPase plays an important role in gamma-irradiation induced G2/M checkpoint activation. Breast cancer research : BCR. 2012;14:R60. doi: 10.1186/bcr3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan Y, Haas JP, Kim M, Sgagias MK, Cowan KH. BRCA1-induced apoptosis involves inactivation of ERK1/2 activities. J Biol Chem. 2002;277:33422–33430. doi: 10.1074/jbc.M201147200. [DOI] [PubMed] [Google Scholar]
- 28.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends in Cell Biology. 2006;16:649–656. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17:1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhomen NS, Mariadason J, Tebbutt N, Scott AM. Therapeutic targeting of the epidermal growth factor receptor in human cancer. Crit Rev Oncog. 2012;17:31–50. doi: 10.1615/critrevoncog.v17.i1.40. [DOI] [PubMed] [Google Scholar]
- 31.Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:2445–2459. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- 32.Goldkorn T, Balaban N, Shannon M, Matsukuma K. EGF receptor phosphorylation is affected by ionizing radiation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1997;1358:289–299. doi: 10.1016/s0167-4889(97)00063-3. [DOI] [PubMed] [Google Scholar]
- 33.Lee HC, An S, Lee H, Woo SH, Jin HO, Seo SK, et al. Activation of Epidermal Growth Factor Receptor and Its Downstream Signaling Pathway by Nitric Oxide in Response to Ionizing Radiation. Molecular Cancer Research. 2008;6:996–1002. doi: 10.1158/1541-7786.MCR-08-0113. [DOI] [PubMed] [Google Scholar]
- 34.Kiyozuka M, Akimoto T, Fukutome M, Motegi A, Mitsuhashi N. Radiation-induced Dimer Formation of EGFR: Implications for the Radiosensitizing Effect of Cetuximab. Anticancer Research. 2013;33:4337–4346. [PubMed] [Google Scholar]
- 35.Nyati MK, Maheshwari D, Hanasoge S, Sreekumar A, Rynkiewicz SD, Chinnaiyan AM, et al. Radiosensitization by Pan ErbB Inhibitor CI-1033 in Vitro and in Vivo. Clinical Cancer Research. 2004;10:691–700. doi: 10.1158/1078-0432.ccr-1041-03. [DOI] [PubMed] [Google Scholar]
- 36.Liang K, Lu Y, Jin W, Ang KK, Milas L, Fan Z. Sensitization of breast cancer cells to radiation by trastuzumab. Molecular Cancer Therapeutics. 2003;2:1113–1120. [PubMed] [Google Scholar]
- 37.Geoerger B, Gaspar N, Opolon P, Morizet J, Devanz P, Lecluse Y, et al. EGFR tyrosine kinase inhibition radiosensitizes and induces apoptosis in malignant glioma and childhood ependymoma xenografts. International journal of cancer Journal international du cancer. 2008;123:209–216. doi: 10.1002/ijc.23488. [DOI] [PubMed] [Google Scholar]
- 38.Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer (Auckl) 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 39.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Current Opinion in Cell Biology. 2009;21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slichenmyer WJ, Elliott WL, Fry DW. CI-1033, a pan-erbB tyrosine kinase inhibitor. Seminars in Oncology. 2001;28:80–85. doi: 10.1016/s0093-7754(01)90285-4. [DOI] [PubMed] [Google Scholar]
- 42.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 43.Arteaga Carlos L, Engelman Jeffrey A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell. 2014;25:282–303. doi: 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jani JP, Finn RS, Campbell M, Coleman KG, Connell RD, Currier N, et al. Discovery and Pharmacologic Characterization of CP-724,714, a Selective ErbB2 Tyrosine Kinase Inhibitor. Cancer Research. 2007;67:9887–9893. doi: 10.1158/0008-5472.CAN-06-3559. [DOI] [PubMed] [Google Scholar]
- 45.Yan Y, Cao PT, Greer PM, Nagengast ES, Kolb RH, Mumby MC, et al. Protein phosphatase 2A has an essential role in the activation of gamma-irradiation-induced G2/M checkpoint response. Oncogene. 2010;29:4317–4329. doi: 10.1038/onc.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MS, Igawa T, Yuan TC, Zhang XQ, Lin FF, Lin MF. ErbB-2 signaling is involved in regulating PSA secretion in androgen-independent human prostate cancer LNCaP C-81 cells. Oncogene. 2003;22:781–796. doi: 10.1038/sj.onc.1206066. [DOI] [PubMed] [Google Scholar]
- 47.Morrison R, Schleicher SM, Sun Y, Niermann KJ, Kim S, Spratt DE, et al. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J Oncol. 2011;2011:941876. doi: 10.1155/2011/941876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deckbar D, Jeggo PA, Lobrich M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit Rev Biochem Mol Biol. 2011;46:271–283. doi: 10.3109/10409238.2011.575764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W. Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Mol Cancer. 2009;8:8. doi: 10.1186/1476-4598-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gewirtz DA. Growth arrest and cell death in the breast tumor cell in response to ionizing radiation and chemotherapeutic agents which induce DNA damage. Breast Cancer Research and Treatment. 2000;62:223–235. doi: 10.1023/a:1006414422919. [DOI] [PubMed] [Google Scholar]
- 51.Schmidt-Ullrich RK, Mikkelsen RB, Dent P, Todd DG, Valerie K, Kavanagh BD, et al. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 1997;15:1191–1197. doi: 10.1038/sj.onc.1201275. [DOI] [PubMed] [Google Scholar]
- 52.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 53.Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Research. 2001;61:3894–3901. [PubMed] [Google Scholar]
- 54.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 55.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Research. 2012;72:3228–3237. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang WH, Hullinger RL, Andrisani OM. Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J Biol Chem. 2008;283:25455–25467. doi: 10.1074/jbc.M801934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou J, Lim CU, Li JJ, Cai L, Zhang Y. The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage. Cancer Lett. 2006;243:9–15. doi: 10.1016/j.canlet.2006.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 59.Hall-Jackson CA, Cross DA, Morrice N, Smythe C. ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene. 1999;18:6707–6713. doi: 10.1038/sj.onc.1203077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.