

Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder that affects some but not all carriers of small, non-coding CGG-repeat expansions (55–200 repeats; premutation) within the fragile X gene (FMR1). Principal features of FXTAS include intention tremor, cerebellar ataxia, Parkinsonism, memory and executive function deficits, autonomic dysfunction, brain atrophy with white matter disease, and cognitive decline. Although FXTAS was originally considered to be confined to the premutation range, rare individuals with a gray zone (45 to 54 repeats) or an unmethylated full mutation (>200 repeats) allele have now been described; the constant feature of the disorder remaining the requirement for FMR1 expression, in contradistinction to the gene silencing mechanism of fragile X syndrome. Although transcriptional activity is required for FXTAS pathogenesis, the specific trigger(s) for FXTAS pathogenesis remains elusive, highlighting the need for more research in this area. This need is underscored by recent neuroimaging findings of changes in the central nervous system that consistently appear well before the onset of clinical symptoms, thus creating an opportunity to delay or prevent the appearance of FXTAS.
Keywords: neurodegeneration, dementia, premutation, RNA toxicity, CGG repeat, FXTAS
Background
Over the last decade, our understanding of fragile X-associated disorders (FXD), which arise from full mutation (>200 CGG repeats in the gene FMR1; fragile X syndrome) or premutation (55 to 200 CGG repeats) alleles, has dramatically evolved. Although premutation disorders were once thought to include only fragile X-associated primary ovarian insufficiency (FXPOI) and fragile X-associated tremor/ataxia syndrome (FXTAS), we now know that carriers of a premutation allele have a variety of medical problems that include psychiatric disorders of anxiety and depression;1–5 chronic pain syndromes, such as fibromyalgia6–8 and chronic migraine;9 hypothyroidism;4,7 hypertension;10 sleep apnea;11 vertigo, olfactory dysfunction, and hearing loss;4,12,13 and enhanced stress.14 In addition, some premutation carriers have neurodevelopmental disorders, such as intellectual disability15 and/or autism spectrum disorder (ASD).16,17 Although most of the premutation problems are thought to relate to elevated FMR1 mRNA,18,19 there is also evidence of FMRP deficits adding to the phenotype, especially when significant cognitive deficits are present; FMRP mRNA levels are lowest in the upper premutation range.20–22 ASD in premutation carriers is also related to the presence of seizures.17 Early life seizures cause FMRP to redistribute from the dendrites to the cell body, rendering FMRP incapable of properly regulating translation at the synapse.23 Therefore, early life seizures can impede development as a consequence of a functional insufficiency of FMRP at the synapse.
Additional factors can influence the phenotype of premutation carriers. In approximately 20% of premutation cases with ASD or neurological problems, a second genetic hit has been identified through either microarray testing or whole exome sequencing.24 Such second hits are thought to contribute to the penetrance and/or severity of the phenotype, thus compounding intellectual disability, ASD, or neurological problems. Environmental toxicity can also cause additive effects to the premutation phenotype because premutation neurons are more vulnerable to toxic insults than are control neurons.25 Specifically, exposure to environmental toxins can lead to a more severe phenotype or earlier onset of FXTAS.26 In this regard, chemotherapy for cancer has been observed to precipitate FXTAS.27 In addition, some patients have reported that surgery involving general anesthesia leads to onset of tremor or ataxia within weeks in those carriers over 60 years of age, suggesting that one or more of the agents used during general anesthesia, or perhaps the surgical procedures themselves (e.g., hypoxia, tissue damage), may exacerbate the premutation-associated disorder. Unfortunately, essentially all of our awareness of a possible association between general surgery and FXTAS is based at present on anecdotal information, underscoring the need for systematic studies in this area.28
Expanding the diagnostic criteria for FXTAS
The standard diagnostic features of FXTAS require a premutation FMR1 allele plus one or more of the following core diagnostic features: intention tremor, cerebellar ataxia (core neurological features), and white matter disease in the middle cerebellar peduncles (MCP sign).29 Additional features contributing to the diagnosis include executive function and memory deficits, Parkinsonism, and additional MRI findings of global brain atrophy and white matter disease.4,12, 22, 30–33
However, recent cases of FXTAS, identified through core diagnostic features, have been found among carriers of gray-zone alleles (45–54 CGG repeats),34,35 and, in rare cases, among those with unmethylated, full mutation or mosaic alleles.34,36–38 These observations underscore the need to develop a broader definition of the disorder, since elevated mRNA and RNA toxicity are expected even outside of the premutation range when mRNA levels are elevated.19
The diagnostic criteria for FXTAS developed in 2003 (Ref. 30) were reviewed by an international research and clinical consortium in 2013, which gave specific recommendations regarding expanding the diagnostic criteria for FXTAS. These recommendations are summarized in Hall et al.33 and include broadening the range of the CGG repeat to incorporate gray-zone alleles on the low end and unmethylated/mosaic full-mutation alleles with elevated mRNA on the high end. In addition to the middle cerebellar peduncle (MCP) sign, several researchers have seen corpus callosum changes, including thinning and white matter disease in the splenium, in approximately 60% of patients, similar to the prevalence of the MCP sign in males with FXTAS.22, 32 Lastly, neuropathy, which is more common in premutation carriers with FXTAS compared to age-matched controls,32, 39 has been added to the diagnostic criteria as a minor sign, rather than a major criterion, since it is common in the aging population.
Several additional medical problems that are more common in carriers than controls include immune-mediated disorders, hypertension, autonomic dysfunction, sleep apnea, hearing loss, and migraines, may also occur in those with FXTAS, but typically start before its onset.22. Wheeler et al.4 carried out a detailed review of all premutation studies to assess the strength of symptom association with the premutation in women. Their review, in addition to the report by Hall et al.,33 represents consensus statements from the 1st International Conference on FMR1 Premutation: Basic Mechanisms and Clinical Involvement, held in Perugia, Italy, in June 2013.
A clear message from premutation research is that forms of clinical involvement occur throughout the life of the carrier—with deficits in visual perceptual abilities in infancy;40 common problems of attention, anxiety, and social interactions in childhood;16,17 psychiatric problems, migraines, hypothyroidism, hypertension and immune-mediated problems in adulthood;4,6, 22, 41,42 and onset of additional medical problems in a significant percentage in aging carriers of the premutation, including neuropathy, pain symptoms, and FXTAS.32,33 Most individuals with the premutation have normal intellectual abilities and often have productive and successful lives until their 60s, when subsequently approximately 40% of males and 16% of females develop FXTAS.7, 43
Why some individuals develop FXTAS and other do not may have to do with additional genetic hits (e.g., the ApoE4 allele44) that are associated with FXTAS, which may include Alzheimer disease.45 Forms of environmental toxicity can also add to the earlier onset or severity of FXTAS, and include smoking, alcoholism, and chemotherapy; or untreated medical problems, such as hypertension, depression, stress, hypothyroidism, cardiac arrhythmia, metabolic syndrome, or sleep apnea with hypoxia.28 However, the problem remains that we do not know who will eventually develop the neurodegenerative disorder. Regarding markers of early processes that will eventually lead to FXTAS—or may be indicators predictive of its eventual onset—neuroimaging studies have demonstrated CNS changes well before the onset of clinically diagnosed FXTAS. These changes are visible as functional MRI (fMRI), diffusion tensor imaging (DTI), and/or fiber track changes46–50 and brain atrophy, particularly in the cerebellum.51,52
Emerging concepts about the molecular mechanism(s) leading to FXTAS
Protein sequestration by the FMR1 CGG-repeat mRNA
Perhaps the singular feature of FXTAS relating to its underlying mechanism is that its phenotypic expression is largely limited to carriers of premutation alleles—and, rarely, to individuals with size- or methylation-mosaicism36–38,53 (however, see Ref. 54) where FMR1 is transcriptionally active. The requirement for mRNA expression has led to a hypothesized mechanism of pathogenesis that involves a toxic gain-of-function of the expanded CGG-repeat mRNA.55–57 As currently envisioned, the gain-of-function arises through the adventitious binding/sequestration by the CGG repeat of one or more proteins, thus at least partially impairing their normal function in the cell. This proposed mechanism is based on an analogous process operating in myotonic dystrophy, where a CUG-repeat expansion in the 3′ UTR of the myotonic dystrophy (DM1) protein kinase gene (DMPK) (or a CCUG repeat in the case of DM2;58, 59) sequesters the splice modulator, muscleblind-like 1 (MBNL1), resulting in altered splicing of proteins that underlie the major phenotypic domains of DM.60–62 For the sequestration model of FXTAS pathogenesis, a number of candidate proteins exist based on evidence for CGG-repeat RNA binding and studies in vitro and in vivo in various animal models (Table 1).
Table 1.
Summary of the models that have been invoked as mechanisms for FXTAS pathogenesis
| Mechanism class | Initiating species | Mechanism | Role in human disease | Citations |
|---|---|---|---|---|
| Protein sequestration by the CGG-repeat RNA | 131,134,135 | |||
| hnRNP A2/B1 | Both hnRNP A2/B1 and Purα appear to bind preferentially, at least in vitro, to shorter (~20 CGG) repeat RNAs, which renders them less likely candidates for FXTAS pathogenesis | Have mediating effects on Drosophila models of CGG-repeat–induced neurodegeneration. Notwithstanding their role(s) in mitigating the phenotypes in animal models, roles for Purα and hnRNP A2/B1 have yet to be demonstrated in human disease or in murine models of FXTAS. | 136–138 | |
| Purα | (see above) Transcriptional activator also thought to be involved in the control of DNA replication |
(see above) | 136–139 | |
| Sam68 | Altered mRNA splicing an RNA-binding protein that belongs to the “signal transduction and activation of RNA” (STAR) family | For Sam68, there is clear evidence of insufficiency in animals and humans; with altered splicing noted in FXTAS patients. However, a critical test of these candidate proteins will be whether they have any direct primary or secondary role in FXTAS pathogenesis in humans. | 63,65 | |
| DGCR8 | With Drosha, reduced processing of microRNA precursors DGCR8, part of the microprocessor complex that processes micro (mi)RNA precursors in the nucleus | 136, 140–143 | ||
| FMRP insufficiency | FMRP | 21,71–74 | ||
| Antisense FMR1 RNAs | RNAs | 144–147 | ||
| RAN translation | polyGly-containing peptides | 78,148 | ||
| R-loop/DNA damage response | R-loops, gH2AX | 83,84 |
All of the sequestration models share a common theme; namely, that the normal function(s) of the candidate protein is diminished through the process of sequestration, with the nature and severity of phenotypic involvement in FXTAS expected to arise through simple insufficiency of the sequestered proteins. For the proteins listed in Table 1, there is evidence of the necessary protein–CGG-repeat RNA interactions (at least in vitro) and the expected functional insufficiency; for example, altered splicing due to lowered Sam68;63,64 and reduced levels of mature miRNAs due to loss of DGCR8/Drosha activity.65
Further, for the specific sequestration mechanisms listed in Table 1, and others not yet identified, two issues should be borne in mind. First, although each of the listed proteins has support from in vitro, in cellulo, and, in some instances, in vivo (Drosophila) data (e.g., Purα;66 hnRNPA267), there are virtually no studies of the consequences to partial depletion of functional protein and, therefore, of the specific role of the protein(s) to the pathogenesis of FXTAS. In this regard, reversal of the model phenotype by the expression of a superabundance of protein (to replace the sequestered species) may reflect mechanisms that are unrelated to the original insufficiency, as might occur through a chaperone function of the added protein. Second, although individual studies have generally focused on individual proteins (e.g., Purα, Sam68, etc.), more than one protein may likely be contributing to the overall disease phenotype. Thus, no single protein candidate for sequestration needs to be responsible for all of the phenotypic domains of FXTAS (e.g., movement disorder, cognitive impairment). In many cases other diseases have also occurred with FXTAS, including multiple sclerosis,68 Lewy body dementia,69 Parkinson disease,70 and Alzheimer disease.45
Reduced FMRP plays only a secondary, non-causative role in the FXTAS phenotype
We and others have observed that FMRP levels trend downward with increasing CGG repeats in the premutation range,21, 71 suggesting that mild-to-moderate FMRP insufficiency is contributing to the reduced cognitive strength observed in a portion of premutation carriers well before the typical age of onset of FXTAS.72–74 Those who have neurodevelopmental problems prior to the onset of FXTAS may be predisposed to an earlier onset of FXTAS70 (and Basuta et al., unpublished results); however, there is no evidence to date suggesting that lowered FMRP levels are causative of FXTAS, and nearly all individuals with fragile X syndrome (with little or no FMRP) do not develop FXTAS.
Antisense FMR1 RNAs
Mounting evidence shows that multiple antisense transcripts are produced from FMR1 in a manner that tracks with the size of the CGG-repeat and, consequently, with the level of expression of the primary sense transcript, which is elevated in the premutation range and generally absent in the full-mutation range,75–77 suggesting that the CGG repeat is capable of regulating bidirectional transcription at the locus. Interestingly, there is a specific antisense splice isoform that is only found with transcripts from premutation alleles,75 raising the possibility that Sam68 and/or MBNL1, both of which are associated with the CGG repeat, could be functionally dysregulated by the expanded CGG repeat.
Repeat-associated non-AUG (RAN) translation
A model for FXTAS pathogenesis has been proposed in which “toxic” peptides are generated by initiating at non-AUG codons located upstream of the CGG-repeat element (Fig. 1);78 RAN translation has been well described in other trinucleotide-repeat disorders.79 In the current instance, Todd et al.78 have provided ample evidence that RAN translation of the FMR1 mRNA generates a poly-glycine peptide that is toxic to cells and is detectable in both the intranuclear inclusions of FXTAS and in the inclusions of the Dutch premutation CGG-repeat mouse model (see Refs 19 and 78). However, the role of the poly-glycine peptides in FXTAS remains an open question, given that a highly related mouse model, termed the “NIH” model,78, 80 displays significant neurodegeneration but apparently does not produce the poly-glycine peptide because of a stop codon just downstream of the initiating codon for the that peptide.78 These seemingly paradoxical observations may indicate that the nuclear inclusions are themselves a consequence of RAN translation, but that the FXTAS-associated neurodegeneration is due to other mechanisms—this is an important area for further study.
Figure 1.
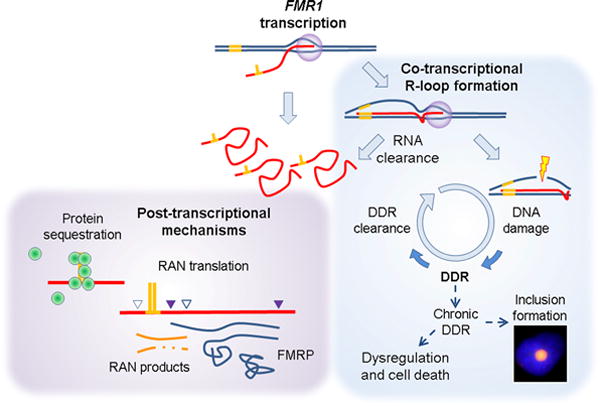
Models for pathogenesis of FXTAS; all are based the requirement for expression of the expanded (premutation) FMR1 mRNA. Post-transcriptional models are based on the properties of the expanded CGG-repeat region within the free mRNA. The CGG-repeat element, likely through its ability to form higher-order structure (e.g., hairpin; depicted in yellow), has been demonstrated to sequester one or more proteins (green spheres), rendering the cell functionally deficient in those proteins; the exemplar for this process is the muscleblind-like 1 protein of myotonic dystrophy. An alternative mechanism has been proposed in which the CGG element leads to aberrant initiation of translation, out of phase with the FMRP coding sequence, and at non-AUG codons. In this model, the short peptides are proposed to be toxic. Despite the focus on post-transcriptional mechanisms, co-transcriptional events (e.g., R-loop formation) can also lead to cellular toxicity, in this instance through failure to clear DNA damage and its repair at/near the R-loop. Although R-loop formation and DNA damage repair (DDR) are occurring normally throughout the genome, occasional failure to clear the DDR response can lead to continued signaling and recruitment of repair proteins. In this regard, we have observed the presence of the DDR signaling protein, gH2AX, in the inclusions, as well as its induction in model systems of CGG-repeat overexpression. None of these models is mutually exclusive.
R-loop formation/DNA damage response
All of the foregoing models for FXTAS pathogenesis––aberrant (RAN) translation, generation of premutation-specific antisense RNAs, and sequestration of one or more RNA binding proteins by the CGG-repeat element in the mRNA—are based on the involvement of the FMR1 mRNA at the post-transcriptional level. However, the requirement for FMR1 mRNA production does not preclude the involvement of co-transcriptional processes (Fig. 1). In this regard, transcription through the highly GC-rich FMR1 5′ UTR region promotes (co-transcriptional) R-loop formation,76 whereby the G-rich RNA transcript reinvades the DNA duplex and forms a stable RNA:DNA hybrid with the C-rich template strand, thereby displacing the non-template DNA strand. Although R-loop formation normally occurs at multiple loci throughout the genome,81 defects in mRNA processing can result in an R-loop–dependent activation of the DNA damage response and lead to the accumulation of the phosphorylated H2A variant, γH2AX, which is associated with the DNA damage repair process.82, 83
We have recently reported R-loop formation both at the endogenous FMR1 locus and in an inducible episomal system in which the expanded CGG-repeat (~95 CGG) is located upstream of a GFP reporter.83 For the episomal system, we had previously reported that high levels of reporter mRNA with the expanded CGG repeat results in the generation of γH2AX,84 which is also found within the intranuclear inclusions of FXTAS.85 Thus, although indirect, current evidence suggests that the increased transcriptional activity associated with premutation alleles may lead to damage at the FMR1 locus and a consequent DNA damage response (DDR; Fig. 1), a possibility that warrants additional study.
For all of the aforementioned mechanisms there remains the task of assessing to what extent they play a role(s) in the development of FXTAS in humans, and whether such mechanisms, singly or in combination, are either sufficient for pathogenesis or secondary (e.g., FMRP deficiency). Moreover, although mitochondrial dysfunction is clearly a component of FXTAS pathogenesis86–89 (reviewed elsewhere, e.g. Ref. 19), neither its linkage to FMR1 mRNA expression nor its specific role in pathogenesis is clear at present.
A nematode model for CGG-repeat–mediated alterations in adaptive behavior
An important feature of the clinical presentation of fragile X syndrome is hyperarousal and an enhanced reaction to various forms of sensory stimuli.90–92 Enhanced response to auditory stimuli was quantified by measuring the electrodermal response (EDR) exhibited by a subject with fragile X syndrome to a defined stimulus91 (see also Ref. 93). The EDRs were significantly enhanced in the subjects relative to controls, with increased magnitude of the response and reduced habituation (i.e., a failure to adapt or attenuate the EDR with continued sensory stimulation). More recently, Schneider et al.92 demonstrated a related feature in carriers of premutation alleles, including FXTAS patients. They applied a low intensity acoustic “prepulse” at varying intervals prior to a main pulse to determine to what extent the prepulse would attenuate (inhibit) the subjects’ reaction to the main pulse (prepulse inhibition, PPI). They observed an impaired PPI that was most pronounced in male FXTAS patients, with a significant correlation between CGG-repeat size and the magnitude of the PPI deficit. Thus, for both premutation and full mutation carriers, there is a failure to habituate to a sensory stimulus.
Remarkably, Juang et al.94 observed an analogous loss of habituation—olfactory habituation—in a nematode (Caenorhabditis elegans) model in which an expanded CGG-repeat expression vector (99 CGG repeats) is transcriptionally active solely in a single pair of olfactory neurons. A noteworthy aspect of the C. elegans model is that it does not involve neuronal death, unlike the neuronal death observed in the Drosophila CGG-repeat–coupled neurodegenerative phenotypes.56, 66, 78, 95, 96
Odor-seeking behavior of C. elegans involves just two pairs of ciliated olfactory neurons, AWA and AWC, to sense many chemoattractants.97 The primary odor-sensory AWC neurons allow worms to chemotax toward attractive volatile chemicals and govern both the primary olfactory response and a secondary adaptive response, which requires neuronal plasticity.97, 98 A single AWC neuron senses the chemoattractant butanone and directs nematodes to move toward this odor source;99 prolonged odor exposure in the absence of food reduces the animal’s attraction to butanone100,101 because of processes that occur within the AWC neuron.94,98,102–104 Remarkably, expression of the expanded CGG-repeat element (but not the normal CGG repeat) in only a single pair of AWC neurons interfered with nematode’s ability to attenuate the prolonged exposure to the odorant, while preserving normal odor detection.94 The authors found that the abnormal response to the expanded CGG repeat is mediated by the miRNA-specific Argonaute (ALG-2), suggesting that this pathway may play a role in the pathogenesis of neuronal function in FXTAS. However, of more immediate interest is the potential of this system for studying the detailed biochemical mechanism for altered neuronal function.
Approaches to treatment of FXTAS
One of the first steps in treating FXTAS is identifying those with neurological problems who also carry premutation FMR1 alleles. For individuals within families with known children or adults with fragile X syndrome (FXS), identification and treatment are generally straightforward; with new options available for targeted treatments to reverse the neurobiological abnormalities that occur with the absence of FMRP.105–108 Those with the premutation and a variety of medical problems have several options for symptomatic treatment,28,109 including the use of selective serotonin reuptake inhibitors (SSRIs) for depression and/or anxiety.3,41
For those with FXTAS, a controlled trial was carried out to assess the benefit of memantine over a 1-year treatment period.110 Although memantine was well tolerated, it did not demonstrate benefit compared to placebo in alleviating the tremor, balance problems, or executive function deficits in those with FXTAS.110 However, a subgroup of patients who participated in the memantine study exhibited significant improvement in a secondary measure, an event-related potential (ERP) paradigm to assess verbal fluency and memory for non-congruent words with memantine.111 Since a subgroup of patients with FXTAS treated with memantine show some improvement,112 further studies are needed to identify this subgroup. ERPs are likely a more sensitive outcome measure for validating improvements in brain processing with targeted treatments, as has been found in treatment of FXS.113
Premutation neuronal cultures have demonstrated that elements of RNA toxicity, such as up-regulation of heat shock proteins and enhanced spike discharges from the neuron, are improved with the addition of allopregnanolone or even an mGluR5 antagonist, MPEP.114 MPEP is neurotoxic in humans, but allopregnanolone is a natural neurosteroid that has neuroprotective features and stimulates neurogenesis; allopregnanolone is now being utilized in trials of traumatic brain injury and Alzheimer disease.115,116 Treatment trials of allopregnanolone will likely begin in the near future for those with FXTAS.
Those with premutation alleles are generally identified as obligate carriers based on the pattern of cases of fragile X syndrome or are identified through cascade testing within the family.17,117,118 However, for isolated individuals with neurological involvement, proper identification of FXTAS cases necessitates the recognition of the phenotypic features of the disorder, which often overlap those of many other disorders with ataxia, tremor, and progressive cognitive impairment. Because of the phenotypic overlap with spinocerebellar ataxias, Parkinson disease, and dementias, screening studies have been performed to assess the extent to which such disorders are actually manifestations of FXTAS.119–128 Results of these studies—and others—have been variable, with the most consistent yield of FXTAS cases (with premutation alleles) found among the ataxias, typically in the range of ~1–2%, with more negative results found in cases with essential tremor, Parkinson disease, Alzheimer disease,128,129 and related disorders. Those studies with negative findings likely reflect the much higher prevalence of these latter disorders compared to FXTAS, necessitating much larger screened populations to determine whether they have any association with the premutation allele.
In a recent screening study in India of a cohort with progressive, late-onset tremor/ataxia (109 patients and 173 control subjects),130 three premutation alleles were detected among the patient group; two of these individuals (96 and 102 CGG repeats) were being evaluated for SCA-12 and another (78 CGG repeats) for progressive gait ataxia. The frequency of FXTAS was 3.3% overall and 9% (2/23 cases) for SCA-12-like presentation, underscoring the need to screen cases of apparent (test-negative) SCA.
Differential diagnosis and testing
Features of FXTAS overlap many of the core features of other neurodegenerative disorders, including Parkinson and Alzheimer diseases and frontotemporal dementia (e.g., progressive cognitive impairment, altered mood and behavior). Therefore, it is important to consider FXTAS in the differential diagnosis of a wide range of neurological or neurodegenerative disorders (e.g., Table 23.2 of Ref. 131). Such overlap also includes disorders with ataxia and/or intention tremor, such as the SCAs, multiple system atrophy (MSA), Parkinson disease (PD), essential tremor, progressive supranuclear palsy (PSP),132 and essential tremor.
However, for most of the above-mentioned disorders, testing for a premutation allele of the FMR1 gene would not generally be productive unless there are additional indicators pointing to that gene. In particular, testing for an expanded allele of the FMR1 gene should be considered in patients who present with cerebellar ataxia and/or action tremor, particularly for those older than 50 years, when the core motor involvement is accompanied by one or more of the features listed in Table 2. Some of these associated conditions—family history of cognitive impairment and/or autism/ASD, primary ovarian insufficiency, family history of fragile X—are indications for fragile X testing that are independent of the presence of motor involvement, but are clear indications for testing with an initial presentation of tremor/ataxia.
Table 2.
Findings associated with cerebellar ataxia and/or action tremor in adultsa that support testing for premutation alleles of FMR1
| Associated feature | Additional comments | Independent indicator for FMR1 testingb |
|---|---|---|
| Family members with intellectual impairment, autism, or autism spectrum disorder (ASD) | Fragile X syndrome is the leading monogenic cause of intellectual impairment and autism/ASD. FXTAS is common among older adults (>50 yr) in fragile X families | Yes |
| Primary ovarian insufficiency (POI) | Premutation alleles of the FMR1 gene constitute the leading monogenic cause of POI, often described in terms of early menopause/infertility (<40 yr) | Yes |
| Hypothyroidism (women) | 50% of females with FXTAS, often associated with other evidence of immune dysfunction7 | No |
| Peripheral neuropathy | 60% of men and 53% of women with FXTAS149 | No |
| Muscle pain / (fibromyalgia) | 76% / (43%) among women with FXTAS7 | No |
| MRI hyperintensities within the middle cerebellar peduncles (MCP) or splenium of the corpus callosum32,150 | The “MCP sign” was one component of the original definition of FXTAS; involvement of the MCP and/or the splenium is highly-characteristic of FXTAS | Yes |
| Family member with the premutation | At risk to be a carrier with no symptoms or any premutation- associated disorder | Yes |
Typical onset > 50 yr
Features that would warrant testing for CGG-repeat expansions of FMR1 irrespective of the presence of tremor or ataxia.
Conclusions
New treatment studies are needed for this relatively common disorder of neurodegeneration—FXTAS. The premutation occurs in 1 in 130–250 females and about half that number in men in the general population (reviewed in Ref. 133). Changes in the CNS, revealed by recent neuroimaging studies, are consistently seen in adulthood well before the onset of the clinical symptoms of FXTAS; this general observation provides the opportunity to treat the medical problems well before the age of typical onset of FXTAS, thus creating a window of therapeutic opportunity to delay or even prevent the neurodegenerative disorder.28
Acknowledgments
This work was funded by NIH Grants (R01 HD040661, R01 HD036071). The authors wish to thank the many families who have supported our work.
Footnotes
Conflicts of Interest
R. Hagerman has received funding from Novartis, Roche, Seaside Therapeutics, Alcobra, and Neuren to carry out treatment studies in fragile X syndrome, autism, or Down syndrome; he has also consulted with Novartis and Roche/Genentech regarding treatment studies in fragile X syndrome. P. Hagerman is an uncompensated collaborator with Pacific Biosciences regarding new FMR1 sequencing strategies; he holds patents for FMR1 genotyping and protein tests.
References
- 1.Roberts JE, Bailey DB, Jr, Mankowski J, et al. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:130–139. doi: 10.1002/ajmg.b.30786. [DOI] [PubMed] [Google Scholar]
- 2.Bourgeois JA, Seritan AL, Casillas EM, et al. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry. 2011;72:175–182. doi: 10.4088/JCP.09m05407blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourgeois JA, Coffey SM, Rivera SM, et al. A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiatry. 2009;70:852–862. doi: 10.4088/JCP.08m04476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler AC, Bailey DB, Jr, Berry-Kravis E, et al. Associated features in females with an FMR1 premutation. J Neurodev Disord. 2014;6:30. doi: 10.1186/1866-1955-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seltzer MM, Baker MW, Hong J, et al. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:589–597. doi: 10.1002/ajmg.b.32065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winarni TI, Chonchaiya W, Sumekar TA, et al. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am J Med Genet A. 2012;158A:2473–2481. doi: 10.1002/ajmg.a.35569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coffey SM, Cook K, Tartaglia N, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A:1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leehey MA, Legg W, Tassone F, et al. Fibromyalgia in fragile X mental retardation 1 gene premutation carriers. Rheumatology (Oxford) 2011;50:2233–2236. doi: 10.1093/rheumatology/ker273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Au J, Akins RS, Berkowitz-Sutherland L, et al. Prevalence and risk of migraine headaches in adult fragile X premutation carriers. Clin Genet. 2013;84:546–551. doi: 10.1111/cge.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamlin AA, Sukharev D, Campos L, et al. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS) Am J Med Genet A. 2012;158A:1304–1309. doi: 10.1002/ajmg.a.35323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamlin A, Liu Y, Nguyen DV, et al. Sleep apnea in fragile X premutation carriers with and without FXTAS. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:923–928. doi: 10.1002/ajmg.b.31237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juncos JL, Lazarus JT, Graves-Allen E, et al. New clinical findings in the fragile X-associated tremor ataxia syndrome (FXTAS) Neurogenetics. 2011;12:123–135. doi: 10.1007/s10048-010-0270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juncos JL, Lazarus JT, Rohr J, et al. Olfactory dysfunction in fragile X tremor ataxia syndrome. Mov Disord. 2012;27:1556–1559. doi: 10.1002/mds.25043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seltzer MM, Barker ET, Greenberg JS, et al. Differential sensitivity to life stress in FMR1 premutation carrier mothers of children with fragile X syndrome. Health Psychol. 2012;31:612–622. doi: 10.1037/a0026528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tassone F, Hagerman RJ, Taylor AK, et al. Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet. 2000;91:144–152. doi: 10.1002/(sici)1096-8628(20000313)91:2<144::aid-ajmg14>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 16.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 17.Chonchaiya W, Au J, Schneider A, et al. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet. 2012;131:581–589. doi: 10.1007/s00439-011-1106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagerman P. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 2013;126:1–19. doi: 10.1007/s00401-013-1138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hessl D, Wang JM, Schneider A, et al. Decreased fragile X mental retardation protein expression underlies amygdala dysfunction in carriers of the fragile X premutation. Biol Psychiatry. 2011;70:859–865. doi: 10.1016/j.biopsych.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ludwig AL, Espinal GM, Pretto DI, et al. CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum Mol Genet. 2014;23:3228–3238. doi: 10.1093/hmg/ddu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013;12:786–798. doi: 10.1016/S1474-4422(13)70125-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernard PB, Castano AM, O’Leary H, et al. Phosphorylation of FMRP and alterations of FMRP complex underlie enhanced mLTD in adult rats triggered by early life seizures. Neurobiol Dis. 2013;59:1–17. doi: 10.1016/j.nbd.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lozano R, Hagerman R, Duyzend M, et al. Genomic studies in fragile X premutation carriers. J Neurodev Disord. 2014;6:27. doi: 10.1186/1866-1955-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Tassone F, Berman RF, et al. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2010;19:196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul R, I, Pessah N, Gane L, et al. Early onset of neurological symptoms in fragile X premutation carriers exposed to neurotoxins. Neurotoxicology. 2010;31:399–402. doi: 10.1016/j.neuro.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Dwyer JP, Clabby C, Crown J, et al. Fragile X-associated tremor/ataxia syndrome presenting in a woman after chemotherapy. Neurology. 2005;65:331–332. doi: 10.1212/01.wnl.0000168865.36352.53. [DOI] [PubMed] [Google Scholar]
- 28.Polussa J, Schneider A, Hagerman R. Molecular advances leading to treatment implications for fragile X premutation carriers. Brain Disord Therapy. 2014;3 doi: 10.4172/2168-975X.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunberg JA, Jacquemont S, Hagerman RJ, et al. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
- 30.Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leehey MA, Berry-Kravis E, Min SJ, et al. Progression of tremor and ataxia in male carriers of the FMR1 premutation. Mov Disord. 2007;22:203–206. doi: 10.1002/mds.21252. [DOI] [PubMed] [Google Scholar]
- 32.Apartis E, Blancher A, Meissner WG, et al. FXTAS: new insights and the need for revised diagnostic criteria. Neurology. 2012;79:1898–1907. doi: 10.1212/WNL.0b013e318271f7ff. [DOI] [PubMed] [Google Scholar]
- 33.Hall D, Birch R, Anheim M, et al. Emerging topics in FXTAS. J Neurodev Disord. 2014;6:31. doi: 10.1186/1866-1955-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall D, Tassone F, Klepitskaya O, et al. Fragile X-associated tremor ataxia syndrome in FMR1 gray zone allele carriers. Mov Disord. 2012;27:296–300. doi: 10.1002/mds.24021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Winarni TI, Zhang L, et al. Fragile X-associated tremor/ataxia syndrome (FXTAS) in grey zone carriers. Clin Genet. 2013;84:74–77. doi: 10.1111/cge.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loesch DZ, Sherwell S, Kinsella G, et al. Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin Genet. 2012;82:88–92. doi: 10.1111/j.1399-0004.2011.01675.x. [DOI] [PubMed] [Google Scholar]
- 37.Pretto DI, Hunsaker MR, Cunningham CL, et al. Intranuclear inclusions in a fragile X mosaic male. Transl Neurodegener. 2013;2:10. doi: 10.1186/2047-9158-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santa Maria L, Pugin A, Alliende MA, et al. FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin Genet. 2014;86:378–382. doi: 10.1111/cge.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soontarapornchai K, Maselli R, Fenton-Farrell G, et al. Abnormal nerve conduction features in fragile X premutation carriers. Arch Neurol. 2008;65:495–498. doi: 10.1001/archneur.65.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallego P, Burris J, Rivera S. Visual motion processing deficits in infants with the fragile X premutation. J Neurodev Disord. 2014;6:29. doi: 10.1186/1866-1955-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Besterman AD, Wilke SA, Mulligan TE, et al. Towards an understanding of neuropsychiatric manifestations in fragile X premutation carriers. Future Neurol. 2014;9:227–239. doi: 10.2217/fnl.14.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17:1359–1362. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 44.Silva F, Rodriguez-Revenga L, Madrigal I, et al. High apolipoprotein E4 allele frequency in FXTAS patients. Genet Med. 2013;15:639–642. doi: 10.1038/gim.2013.12. [DOI] [PubMed] [Google Scholar]
- 45.Tassone F, Greco CM, Hunsaker MR, et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012;1:577–585. doi: 10.1111/j.1601-183X.2012.00779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang JY, Hagerman RJ, Rivera SM. A multimodal imaging analysis of subcortical gray matter in fragile X premutation carriers. Mov Disord. 2013;28:1278–1284. doi: 10.1002/mds.25473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JY, Hessl D, Iwahashi C, et al. Influence of the fragile X mental retardation (FMR1) gene on the brain and working memory in men with normal FMR1 alleles. Neuroimage. 2013;65:288–298. doi: 10.1016/j.neuroimage.2012.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang JY, Hessl D, Schneider A, et al. Fragile X-associated tremor/ataxia syndrome: influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles. JAMA Neurol. 2013;70:1022–1029. doi: 10.1001/jamaneurol.2013.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JY, Hessl DH, Hagerman RJ, et al. Age-dependent structural connectivity effects in fragile X premutation. Arch Neurol. 2012;69:482–489. doi: 10.1001/archneurol.2011.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Battistella G, Niederhauser J, Fornari E, et al. Brain structure in asymptomatic FMR1 premutation carriers at risk for fragile X-associated tremor/ataxia syndrome. Neurobiol Aging. 2013;34:1700–1707. doi: 10.1016/j.neurobiolaging.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Hashimoto R, Javan AK, Tassone F, et al. A voxel-based morphometry study of grey matter loss in fragile X-associated tremor/ataxia syndrome. Brain. 2011;134:863–878. doi: 10.1093/brain/awq368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen S, Masyn K, Adams J, et al. Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology. 2006;67:1426–1431. doi: 10.1212/01.wnl.0000239837.57475.3a. [DOI] [PubMed] [Google Scholar]
- 53.Hunsaker MR, Greco CM, Tassone F, et al. Rare intranuclear inclusions in the brains of 3 older adult males with fragile X syndrome: implications for the spectrum of fragile X-associated disorders. J Neuropathol Exp Neurol. 2011;70:462–469. doi: 10.1097/NEN.0b013e31821d3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Utari A, Adams E, Berry-Kravis E, et al. Aging in fragile X syndrome. J Neurodev Disord. 2010;2:70–76. doi: 10.1007/s11689-010-9047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 56.Jin P, Zarnescu DC, Zhang F, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 57.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–816. doi: 10.1086/386296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, Jin P. RNA-mediated neurodegeneration in fragile X-associated tremor/ataxia syndrome. Brain Res. 2012;1462:112–117. doi: 10.1016/j.brainres.2012.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin M, Entezam A, Usdin K, et al. A mouse model of the fragile X premutation: Effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol Dis. 2011;44:85–89. doi: 10.1016/j.nbd.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Echeverria GV, Cooper TA. RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res. 2012;1462:100–111. doi: 10.1016/j.brainres.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahadevan MS. Myotonic dystrophy: is a narrow focus obscuring the rest of the field? Curr Opin Neurol. 2012;25:609–613. doi: 10.1097/WCO.0b013e328357b0d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Timchenko L. Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int J Biochem Cell Biol. 2013;45:2280–2287. doi: 10.1016/j.biocel.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bielli P, Busa R, Paronetto MP, et al. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr Relat Cancer. 2011;18:R91–R102. doi: 10.1530/ERC-11-0041. [DOI] [PubMed] [Google Scholar]
- 64.Sellier C, Rau F, Liu Y, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sellier C, Freyermuth F, Tabet R, et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013;3:869–880. doi: 10.1016/j.celrep.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin P, Duan R, Qurashi A, et al. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sofola OA, Jin P, Qin Y, et al. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greco CM, Tassone F, Garcia-Arocena D, et al. Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol. 2008;65:1114–1116. doi: 10.1001/archneur.65.8.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 70.Loesch DZ, Kotschet K, Trost N, et al. White matter changes in basis pontis in small expansion FMR1 allele carriers with parkinsonism. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:502–506. doi: 10.1002/ajmg.b.31189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pretto DI, Mendoza-Morales G, Lo J, et al. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet. 2014;51:309–318. doi: 10.1136/jmedgenet-2013-102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tassone F, Hagerman RJ, Taylor AK, et al. A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J Med Genet. 2001;38:453–456. doi: 10.1136/jmg.38.7.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goodlin-Jones BL, Tassone F, Gane LW, et al. Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr. 2004;25:392–398. doi: 10.1097/00004703-200412000-00002. [DOI] [PubMed] [Google Scholar]
- 74.Hessl D, Tassone F, Loesch DZ, et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am J Med Genet B Neuropsychiatr Genet. 2005;139B:115–121. doi: 10.1002/ajmg.b.30241. [DOI] [PubMed] [Google Scholar]
- 75.Ladd PD, Smith LE, Rabaia NA, et al. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007;16:3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- 76.Reddy K, Tam M, Bowater RP, et al. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 2011;39:1749–1762. doi: 10.1093/nar/gkq935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pastori C, V, Peschansky J, Barbouth D, et al. Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Hum Genet. 2014;133:59–67. doi: 10.1007/s00439-013-1356-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Todd PK, Oh SY, Krans A, et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cleary JD, Ranum LP. Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev. 2014;26C:6–15. doi: 10.1016/j.gde.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Entezam A, Biacsi R, Orrison B, et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ginno PA, Lim YW, Lott PL, et al. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013;23:1590–1600. doi: 10.1101/gr.158436.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clifford S, Dissanayake C, Bui QM, et al. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37:738–747. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 83.Loomis EW, Sanz LA, Chedin F, et al. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014;10:e1004294. doi: 10.1371/journal.pgen.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoem G, Raske CR, Garcia-Arocena D, et al. CGG-repeat length threshold for FMR1 RNA pathogenesis in a cellular model for FXTAS. Hum Mol Genet. 2011;20:2161–2170. doi: 10.1093/hmg/ddr101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iwahashi CK, Yasui DH, An HJ, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 86.Ross-Inta C, Omanska-Klusek A, Wong S, et al. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J. 2010;429:545–552. doi: 10.1042/BJ20091960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Napoli E, Ross-Inta C, Wong S, et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum Mol Genet. 2011;20:3079–3092. doi: 10.1093/hmg/ddr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaplan ES, Cao Z, Hulsizer S, et al. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J Neurochem. 2012;123:613–621. doi: 10.1111/j.1471-4159.2012.07936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Loesch DZ, Godler DE, Evans A, et al. Evidence for the toxicity of bidirectional transcripts and mitochondrial dysfunction in blood associated with small CGG expansions in the FMR1 gene in patients with parkinsonism. Genet Med. 2011;13:392–399. doi: 10.1097/GIM.0b013e3182064362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hagerman RJ. Physical and behavioral phenotype. In: Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome: Diagnosis, Treatment and Research. The Johns Hopkins University Press; Baltimore: 2002. pp. 3–109. [Google Scholar]
- 91.Miller LJ, McIntosh DN, McGrath J, et al. Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet. 1999;83:268–279. [PubMed] [Google Scholar]
- 92.Schneider A, Ballinger E, Chavez A, et al. Prepulse inhibition in patients with fragile X-associated tremor ataxia syndrome. Neurobiol Aging. 2012;33:1045–1053. doi: 10.1016/j.neurobiolaging.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams TA, Langdon R, Porter MA. Hyper-reactivity in fragile X syndrome females: generalised or specific to socially-salient stimuli? A skin conductance study. Int J Psychophysiol. 2013;88:26–34. doi: 10.1016/j.ijpsycho.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 94.Juang BT, Ludwig AL, Benedetti KL, et al. Expression of an expanded CGG-repeat RNA in a single pair of primary sensory neurons impairs olfactory adaptation in Caenorhabditis elegans. Hum Mol Genet. 2014;23:4945–4959. doi: 10.1093/hmg/ddu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qurashi A, Li W, Zhou JY, et al. Nuclear accumulation of stress response mRNAs contributes to the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 2011;7:e1002102. doi: 10.1371/journal.pgen.1002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.He F, Krans A, Freibaum BD, et al. TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum Mol Genet. 2014;23:5036–5051. doi: 10.1093/hmg/ddu216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bargmann CI, Hartwieg E, Horvitz HR. Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell. 1993;74:515–527. doi: 10.1016/0092-8674(93)80053-h. [DOI] [PubMed] [Google Scholar]
- 98.L’Etoile ND, Coburn CM, Eastham J, et al. The cyclic GMP-dependent protein kinase EGL-4 regulates olfactory adaptation in C. elegans. Neuron. 2002;36:1079–1089. doi: 10.1016/s0896-6273(02)01066-8. [DOI] [PubMed] [Google Scholar]
- 99.Wes PD, Bargmann CI. C. elegans odour discrimination requires asymmetric diversity in olfactory neurons. Nature. 2001;410:698–701. doi: 10.1038/35070581. [DOI] [PubMed] [Google Scholar]
- 100.Colbert HA, Bargmann CI. Odorant-specific adaptation pathways generate olfactory plasticity in C. elegans. Neuron. 1995;14:803–812. doi: 10.1016/0896-6273(95)90224-4. [DOI] [PubMed] [Google Scholar]
- 101.L’Etoile ND, Bargmann CI. Olfaction and odor discrimination are mediated by the C. elegans guanylyl cyclase ODR-1. Neuron. 2000;25:575–586. doi: 10.1016/s0896-6273(00)81061-2. [DOI] [PubMed] [Google Scholar]
- 102.Kaye JA, Rose NC, Goldsworthy B, et al. A 3′UTR pumilio-binding element directs translational activation in olfactory sensory neurons. Neuron. 2009;61:57–70. doi: 10.1016/j.neuron.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee JI, O’Halloran DM, Eastham-Anderson J, et al. Nuclear entry of a cGMP-dependent kinase converts transient into long-lasting olfactory adaptation. Proc Natl Acad Sci U S A. 2010;107:6016–6021. doi: 10.1073/pnas.1000866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.O’Halloran DM, Altshuler-Keylin S, Lee JI, et al. Regulators of AWC-mediated olfactory plasticity in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000761. doi: 10.1371/journal.pgen.1000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hagerman RJ, Des-Portes V, Gasparini F, et al. Translating molecular advances in fragile X syndrome into therapy: a review. J Clin Psychiatry. 2014;75:e294–307. doi: 10.4088/JCP.13r08714. [DOI] [PubMed] [Google Scholar]
- 106.Leigh MJ, Nguyen DV, Mu Y, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile X syndrome. J Dev Behav Pediatr. 2013;34:147–155. doi: 10.1097/DBP.0b013e318287cd17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Winarni TI, Schneider A, Borodyanskara M, et al. Early intervention combined with targeted treatment promotes cognitive and behavioral improvements in young children with fragile X syndrome. Case Rep Genet. 2012;2012:280813. doi: 10.1155/2012/280813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hare EB, Hagerman RJ, Lozano AM. Targeted treatments in fragile X syndrome. Expert Opin Orphan Drugs. 2014;2 [Google Scholar]
- 109.Berry-Kravis E, Hall D, Leehey M, et al. Treatment and Management of FXTAS. In: Tassone F, Berry-Kravis E, editors. The Fragile X-Associated Tremor Ataxia Syndrome (FXTAS) Springer; New York: 2011. pp. 137–154. [Google Scholar]
- 110.Seritan AL, Nguyen DV, Mu Y, et al. Memantine for fragile X-associated tremor/ataxia syndrome: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2014;75:264–271. doi: 10.4088/JCP.13m08546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang JC, Niu YQ, Simon C, et al. Memantine Effects on Verbal Memory in Fragile X-associated Tremor/Ataxia Syndrome (FXTAS): a Double-Blind Brain Potential Study. Neuropsychopharmacology. 2014;39:2760–2768. doi: 10.1038/npp.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ortigas MC, Bourgeois JA, Schneider A, et al. Improving fragile X-associated tremor/ataxia syndrome symptoms with memantine and venlafaxine. J Clin Psychopharmacol. 2010;30:642–644. doi: 10.1097/JCP.0b013e3181f1d10a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schneider A, Leigh MJ, Adams P, et al. Electrocortical changes associated with minocycline treatment in fragile X syndrome. J Psychopharmacol. 2013;27:956–963. doi: 10.1177/0269881113494105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cao Z, Hulsizer S, Cui Y, et al. Enhanced asynchronous Ca(2+) oscillations associated with impaired glutamate transport in cortical astrocytes expressing Fmr1 gene premutation expansion. J Biol Chem. 2013;288:13831–13841. doi: 10.1074/jbc.M112.441055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reddy DS, Rogawski MA. Neurosteroids – endogenous regulators of seizure susceptibility and role in the treatment of epilepsy. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- 116.Brinton RD. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol. 2013;9:241–250. doi: 10.1038/nrendo.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sorensen PL, Gane LW, Yarborough M, et al. Newborn screening and cascade testing for FMR1 mutations. Am J Med Genet A. 2013;161A:59–69. doi: 10.1002/ajmg.a.35680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yrigollen CM, Mendoza-Morales G, Hagerman R, et al. Transmission of an FMR1 premutation allele in a large family identified through newborn screening: the role of AGG interruptions. J Hum Genet. 2013;58:553–559. doi: 10.1038/jhg.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Macpherson J, Waghorn A, Hammans S, et al. Observation of an excess of fragile-X premutations in a population of males referred with spinocerebellar ataxia. Hum Genet. 2003;112:619–620. doi: 10.1007/s00439-003-0939-z. [DOI] [PubMed] [Google Scholar]
- 120.Garcia Arocena D, Louis ED, Tassone F, et al. Screen for expanded FMR1 alleles in patients with essential tremor. Mov Disord. 2004;19:930–933. doi: 10.1002/mds.20043. [DOI] [PubMed] [Google Scholar]
- 121.Van Esch H, Dom R, Bex D, et al. Screening for FMR-1 premutations in 122 older Flemish males presenting with ataxia. Eur J Hum Genet. 2005;13:121–123. doi: 10.1038/sj.ejhg.5201312. [DOI] [PubMed] [Google Scholar]
- 122.Rodriguez-Revenga L, Gomez-Anson B, Munoz E, et al. FXTAS in spanish patients with ataxia: support for female FMR1 premutation screening. Mol Neurobiol. 2007;35:324–328. doi: 10.1007/s12035-007-0020-3. [DOI] [PubMed] [Google Scholar]
- 123.Kraff J, Tang HT, Cilia R, et al. Screen for excess FMR1 premutation alleles among males with parkinsonism. Arch Neurol. 2007;64:1002–1006. doi: 10.1001/archneur.64.7.1002. [DOI] [PubMed] [Google Scholar]
- 124.Rajkiewicz M, Sulek-Piatkowska A, Krysa W, et al. Screening for premutation in the FMR1 gene in male patients suspected of spinocerebellar ataxia. Neurol Neurochir Pol. 2008;42:497–504. [PubMed] [Google Scholar]
- 125.Hall DA, Howard K, Hagerman R, et al. Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson disease. Parkinsonism Relat Disord. 2009;15:156–159. doi: 10.1016/j.parkreldis.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Groen EJ, van Rheenen W, Koppers M, et al. CGG-repeat expansion in FMR1 is not associated with amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:1852 e1851–1853. doi: 10.1016/j.neurobiolaging.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 127.Cilia R, Kraff J, Canesi M, et al. Screening for the presence of FMR1 premutation alleles in women with parkinsonism. Arch Neurol. 2009;66:244–249. doi: 10.1001/archneurol.2008.548. [DOI] [PubMed] [Google Scholar]
- 128.Hall DA, Bennett DA, Filley CM, et al. Fragile X gene expansions are not associated with dementia. Neurobiol Aging. 2014;35:2637–2638. doi: 10.1016/j.neurobiolaging.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Seritan AL, Nguyen DV, Farias ST, et al. Dementia in fragile X-associated tremor/ataxia syndrome (FXTAS): comparison with Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1138–1144. doi: 10.1002/ajmg.b.30732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Faruq M, Srivastava AK, Suroliya V, et al. Identification of FXTAS presenting with SCA 12 like phenotype in India. Parkinsonism Relat Disord. 2014;20:1089–1093. doi: 10.1016/j.parkreldis.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 131.Leehey MA, Hagerman PJ. Fragile X-associated tremor/ataxia syndrome. In: Vinken PJ, Bruyn GW, editors. Handb Clin Neurol. Vol. 103. 2012. pp. 373–386. [Google Scholar]
- 132.Fraint A, Vittal P, Szewka A, et al. New observations in the fragile X-associated tremor/ataxia syndrome (FXTAS) phenotype. Front Genet. 2014;5:365. doi: 10.3389/fgene.2014.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tassone F, Iong KP, Tong TH, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012;4:100. doi: 10.1186/gm401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Renda MM, Voigt RG, Babovic-Vuksanovic D, et al. Neurodevelopmental disabilities in children with intermediate and premutation range fragile X cytosine-guanine-guanine expansions. J Child Neurol. 2014;29:326–330. doi: 10.1177/0883073812469723. [DOI] [PubMed] [Google Scholar]
- 134.Hagerman PJ. Current Gaps in Understanding the Molecular Basis of FXTAS. Tremor Other Hyperkinet Mov (N Y) 2012:2. doi: 10.7916/D80C4TH0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schofield CM, Hsu R, Barker AJ, et al. Monoallelic deletion of the microRNA biogenesis gene Dgcr8 produces deficits in the development of excitatory synaptic transmission in the prefrontal cortex. Neural Dev. 2011;6:11. doi: 10.1186/1749-8104-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Iliff AJ, Renoux AJ, Krans A, et al. Impaired activity-dependent FMRP translation and enhanced mGluR-dependent LTD in Fragile X premutation mice. Hum Mol Genet. 2013;22:1180–1192. doi: 10.1093/hmg/dds525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sobesky WE, Taylor AK, Pennington BF, et al. Molecular/clinical correlations in females with fragile X. Am J Med Genet. 1996;64:340–345. doi: 10.1002/(SICI)1096-8628(19960809)64:2<340::AID-AJMG21>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 138.Aumiller V, Graebsch A, Kremmer E, et al. Drosophila Pur-alpha binds to trinucleotide-repeat containing cellular RNAs and translocates to the early oocyte. RNA Biol. 2012;9:633–643. doi: 10.4161/rna.19760. [DOI] [PubMed] [Google Scholar]
- 139.Faller M, Toso D, Matsunaga M, et al. DGCR8 recognizes primary transcripts of microRNAs through highly cooperative binding and formation of higher-order structures. RNA. 2010;16:1570–1583. doi: 10.1261/rna.2111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Perron MP, Provost P. Protein components of the microRNA pathway and human diseases. Methods Mol Biol. 2009;487:369–385. doi: 10.1007/978-1-60327-547-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stark KL, Xu B, Bagchi A, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- 142.Disney MD, Liu B, Yang WY, et al. A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem Biol. 2012;7:1711–1718. doi: 10.1021/cb300135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kenneson A, Zhang F, Hagedorn CH, et al. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- 144.Kunst CB, Leeflang EP, Iber JC, et al. The effect of FMR1 CGG repeat interruptions on mutation frequency as measured by sperm typing. J Med Genet. 1997;34:627–631. doi: 10.1136/jmg.34.8.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Loesch DZ, Cook M, Litewka L, et al. A low symptomatic form of neurodegeneration in younger carriers of the FMR1 premutation, manifesting typical radiological changes. J Med Genet. 2008;45:179–181. doi: 10.1136/jmg.2007.054171. [DOI] [PubMed] [Google Scholar]
- 146.Filipovic-Sadic S, Sah S, Chen L, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010;56:399–408. doi: 10.1373/clinchem.2009.136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Todd PK, Ackall FY, Hur J, et al. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis Model Mech. 2014;7:143–155. doi: 10.1242/dmm.012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Leehey MA. Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med. 2009;57:830–836. doi: 10.231/JIM.0b013e3181af59c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Adams JS, Adams PE, Nguyen D, et al. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS) Neurology. 2007;69:851–859. doi: 10.1212/01.wnl.0000269781.10417.7b. [DOI] [PubMed] [Google Scholar]