

Abstract
A number of studies have indicated that antagonists of the N-methyl-d-aspartate (NMDA) subtypes of glutamate receptors can cause schizophrenia-like symptoms in healthy individuals and exacerbate symptoms in individuals with schizophrenia. These findings have led to the glutamate hypothesis of schizophrenia. Here we review the evidence for this hypothesis in postmortem studies of brain tissue from individuals affected by schizophrenia, summarizing studies of glutamate neuron morphology, of expression of glutamate receptors and transporters, and of the synthesizing and metabolizing enzymes for glutamate and its co-agonists. We found consistent evidence of morphological alterations of dendrites of glutamatergic neurons in the cerebral cortex of subjects with schizophrenia and of reduced levels of the axon bouton marker synaptophysin. There were no consistent alterations of mRNA expression of glutamate receptors, although there has been limited study of the corresponding proteins. Studies of the glutamate metabolic pathway have been limited, although there is some evidence that excitatory amino acid transporter-2, glutamine synthetase, and glutaminase have altered expression in schizophrenia. Future studies would benefit from additional direct examination of glutamatergic proteins. Further advances, such as selective testing of synaptic microdomains, cortical layers, and neuronal subtypes, may also be required to elucidate the nature of glutamate signaling impairments in schizophrenia.
Introduction
For more than two decades phencyclidine (PCP), ketamine, and other antagonists of the N-methyl-d-aspartate (NMDA) subtypes of glutamate receptors have been known to induce schizophrenia-like, positive, negative, and cognitive symptoms.1–4 Similarly, NMDA receptor antagonist administration in schizophrenia subjects exacerbates these symptoms for prolonged periods.5,6 These observations have formed the bedrock of the glutamate hypothesis of schizophrenia, which, most broadly, posits that dysfunction of glutamatergic neurotransmission may be involved in the etiology of schizophrenia.7
This hypothesis has motivated a large number of animal model studies that have provided substantial support. For example, NMDA receptor antagonists have been found to induce schizophrenia-associated phenotypes in non-human primates and rodents, such as cognitive and sensorimotor gating impairments, hyperlocomotion, and social impairments.8 Animal model studies have also shown that the application of NMDA receptor autoantibodies, which in human patients induce a schizophrenia-like syndrome that is completely resolved following dialysis of the antibodies,9 cause NMDA receptor internalization, decrease NMDA receptor-mediated currents, and impair learning and memory.10 Supporting findings have also arisen from animal models that reduce levels of the NMDA receptor co-agonist d-serine, in which impaired long-term potentiation, reduced dendritic spine density, reduced hippocampal volume, and impaired memory performance have been observed.11 Though not yet firmly established, clinical trials in subjects with schizophrenia suggest that enhancing NMDA receptor function via increasing availability of co-agonists has some efficacy.7
These findings, however, raise an important question. Is there direct evidence of alterations to glutamate signaling within individuals with schizophrenia? One approach to answering this question has been to measure glutamate indices using magnetic resonance spectroscopy in affected individuals. A recent review of many such studies concluded that elevated, not reduced, tissue levels of glutamate indices are present in medial prefrontal cortex in medication-naive and medication-free patients.12 Another widely used approach has been evaluation of genetic variation in components of glutamate signaling pathways. Although initial studies of positional and functional candidate genes suggested that many schizophrenia risk variants would be located in glutamate signaling genes,13 these initial findings were not clearly supported in large scale assessments of common variants.14 More recent studies that have examined common genetic variation have found genome-wide significant evidence of support for a number of glutamate signaling genes, including GRIA1, GRIN2A, GRM3 and SRR.181 In contrast, recent studies of rare and de novo mutations suggest that mutations in signaling molecules downstream of glutamate receptors, rather than in the receptors themselves, may contribute to schizophrenia risk.15,16
In the current review, we examine the evidence for alterations in components of glutamate circuits and signaling pathways, as assessed in studies of postmortem brain tissue obtained from individuals diagnosed with schizophrenia and schizoaffective disorder during life. We review evidence for structural alterations in glutamatergic neurons. In addition, we review studies of mRNA and protein expression of molecules involved in glutamate signaling, specifically glutamate receptors, glutamate transporters, glutamate synthesizing enzymes, and glutamate receptor co-agonists.
Structural alterations in glutamate neurons (Table 1)
Table 1.
Summary of studies of structural alterations in glutamate (pyramidal) neurons
| Reference | Brain region | SZ/Cntl | Methods | Difference in SZ |
|---|---|---|---|---|
| Somal volume | ||||
| 19 | BA9 | 9/10 | Nissl | 7% decrease in layer III |
| 17 | BA9 | 28/28 | Nissl | 9.2% decrease in deep layer III |
| 18 | BA9 | 13/13 | Nissl | 14.2% decrease in deep layer III |
| 24 | BA9 | 11/10 | Nissl | No differences in layer IIIb-c |
| 18 | BA9 | 9/9 | IHC | No difference in NNFP-IR neurons in deep layer III |
| 20 | BA9 | 11/13 | IHC | 59% increase in layer III NF200-IR pyramidal neuron volume |
| 23 | BA41 | 16/16 | Nissl | 10.4% decrease in deep layer III |
| 22 | BA42 | 18/18 | Nissl | 13.1% decrease in deep layer III |
| 23 | BA42 | 18/18 | Nissl | No difference in layer V |
| 24 | PT (~BA41, BA42, BA22) | 11/10 | Nissl | No difference in layers II, IIIc, or VI |
| 25 | Insula (~BA13) | 15/15 | Nissl | 16.2% decrease in layer II but not in layer III |
| 26 | Fusiform cortex (~BA37) | 11/13 | Nissl | No difference in pyramidal cells of layers III or V |
| 27 | ~BA40 | 24/24 | Nissl | No difference in upper or lower cortical layers or lower layer III |
| 19 | BA17 | 7/7 | Nissl | No difference |
| Dendritic length and number | ||||
| 31 | BA46 | 15/15 | Golgi | 14% decrease in deep, but not superficial, layer III |
| 32 | BA 46 | 14/15 | Golgi | No difference in layers V or VI |
| 30 | BA11 | 5/5 | Golgi | 29% decrease in layer III |
| 28 | BA10 | 15/18 | Golgi | 40% decrease in layer V |
| 29 | BA32 | 11/11 | Golgi | 29% and 46% decrease in primary and secondary dendrite numbers respectively in layer V; 17% and 15% decrease in layer III |
| 31 | BA17 | 13/15 | Golgi | No difference in layer III |
| Dendritic spine density | ||||
| 31 | BA46 | 15/15 | Golgi | 23% decrease in deep, but not superficial, layer III |
| 32 | BA 46 | 14/15 | Golgi | No difference in layers V and VI |
| 33 | BA10, BA11, BA45 | 13/11 | Golgi | 66% decrease in layer III |
| 33 | BA38, BA20, BA21, BA22 | 13/11 | Golgi | 59% decrease in layer III |
| 35 | BA41 | 15/15 | IHC | 27% decrease in spinophilin-IR puncta in deep layer III |
| 35 | BA42 | 15/15 | IHC | 22% decrease in spinophilin-IR puncta in deep layer III |
| 34 | Subiculum | 13/8 | Golgi | 35% decrease |
| 31 | BA17 | 13/15 | Golgi | No difference in layer III |
| Synaptophysin protein expression | ||||
| 52 | BA 8 | 10/13 | IB | No difference |
| 47 | BA9 | 5/4 | IB | Decrease |
| 43 | BA9 | 10/10 | IHC | Decrease |
| 50 | BA9 | 19/19 | IB, IAR | No difference |
| 45 | ~BA10 | 13/10 | ELISA | No difference |
| 47 | BA10 | 6/6 | IB | Decrease |
| 44 | BA10 | 14/12 | IB | Decreases |
| 48 | BA10 | 10/10 | IB | Decrease |
| 46 | BA45, BA46 | 5/6 | IB | Trend of decrease |
| 53 | PFC | 18/23 | IB | No difference |
| 43 | BA46 | 10/10 | IHC | Decrease |
| 50 | BA46 | 19/19 | IB, IAR | No difference |
| 46 | BA32, BA33 | 18/12 | IB | Decrease |
| 176 | ~BA32, BA33 | 11/13 | IB | Decrease |
| 54 | ~BA24 | 18/24 | ELISA | No difference |
| 51 | BA24 | 15/14 | IB | No difference |
| 52 | BA24 | 17/15 | IB | Increase |
| 42 | Medial temporal lobe | 11/14 | IAR | Decrease |
| 49 | Medial temporal lobe | 7/13 | IR | No difference |
| 46 | Hippocampus | 13/10 | IB | Decrease |
| 52 | BA20 | 19/12 | IB | No difference |
| 47 | BA20 | 6/4 | IB | Decrease |
| 46 | BA21, BA22 | 13/9 | IB | No difference |
| 46 | BA39, BA40 | 15/10 | IB | Trend of decrease |
| 52 | BA7 | 8/12 | IB | No difference |
| 43 | BA17 | 10/10 | IHC | No difference |
| 47 | BA17 | 10/6 | IB | Decrease |
| Axon boutons | ||||
| 55 | BA41 | 15/15 | IHC | 14% decreases of synaptophysin-IR puncta density in deep layer III |
| 56 | BA41 | 27/27 | IHC | No differences in VGluT1-, VGluT2-IR bouton density or number in deep layer III |
| 55 | BA42 | 15/15 | IHC | No differences in synaptophysin-IR puncta density |
Abbreviations: SZ, schizophrenia; Cntl, control; BA, Brodmann area; PT, planum temporale; IHC, immunohistochemistry; IB, immunoblotting; ELISA, enzyme-linked immunosorbent assay; IR, immunoreactivity; IAR, immunoautoradiography; NNFP, nonphosphorylated neurofilament protein; NF200, neurofilament 200. ~indicates approximate region based on description of location in published report. Red, yellow, and green colors represent decreases, no changes, and increases respectively in structural measures in schizophrenia.
Somal volume
Somal volume has been evaluated in ten studies, including eight cortical areas. Because some studies tested multiple areas, and/or multiple cortical layers within a region, in all fourteen comparisons between schizophrenia and control subjects have been reported. Somal volume was decreased in SZ relative to control subjects in six comparisons, unchanged relative to control subjects in seven, and greater than control subjects in one. Somal size of pyramidal neurons decreased by 9.2–14.2% in deep layer III of DLPFC in SZ.17,18 Rajkowska et al.19 reported a 7.1% decrease in somal volume when both pyramidal neurons and interneurons are counted. Studies also found that pyramidal neurons staining positive for neurofilament 200 or nonphosphorylated epitopes of neurofilament have either larger or unchanged somal size,18,20 possibly indicating that other pyramidal neuronal subpopulations contribute to the lower average somal volume. However, a potential confound of these latter studies is that the immunostaining protocol used to label neurofilament epitopes in these studies over-estimates somal sizes, especially in SZ.21
In deep layer III of the primary and association auditory cortices, somal volume of pyramidal neurons were also decreased by 10.4% and 13.1%, respectively.22,23 However, such a decrease was not present in layer V,23 consistent with layer specificity. One study, with a smaller sample size, did not find somal size differences, counting all neurons in the planum temporale, a structure containing the auditory association cortex.24 An important consideration here is that when all neurons are counted, a small difference in pyramidal neurons, if present, would be masked by inclusion of numbers of interneurons. Decreased somal volume is also found in the insula,25 but not in other brain areas such as fusiform cortex, primary visual cortex, and inferior parietal lobe.19,26,27 Whether chronic antipsychotic exposure affects somal volume has not been studied in animal models.
Dendrite arborization
Dendrite extent has been evaluated in five studies, including five cortical areas, in which dendritic arbors were found to be reduced in SZ relative to control subjects in four regions and unchanged relative to control subjects in one region. Several studies have found decreased basilar dendrite length by 14–29% or dendrite number in the prefrontal cortex, especially layer III.28–31 However, the decrease was not found in the primary visual cortex.31 One study found no difference in layer V or VI32 in the prefrontal cortex but two other studies found a decrease in either dendritic field size28 or dendrite number29 in layer V. Black et al.28 reported a 40% decrease in basilar dendritic field size, a measure of both dendrite length and branching, in layer V pyramidal neurons. Whether chronic antipsychotic exposure affects dendrite arborization has not been studied in animal models.
Dendritic spine density
Dendritic spine density has been evaluated in five studies, including tissue from thirteen cortical areas in six brain regions (dorsolateral prefrontal cortex, orbitofrontal cortex, temporal association cortex, auditory cortex, hippocampal formation, visual cortex). Spine density was reduced in SZ relative to control subjects in five regions and unchanged relative to control subjects in one. Decreases in spine density have been found in layer III of prefrontal, temporal, primary auditory, and auditory association cortices,31,33–35 but not in the primary visual cortex.31 The reported decreases ranged from 22–66% (median 27%) and included both estimates of decreased density of spines per dendritic length31,33,34 and decreased density of spines per µm3 of gray matter.35 In the one study evaluating deeper layers there was no decrease found in layers V or VI in prefrontal cortex.32 One study found a 35% decrease in spine density in the subiculum.34 However, dendritic spine density was not altered either in animal models with antipsychotic exposure35 or in human subjects with a non-schizophrenia psychiatric illness exposed to antipsychotics,31 suggesting the reduction in spine density observed in human SZ subjects was not related to antipsychotic use.
Axon boutons
Synaptophysin is a 38-kd synaptic vesicles protein present in virtually all presynaptic boutons.36–38 As a general marker of synapses, synaptophysin protein expression has been shown to reliably measure synaptic density.39–41 Synaptophysin protein expression has been evaluated in sixteen studies, including sixteen cortical areas. Because some studies tested multiple areas, in all twenty-seven comparisons between schizophrenia and control subjects have been reported. Synaptophysin protein expression was decreased in SZ relative to control subjects in twelve studies, unchanged relative to control subjects in fourteen studies, and greater than control subjects in one study. Decreases in synaptophysin protein expression have been found in frontal cortex in multiple studies, and in temporal and cingulate cortices in some studies.42–48 However, other studies do not find reductions in the same regions.49–54 Chronic antipsychotic exposure does not appear to affect synaptophysin expression in animal models.55
The potential reduction in synaptophysin protein expression could reflect reduced synaptophysin protein per bouton, reduced density of axon boutons, or their combination. Interestingly, Sweet et al.55 found that in deep layer III, but not layer I, of the primary auditory cortex the density of synaptophysin-immunoreactive puncta, i.e., presumptive axon boutons, was decreased by 14%. This reduction did not extend to the nearby auditory association cortex.55 However, presumptive axon boutons labeled with antibodies directed against vesicular glutamate transporter 1 (VGluT1) and VGluT2, indicative of excitatory intracortical and thalamocortical axon boutons, respectively, were not altered in density or number in deep layer III of the primary auditory cortex in SZ.56 This combination of findings––no change in VGluT-immunoreactive bouton density in the presence of reduced density of synaptophysin-immunoreactive bouton density––suggests one of two interpretations: either there are reductions in the density of non-glutamatergic boutons labeled by synaptophysin or synaptophysin protein levels within glutamate boutons is reduced (leading to reduced detectability of boutons when only labeled with synaptophysin). The latter alternative would have important implications in disease, as it would suggest that glutamatergic boutons are functionally impaired but structurally preserved, rendering them potential targets for treatments designed to enhance their function.
Molecular alterations in glutamate receptors
mRNA expression of glutamate receptors (Table 2)
Table 2.
Summary of studies of mRNA expression of glutamate receptors in brain regions in schizophrenia
| Region (Reference) |
NMDA | AMPA | Kainate | Metabotropic | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frontal cortex | n (SZ/Cntl) | GRIN1 | GRIN2A | GRIN2B | GRIN2C | GRIN2D | GRIN3A | GRIA1 | GRIA2 | GRIA3 | GRIA4 | GRIK1 | GRIK2 | GRIK3 | GRIK4 | GRIK5 | MGLUR1 | MGLUR2 | MGLUR3 | MGLUR4 | MGLUR5 |
| 66 | 15/15 | ||||||||||||||||||||
| 64 | 21/9 | & | & | & | & | ||||||||||||||||
| 73 | 16/9 | ||||||||||||||||||||
| 77 | 6/10 | * | |||||||||||||||||||
| 68 | 6/6 | ||||||||||||||||||||
| 67 | 10/10 | ||||||||||||||||||||
| 75 | 15/9 | ||||||||||||||||||||
| 70 | 15/15 | ||||||||||||||||||||
| 72 | 36/26 | ||||||||||||||||||||
| 74 | 20/20 | ||||||||||||||||||||
| 71 | 15/15 | ||||||||||||||||||||
| 63 | 15/15 | ||||||||||||||||||||
| 155 | 15/15 | ||||||||||||||||||||
| 79 | 14/23 | && | |||||||||||||||||||
| 78 | 7/7 | ||||||||||||||||||||
| 76 | 42/42 | ** | |||||||||||||||||||
| 48 | 10/10 | ||||||||||||||||||||
| 65 | 37/37 | ||||||||||||||||||||
| Temporal cortex | GRIN1 | GRIN2A | GRIN2B | GRIN2C | GRIN2D | GRIN3A | GRIA1 | GRIA2 | GRIA3 | GRIA4 | GRIK1 | GRIK2 | GRIK3 | GRIK4 | GRIK5 | MGLUR1 | MGLUR2 | MGLUR3 | MGLUR4 | MGLUR5 | |
| 81 | 9/14 | ||||||||||||||||||||
| 80 | 8/7 | # | |||||||||||||||||||
| 66 | 15/15 | ||||||||||||||||||||
| 68 | 6/6 | ## | |||||||||||||||||||
| 70 | 15/15 | ||||||||||||||||||||
| 82 | 15/15 | ||||||||||||||||||||
| Occipital cortex | GRIN1 | GRIN2A | GRIN2B | GRIN2C | GRIN2D | GRIN3A | GRIA1 | GRIA2 | GRIA3 | GRIA4 | GRIK1 | GRIK2 | GRIK3 | GRIK4 | GRIK5 | MGLUR1 | MGLUR2 | MGLUR3 | MGLUR4 | MGLUR5 | |
| 73 | 16/9 | ||||||||||||||||||||
| 68 | 6/6 | ||||||||||||||||||||
| 75 | 15/9 | ||||||||||||||||||||
| 67 | 10/10 | ||||||||||||||||||||
| 72 | 36/26 | ||||||||||||||||||||
| Hippocampus | GRIN1 | GRIN2A | GRIN2B | GRIN2C | GRIN2D | GRIN3A | GRIA1 | GRIA2 | GRIA3 | GRIA4 | GRIK1 | GRIK2 | GRIK3 | GRIK4 | GRIK5 | MGLUR1 | MGLUR2 | MGLUR3 | MGLUR4 | MGLUR5 | |
| 85 | 6/8 | ||||||||||||||||||||
| 81 | 9/14 | ||||||||||||||||||||
| 87 | 11/13 | ||||||||||||||||||||
| 83 | 11/11 | ||||||||||||||||||||
| 84 | 24/21 for NR1, 8/4 for NR2A, 2B |
||||||||||||||||||||
| 82 | 15/15 | ||||||||||||||||||||
| 86 | 30/31 | *** | |||||||||||||||||||
| Thalamus | GRIN1 | GRIN2A | GRIN2B | GRIN2C | GRIN2D | GRIN3A | GRIA1 | GRIA2 | GRIA3 | GRIA4 | GRIK1 | GRIK2 | GRIK3 | GRIK4 | GRIK5 | MGLUR1 | MGLUR2 | MGLUR3 | MGLUR4 | MGLUR5 | |
| 93 | 12/8 | ||||||||||||||||||||
| 89 | 12/8 | ||||||||||||||||||||
| 88 | 13/8 | &&& | |||||||||||||||||||
| 91 | 15/15 | ||||||||||||||||||||
| 92 | 14/16 | ||||||||||||||||||||
| 90 | 14/20 | ### | ### | ### | ### | ||||||||||||||||
significant difference was found only in SZ subjects who were neuroleptic free >6 months, not in those receiving neuroleptic until death;
white matter only;
exon 22 containing NR1 splice isoform;
pyramidal cell layers of BA11 only;
MGLUR1alpha only;
there was no change in total NR1 level, while the NR1-2b splice form was decreased in the right hippocampus and the NR1-4b splice form was decreased in the left hippocampus;
GRIN1 was decreased only in cognitively impaired SZ subjects.
the NR1 splice isoform containing neither C-terminal C1 nor C2 cassette only;
relay neurons of medial dorsal thalamus only.
Red, yellow and green colors represent decreases, no changes, and increases respectively in gene expression in schizophrenia.
Glutamate receptors are classified into two functional groups, ionotropic receptors, including NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors, and metabotropic receptors.57 NMDA receptors are tetramers, composed of 2 obligatory GRIN1 subunits and two regulatory GRIN2 or GRIN3 subunits.58 AMPA receptors are tetramers, encoded by four genes (GRIA1–4). The receptors are assembled as dimers of homodimers of subunits.58 Kainate receptors, encoded by five genes (GRIK1–5), are primarily located presynaptically and modulate synaptic activities.59 Metabotropic glutamate receptors, which are coupled with G proteins, are encoded by eight genes.60
Messenger RNA abundance has been studied by in situ hybridization, northern blotting, quantitative PCR, and other techniques, either individually or with microarray analysis that measures up to the entire transcriptome simultaneously. A number of microarray studies, which usually report genes with top changes, have been used to identify differentially expressed genes in SZ (reviewed in Sequeira et al.61). Genes involved in the glutamate neurotransmission pathway have been reported; however, glutamate receptor genes themselves generally do not stand out.61 It is to be remembered that microarray analysis is a screening test with false positives and false negatives and therefore results generally need to be verified with other methods. Individual glutamate receptor mRNA expression has been studied extensively in various brain areas. Changes in expression of glutamate receptors in SZ were previously reviewed by Rubio et al.62 and here we have also included new studies, as described below.
Several studies found decreases in mRNA expression of NMDA receptor subunits, such as GRIN1, GRIN2A, and GRIN2C, in the frontal cortex of schizophrenia subjects.63–65 However, this was not verified by other studies.66–69 One study reported increases in GRIN3A expression in schizophrenia,70 while another study involving more subjects reported no increased expression.65 Two studies found decreases in GRIA1, GRIA2, or GRIA4 mRNA expression in SZ, 64,71 which were not found by two other studies.72,73 Decreases in expression of GRIK1,74 GRIK3, GRIK4,64 and GRIK575 have each been shown in only one of three studies above. One study reported increases in mGluR1α mRNA expression in the PFC in SZ76 and another study found increases in mGluR5 expression in BA11, but not in other prefrontal cortices such as BA9 and BA10.77 One study found decreases in mGluR2 expression78 in SZ, while another study showed no difference.79
In the temporal cortex, decreases in mRNA expression of GRIN1,80 GRIA2,81 or GRIK182 have been reported in single studies, but not by others.66,82 In the occipital cortex, increases in mRNA expression of GRIN1 and GRIN2 have been reported,67 which were not found by another study.68 Increases in GRIA4 expression have been reported in one study,72 but not another.73 In the hippocampus, decreases in GRIN1, GRIA1, and GRIA2 have been reported in two studies,81,83–86 while decreases in GRIK3 and GRIK5 were reported in a single study.87 For GRIK1, one study reported a finding of decreased expression in the perirhinal cortex.82 In the thalamus, lower expression of NMDA, AMPA, or GRIK receptor subunits has been reported in one or two studies,88–90 but not verified by others.91–93 Two studies found no change in MGLURs.90,93
In contrast to structural studies indicating impairments of glutamatergic pyramidal neurons, the results above do not indicate any clear change in transcriptional control of glutamate receptor subunits in schizophrenia. For example, expression of the most studied mRNA, the GRIN1 subunit of the NMDA receptor, was reduced in four studies, unchanged in five studies, and increased in three of twelve studies. The lack of consistent findings could reflect a true negative. Other possibilities remain open, however. For example, studies of tissue homogenates, which include glutamate receptor RNA obtained from both pyramidal neurons and interneurons, might obscure findings limited to just one of those populations. Variable results could also reflect cohort-specific factors, including technical factors that affect RNA expression (see, for example, Sequeira et al.61 for a discussion of these issues). To the extent that functional changes in glutamate signaling ultimately require alterations in synaptic receptor protein levels, an alternative approach would be to assay receptor proteins directly.
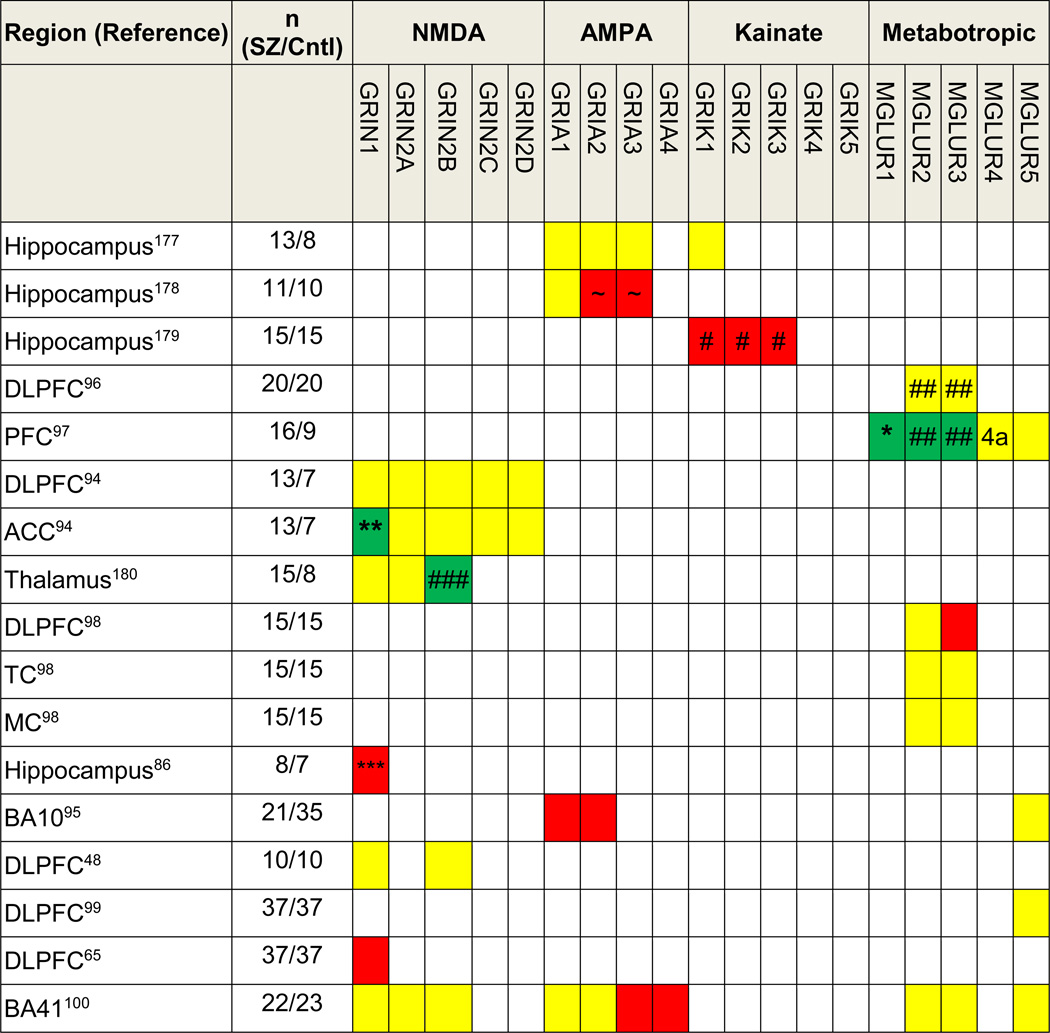
Protein expression of glutamate receptors (Table 3)
Table 3.
Summary of studies of protein expression of glutamate receptors in brain regions in schizophrenia
 |
single antibody raised against common epitope of GRIA2 and GRIA3;
MGLUR1alpha only;
C2’ isoform only;
expression was lower in the left hippocampus of male subjects only.
single antibody against GRIK1/2/3;
single antibody against both MGLUR2 and MGLUR3;
dorsomedial thalamus only;
Abbreviations: DLPFC, dorsolateral prefrontal cortex; PFC, prefrontal cortex; ACC, anterior cingulate cortex; TC, temporal cortex, MC, motor cortex. Red, yellow and green colors represent decreases, no changes, and increases respectively in gene expression in schizophrenia, 4a, MGLUR4 subtype alpha.
There are fewer studies of glutamate receptors in SZ at the protein level than of mRNA, and these, generally speaking, are more recent than mRNA studies. Similar to the mRNA data, however, no clear trends have emerged. Kristiansen et al.94 found no changes in expression of NMDA receptor subunits (GRIN1, GRIN2A–2D) in either the DLPFC or the ACC, except the C2′ isoform of GRIN1 was higher in the ACC of SZ subjects. Rao et al.48 found that GRIN1 and GRIN2B were unchanged in the DLPFC in SZ. In contrast to Kristiansen, Weickert et al.65 showed that GRIN1 expression was lower in SZ in a larger study of an Australian sample. One study showed that GRIA1 and GRIA2 were lower in the DLPFC in SZ.95 Using an antibody that identifies both mGluR2 and 3, Crook et al.96 reported no differences in the DLPFC in SZ, but Gupta et al.97 reported increases of both mGluR1α (consistent with an mRNA study76) and mGluR2/3 in SZ. However, using subtype-specific antibodies, Ghose et al.98 reported decreases in mGluR3 but no changes in mGluR2. Three studies reported no changes in mGluR5 expression.95,97,99 We found that in animal models chronic antipsychotic exposure did not appear to affect expression of some of the receptor subunits, such as GRIA3 and GRIA4.100
Molecular alterations in glutamate transport, synthesis, and co-agonists (Table 4)
Table 4.
Summary of alterations in glutamate transport, synthesis, and co-agonist in brain regions in schizophrenia
| Reference | Brain region |
n (SZ/Cntl) |
Methods | Difference in SZ | |||
|---|---|---|---|---|---|---|---|
| EAATs | EAAT1 | EAAT2 | EAAT3 | EAAT4 | |||
| 77 | BA9, BA10 | 6/10 | mRNA | ||||
| 119 | DLPFC | 20/11 | mRNA | ||||
| 119 | DLPFC | 13/8 | protein | monomer | |||
| 119 | ACC | 20/11 | mRNA | ||||
| 119 | ACC | 13/8 | protein | multimer | |||
| 69 | BA10 | 10/10 | mRNA | ||||
| 69 | BA10 | 10/10 | protein | ||||
| 108 | STG | 23/27 | protein | ||||
| 108 | HC | 23/27 | protein | ||||
| 120 | DLPFC | 33/32 | glycosylation | multimer | |||
| 120 | ACC | 34/34 | glycosylation | monomer | |||
| VGluT | VGluT1 | VGluT2 | |||||
| 110 | DGiml | 17/17 | protein | ||||
| 105 | HC | 13/12 | mRNA | ||||
| 105 | DLPFC | 10/10 | mRNA | ||||
| 105 | STG | 11/12 | mRNA | ||||
| 107 | DLPFC | 18/11 | mRNA | ||||
| 107 | ACC | 18/11 | mRNA | ||||
| 107 | DLPFC | 23/27 | protein | ||||
| 107 | ACC | 23/27 | protein | ||||
| 109 | HC, EC, MTG | 13/13 | mRNA | ||||
| 109 | ITG | 13/13 | mRNA | ||||
| 106 | DLPFC | 37/37 | mRNA | ||||
| 108 | HC | 23/27 | protein | ||||
| 108 | STG | 23/27 | protein | ||||
| 56 | Deep layer 3 of primary auditory cortex | 27/27 | protein (IHC) | ||||
|
Glutamine synthetase |
Brain region |
n (SZ/Cntl) |
Measure | Difference in SZ | |||
| 129 | DLPFC | 27/13 | enzymatic activity | ||||
| 125 | Prefrontal cortex | 8/9 | protein | ||||
| 126 | Prefrontal cortex | 10/10 | protein | ||||
| 130 | Thalamus | 13/8 | mRNA | ||||
| 127 | PFC | 15/15 | protein | ||||
| 124 | ACC, STG | 23/27 | protein | ||||
| 124 | DLPFC, PVC, HC | 23/27 | protein | ||||
| 128 | ACC | 11/8 | protein | Female only | |||
| 123 | Deep layers of ACC | 18/21 | mRNA | ||||
| Phosphate-activated glutaminase | |||||||
| 129 | DLPFC | 27/13 | enzyme activity | ||||
| 130 | Thalamus | 13/8 | mRNA | ||||
| 123 | ACC | 18/21 | mRNA | ||||
| Serine racemase | |||||||
| 138 | Cerebellum, parietal cortex | 4/5 | mRNA | ||||
| 139 | HC | 23/27 | protein | ||||
| 139 | DLPFC, ACC, STG PVC | 23/27 | protein | ||||
| 140 | Cerebellum | 15/15 | mRNA and protein | ||||
| 140 | DPFC | 15/15 | mRNA | ||||
| 140 | DPFC | 15/15 | protein | ||||
| 137 | BA9 | 13/14 | protein | ||||
| 137 | HC | 15/14 | protein | ||||
| DAAO | |||||||
| 138 | cerebellum | 4/5 | mRNA and enzyme activity | * | |||
| 140 | cerebellum | 14/14 | mRNA | ||||
| 140 | cerebellum | 14/14 | protein | Trend of increase | |||
| 138 | Parietal cortex | 4/5 | mRNA | ||||
| 140 | DPFC | 14/14 | mRNA | ||||
| 137 | BA9 and HC | 15/14–15 | protein | ||||
| 143 | Parietal cortex | 15/15 | enzyme activity | ||||
| KMO | |||||||
| 147 | BA6 | 32/32 | mRNA and enzyme activity | ||||
| KAT-1 | |||||||
| 138 | cerebellum | 4/5 | Enzyme activity | ||||
| 138 | cerebellum | 4/5 | mRNA | ||||
only hDAAOl isoform mRNA was increased in SZ without a change in total DAAO mRNA.
Abbreviations: DPFC, dorsal prefrontal cortex; DLPFC, dorsolateral prefrontal cortex; ACC, anterior cingulate cortex; HC, hippocampus; EC, entorhinal cortex; DGiml, the inner molecular layer of the dentate gyrus; PVC, primary visual cortex; STG, superior temporal gyrus; ITG, inferior temporal gyrus; MTG, middle temporal gyrus; DAAO, D-amino acid oxidase; KMO, kynurunine 3-monooxygenase; KAT-1, kynurenine aminotransferase-1. Red, yellow and green colors represent decreases, no changes, and increases respectively in schizophrenia.
Vesicular glutamate transporter (VGluT) is involved in packaging of glutamate in synaptic vesicles.101 VGluT1 is preferentially expressed in neurons from the cortex and hippocampus, VGluT2 in neurons from subcortical structures.101–103 It has been reported that VGluT expression level correlates with synaptic glutamate release.104 Eastwood and Harrison105 found decreased VGluT1 mRNA expression in the prefrontal cortex and hippocampus in SZ; however, two other studies reported no such changes in the prefrontal cortex.106,107 In contrast, increased VGluT1 mRNA and protein was found in the ACC.107 Oni-Orisan et al.107 also found VGluT2 mRNA and protein levels unchanged in the ACC and PFC. Shan et al.108 found no changes in either VGluT1 or VGluT2 protein expression in the superior temporal gyrus and hippocampus. Consistent with that report, Moyer et al.56 found no differences in the number of VGluT1 and VGluT2 containing axon boutons or in the mean within-bouton levels of VGluT1 and VGluT2 protein in the primary auditory cortex located in the superior temporal gyrus. Uezato et al.109 also found no differences in expression of VGluT1 and VGluT2 mRNA in the medial temporal lobe, although they reported that VGluT2 mRNA expression was lower in the inferior temporal gyrus in SZ. However, in the inner molecular layer of the dentate gyrus, Talbot et al.110 found that VGluT1 protein expression was increased.
Glutamate is eliminated from the synaptic cleft by excitatory amino acid transporters (EAATs).111 EAAT1 and 2 are expressed primarily in glial cells,112,113 while EAAT3 and 4 are primarily expressed by neurons.114–116 Around 90% of glutamate at the synaptic cleft is cleared by EAAT2.117,118 Thus, changes in EAAT expression can potentially alter synaptic glutamate levels. Bauer et al.119 found that in SZ EAAT1 protein expression was lower in the DLPFC, EAAT2 was unchanged, and EAAT3 was higher in the ACC but not in the DLPFC. Bauer et al.120 later found less glycosylation of EAAT1 and 2 in SZ, a process important for plasma membrane localization. The same group also found lower protein expression of EAAT1 and 2 in the superior temporal gyrus, lower EAAT2 in the hippocampus, with EAAT3 level unchanged.108 Rao69 reported that EAAT1, EAAT3, and EAAT4 protein expression were higher, while EAAT2 was unchanged in the DLPFC in SZ. Therefore, conflicting results have been reported from studies by the two groups, although both found no changes of EAAT2 in the DLPFC.
After synaptic uptake into astrocytes by EAATs, glutamate is turned into glutamine by glutamine synthetase (GS).121,122 Glutamine is subsequently delivered to neurons where it is turned into glutamate by phosphate-activated glutaminase (PAG). Therefore, GS and PAG expression levels can potentially affect glutamate cycling and availability to neurons. Katsel et. al.123 found that GS mRNA expression was lower in the deep, but not superficial, layers of the ACC, indicating layer specificity. Steffek et al.124 found that GS protein expression was lower in the superior temporal gyrus (STG) and ACC, but not in the DLPFC, primary visual cortex, or hippocampus of subjects with SZ. However, two other studies showed decreased GS protein level in the frontal cortex in SZ,125,126 while a third study was negative.127 One study found that GS protein was higher in the ACC of female SZ patients only, not in males.128 In addition, Gluck129 found that GS enzymatic activity was unchanged in the DLPFC in SZ. Only a few postmortem studies investigated PAG in SZ. Bruneau et al.130 found that transcripts of both PAG and glutamine synthetase were increased in the thalamus of SZ subjects. Katsel et al.123 found PAG transcript was unchanged in the ACC of SZ subjects. One study found that PAG activity was 4-fold greater in the DLPFC of subjects with SZ.129
D-Serine, an NMDA receptor co-agonist,131 is synthesized by serine racemase from l-serine,132,133 and it can be degraded by d-amino acid oxidase (DAAO).134,135 Labrie et al.136 reviewed d-serine in schizophrenia, and found some evidence of decreased levels of d-serine or ratio of d-serine to total serine in serum or cerebrospinal fluid. Studies have shown decreased,137 unchanged,138 and increased levels of serine racemase139,140 in SZ. DAAO expression was higher in the hindbrain than forebrain.141,142 Increased DAAO expression and activity have been reported in the cerebellum in SZ.138,140 However, most studies in the cortex were negative137,138,140 although one study did show increased DAAO activity in the parietal cortex in SZ.143 Kynurenic acid (KYNA), a metabolite of kynurenine arising in the tryptophan degradation pathway, is an antagonist at the glycine site of NMDA receptors.144 KYNA levels were reported higher in the prefrontal cortex145 and cerebrospinal fluid146 of SZ patients. Kynurenine 3-monooxygenase (KMO) decreases KYNA synthesis by metabolizing kynurenine into products other than KYNA.147 In one study, Wonodi et al.147 found that KMO expression and enzymatic activity were lower in the frontal eye field of the cortex, potentially contributing to a higher level of KYNA. Kynurenine aminotransferase-1, involved in KYNA synthesis, was found to have higher enzyme activity in the cerebellum without change in mRNA expression.138
Conclusions and recommendations for future progress
The glutamate hypothesis of schizophrenia has been elaborated largely on the basis of strong support from pharmacologic challenge studies in humans and in animal models, although other evidence, such as the ability of anti-NMDA receptor autoantibodies to induce psychotic symptoms, is also supportive. In the current review we evaluated whether there is direct evidence of glutamatergic impairments in brain tissue obtained from individuals who experienced schizophrenia during life. These studies reveal several clear conclusions. First, there is consistent evidence for morphological alterations of dendrites of glutamatergic neurons in the cerebral cortex of subjects with schizophrenia, and of reduced levels of the axon bouton marker, synaptophysin. Second, despite their expression in these affected structures, there is no clear evidence for reduction of mRNA expression of glutamate receptors and vesicular transporters, although there has been limited study of the corresponding proteins. Third, while the number of studies of molecules regulating the metabolism of glutamate and its co-agonists has been limited, there is some evidence that the key components of the glutamate metabolic cycle, EAAT2, glutamine synthetase, and glutaminase, have altered expression in schizophrenia.
The study of glutamatergic, or other alterations, in postmortem brain tissue from affected individuals has several distinctive advantages and limitations that need to be considered in order to evaluate the findings above. Although in vivo imaging methods are expanding the number of molecules that can be investigated, at present brain tissue studies are the only approach by which alterations in large numbers of molecules within specific cerebral cortex layers, cells, and cellular compartments can be detected. With regard to brain tissue studies in affected individuals, to the extent that schizophrenia is an illness of the brain, the relevant pathologies leading to schizophrenia must be manifest, and therefore detectable, in the circuits, cells, cellular compartments, and molecules within the brains of affected individuals. Importantly, for a number of reasons it cannot be assumed that the same pathologies can be more readily created and evaluated in animal models. Although there is substantial conservation of neuronal genes, proteins and cell types from mice to men, the differences in molecules, architecture, and connectivity can have substantial impact. This has been very evident in the study of Alzheimer’s disease, where small amino acid sequence differences in mouse amyloid-β protein prevent it from aggregating into the toxic assemblies present in human.148 Even when transgenic human amyloid-β sequences containing mutations that cause Alzheimer’s disease in humans are introduced into mice, many key aspects of the neurodegenerative pathology are not reproduced.148 It is not yet clear if the advent of transgenic approaches in other mammals will solve these problems for some brain disorders.149 More recently, an intriguing observation indicated that there is a high rate of somatic mosaicism (i.e., the presence of genetic variation not transmitted through the germ line) among human neurons.150 If confirmed, one implication would be that it may be only possible to study the molecular manifestations of human brain disease in human brain tissue itself.
There are also disadvantages to the use of human brain tissue to study a chronic disease with onset early in life, such as schizophrenia. Many of these factors are technical in nature, and can be mitigated with rigorous attention to the methodology for tissue harvesting, preservation, characterization, and diagnostic evaluation.151 However, a fundamental limitation arises from the fact that most individuals who die with disease do so years, and more commonly decades, after onset. Thus, aging itself, which can affect neuronal morphology and gene expression, might impact the differences between SZ and normal subjects (i.e., an age by disease interaction). The mean ages of subjects included in the studies summarized above vary broadly from mid-life to late-life. However, the many additional cohort and technical factors that differ between reports are confounded by differences in subject ages, precluding any conclusion that differences between studies can be reconciled as due to an interaction of aging with the disease process.
In addition to possible effects of aging, the long illness durations of most subjects prior to death make it difficult to disentangle those alterations that may have contributed to the cause and persistence of symptoms of schizophrenia from those that may be either compensatory or the result of chronic disease, of co-morbid conditions such as nicotine and other substance use, or of long term medication treatment. It is possible in some cases to evaluate the impact of substances and medications via construction of appropriate contrasts among human subjects in post-mortem studies. However, post-mortem human studies are not that useful for assessing long term effects of antipsychotics because the vast majority of subjects have received such treatment for a long period of time. The best alternative approach to evaluate this effect in tissue studies is to use animal models chronically treated with antipsychotics, as described above, e.g., regarding the effect of antipsychotic exposure on dendritic spine density and axon boutons). Nevertheless, the problem of cause versus compensation often remains problematic. This concern can be seen in the findings regarding components of the glutamate cycle, in which postmortem tissue studies suggest reductions in EAAT2 and glutamine synthetase, and increased PAG. These findings might predict indices of glutamate would be altered in vivo in chronic subjects with schizophrenia. In contrast, in vivo studies suggest overall that tissue levels of glutamate indices are unchanged in chronic, treated schizophrenia, but instead are elevated at first episode of illness.12 This raises the possibility that the postmortem findings reflect a compensation in the face of an earlier, sustained glutamate elevation or even a medication effect, although animal model studies largely do not indicate an effect of long term antipsychotic treatment.108,123,124,152 However, it should be noted that the relative contribution of neuronal somata, neuronal presynaptic boutons, astrocytes, and extracellular glutamate to the tissue levels of glutamate indices detected by in vivo MRS is not known.
One approach to overcome most of the above concerns would be to develop a sufficiently large collection of samples from individuals who died very early in disease- a necessary but challenging enterprise. Alternatively, an approach is to relate identified alterations in postmortem brain tissue to potentially causal genetic variations (ideally mutations of moderate to high penetrance). For example, we recently reported reduced protein levels of ATP1A3 in auditory cortex of subjects with schizophrenia.100 Rare mutations in ATP1A3 have been identified as contributing to a polygenic burden for schizophrenia risk,16 and mutations in ATP1A3 that reduce protein expression are also strongly linked to rapid-onset dystonia-parkinsonism, a movement disorder in which individuals with ATP1A3 mutations also display significant impairments in memory, attention, and executive function, and in which 19% develop a psychotic syndrome characterized by auditory hallucinations prior to or at onset of the motor symptoms.153 Thus, the genetic findings strongly support the interpretation that the altered ATP1A3 expression in postmortem brain could contribute to onset of symptoms (rather than result from chronic illness), although confirmatory studies in model systems are also needed.
Another potential technical issue affecting postmortem studies that attempt to identify the molecular correlates of the structural alterations in glutamate neurons in schizophrenia brain tissue has been the preponderance of studies measuring mRNA expression. As summarized in Table 2, investigation of mRNA transcript levels of the glutamate receptors found in specific brain structures has not provided clear consensus on the extent of change (if any) or direction of change between disease and control tissues. The lack of correspondence between the structural and molecular studies could be one of spatial resolution. One advantage histological studies have over many mRNA studies is the ability of the former to resolve the physical unit being quantified, e,g, dendritic spines. The ability to differentiate between cortical layers, neuronal subtypes, and synaptic compartments has been essential to the successful detection of alterations in neuronal morphology. Thus, differences measured in mRNAs between schizophrenia and control tissues localized to a specific layer, neuronal type, or synaptic compartment could be lost in the noise of a homogenized tissue sample. The use of in situ hybridization and, more recently, the introduction of laser-capture and linear amplification strategies have mitigated this issue somewhat, allowing for quantification of mRNAs in discreet cortical layers and specific neuronal populations.90,154,155
Some of the variability in the molecular findings might also reflect the more general problem that mRNA levels explain only a minority of the variance in protein level for many proteins.156 Unfortunately, to date only limited analysis of glutamate receptor protein levels in schizophrenia has been reported, the number of evaluations too few to draw clear conclusions (Table 3). Moreover, the needed additional investigation of protein levels would benefit from an approach that advances traditional strategies (such as immunoblotting) with the ability to provide multiplex quantification so as to more comprehensively survey the glutamatergic protein network within individuals, to generate results with high precision and accuracy, and to have the necessary sensitivity for measurement of synaptic microdomains, cortical layers, and neuronal subtypes.
Mass spectrometry (MS) proteomic approaches have been used extensively to catalog protein expression in mammalian neuronal systems, and are beginning to see application in the investigation of human brain disease. MS approaches to protein analysis vary, but most workflows fall into one of three types: multi-dimensional protein identification technology (MudPIT), differential MS, and selected reaction monitoring. In MudPIT, proteins are cleaved in to predictable peptide components which are then separated into different fractions, to overcome the dynamic range of protein expression, often by reverse phase chromatography or SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis). These fractions are then analyzed one at a time by high mass accuracy ion trap or time-of-flight instrumentation, to identify and quantify the tryptic peptides. In differential MS, the peptide fragments are instead quantified by ion trap or time-of-flight instrumentation without first being sequenced for identification. Peptides found to be differentially expressed are then sequenced for identification in subsequent analyses. In selected reaction monitoring, preselected peptides are targeted for quantification. The first two methods are capable of quantifying 1000s of proteins in complex mixtures but often have high tissue and instrument time requirements and are well suited for describing complex proteomes and hypothesis generation. In contrast to these approaches, selected reaction monitoring is capable of quantifying fewer proteins, 100’s, but does have several notable advantages. First, it is not confounded by dynamic range of protein expression, thus sample enrichment/fractionation is often unnecessary and far less instrument time is required allowing for higher throughput. Second, it is extremely sensitive, and thus protein requirements can be as low as 10–100 nanograms. This allows for the analysis of biochemical fractions such as synaptic microdomains,157 and should similarly facilitate approaches such as laser capture microdissection of cortical layers and potentially cell types. As new instrumentation is introduced the breadth, throughput, and sensitivity of these approaches will continue to advance allowing for more accurate and comprehensive investigations of glutamate signaling protein abnormalities in schizophrenia.
Despite these potential interpretive and technical issues, some conclusions about the postmortem findings regarding glutamatergic measures in schizophrenia are possible. As a whole, these studies reveal strong evidence for structural alterations of dendrites of glutamatergic neurons in schizophrenia, with reduced dendrite length and complexity, and reduced dendritic spine density each reported in multiple regions, from multiple cohorts, by multiple labs (Table 1). Although some reports found these parameters unchanged, opposing findings (i.e., increased dendritic measures) have not been reported. While not as consistent, reductions in pyramidal neuron somal volume have also been reported in many studies. When assessments of pyramidal neuron structure have been made across multiple cortical layers, alterations affecting layer III pyramidal neurons have been most commonly, but not exclusively, reported. Finally, both levels of the axon bouton marker synaptophysin (a protein that is predominantly, but not specifically, expressed within glutamatergic boutons in cerebral cortex) and synaptophysin immunoreactive puncta density are also widely reported to be decreased in schizophrenia (Table 1). While these structural alterations could represent consequences of long term illness, there is now a substantial body of longitudinal in vivo imaging evidence indicating that progressive reductions in cortical gray matter volume occur in subjects with schizophrenia around the onset of overt psychosis, suggesting these structural alterations are an early disease-related alteration.158 Similarly, animal model studies do not indicate that either dendritic spine or bouton loss is caused by long term antipsychotic exposure.35,55
The findings of structural alterations in at least some cortical pyramidal neurons provides compelling support for the presence of glutamatergic alterations in schizophrenia, and provides some links to other elements of the glutamate hypothesis of schizophrenia. For example, it is well established that activity-dependent glutamate signaling has been shown to modify spine and dendrite structure.159–161 While it cannot be concluded that the observed alterations in pyramidal neuron structure in subjects with SZ arise as a result of impaired glutamate signaling, it is at least possible that they do, as there is evidence that disruptions at various points within the glutamate signaling pathway may cause alterations in pyramidal cell morphology, mimicking those found in SZ. For example, in mature neural systems, either pharmacological blockade of GRIAs162 or deafferentation of glutamatergic projections162–164 decreases density of spines. Reduction in NMDA receptor activity by knockout of serine racemase results in reduced dendritic length, branching, and spines.165,166
Similarly, we have demonstrated reduced dendritic spine density and reduced pyramidal cell somal volume in layer III of the primary auditory cortex (AI). It is largely the reciprocal connections of these same layer III pyramidal cells in AI that sharpen frequency tuning to selectively enhance the preferred frequency during auditory perception,167,168 a necessary prerequisite for tone discrimination. Individuals with SZ demonstrate impairments of tone discrimination169,170 and of the generation of auditory event-related potentials that localize to AI, such as mismatch negativity (MMN).171,172 MMN and tone discrimination likely tap the same underlying intracortical mechanisms,173 and the degree of impairment in MMN and tone discrimination are correlated.169 Reductions of MMN similar to those in subjects with SZ can be modeled by infusing NMDA antagonists into the auditory cortex,173 an observation paralleled by systemic administration of NMDA antagonists in mice174 and in normal humans,175 thus linking the observation of altered cortical pyramidal neuron structure to one of the key historical observations underlying the glutamate hypothesis of schizophrenia. Whether dendritic spine loss in itself functionally mimics the effects of NMDA receptor antagonism in intact tissue will require investigation in disease-relevant models of dendritic spine loss. Alternatively, whether the remaining dendritic spines in SZ subjects are additionally affected by deficits in glutamate receptors or signaling molecules that could be targeted therapeutically will benefit from additional studies of postmortem tissue that address some of the technical limitations reviewed above.
Acknowledgments
This work was supported by National Institutes of Health grants MH 071533, MH 103204, and MH 16804. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health, the National Institutes of Health, the Department of Veterans Affairs, or the United States Government.
Footnotes
Conflicts of interest
The authors report no conflicts of interest.
References
- 1.Javitt DC. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia Hillside. J. Clin. Psychiatry. 1987;9:12–35. [PubMed] [Google Scholar]
- 2.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 3.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 4.Coyle JT. The glutamatergic dysfunction hypothesis for schizophrenia. Harv. Rev. Psychiatry. 1996;3:241–253. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- 5.Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 6.Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–150. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- 7.Javitt DC, Zukin SR, Heresco-Levyt U, et al. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr. Bull. 2012;38:958–966. doi: 10.1093/schbul/sbs069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology. 2000;23:223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 9.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–1098. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010;30:5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balu DT, Li Y, Puhl MD, et al. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2400–E2409. doi: 10.1073/pnas.1304308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poels EM, Kegeles LS, Kantrowitz JT, et al. Glutamatergic abnormalities in schizophrenia: a review of proton MRS findings. Schizophr. Res. 2014;152:325–332. doi: 10.1016/j.schres.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 14.Ripke S, O'Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierri JN, Volk CL, Auh S, et al. Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch. Gen. Psychiatry. 2001;58:466–473. doi: 10.1001/archpsyc.58.5.466. [DOI] [PubMed] [Google Scholar]
- 18.Pierri JN, Volk CL, Auh S, et al. Somal size of prefrontal cortical pyramidal neurons in schizophrenia: differential effects across neuronal subpopulations. Biol. Psychiatry. 2003;54:111–120. doi: 10.1016/s0006-3223(03)00294-4. [DOI] [PubMed] [Google Scholar]
- 19.Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch. Gen. Psychiatry. 1998;55:215–224. doi: 10.1001/archpsyc.55.3.215. [DOI] [PubMed] [Google Scholar]
- 20.Miguel-Hidalgo JJ, Dubey P, Shao Q, et al. Unchanged packing density but altered size of neurofilament immunoreactive neurons in the prefrontal cortex in schizophrenia and major depression. Schizophr. Res. 2005;76:159–171. doi: 10.1016/j.schres.2005.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maldonado-Aviles JG, Wu Q, Sampson AR, et al. Somal size of immunolabeled pyramidal cells in the prefrontal cortex of subjects with schizophrenia. Biol. Psychiatry. 2006;60:226–234. doi: 10.1016/j.biopsych.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 22.Sweet RA, Pierri JN, Auh S, et al. Reduced pyramidal cell somal volume in auditory association cortex of subjects with schizophrenia. Neuropsychopharmacology. 2003;28:599–609. doi: 10.1038/sj.npp.1300120. [DOI] [PubMed] [Google Scholar]
- 23.Sweet RA, Bergen SE, Sun Z, et al. Pyramidal cell size reduction in schizophrenia: evidence for involvement of auditory feedforward circuits. Biol. Psychiatry. 2004;55:1128–1137. doi: 10.1016/j.biopsych.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Smiley JF, Rosoklija G, Mancevski B, et al. Hemispheric comparisons of neuron density in the planum temporale of schizophrenia and nonpsychiatric brains. Psychiatry Res. 2011;192:1–11. doi: 10.1016/j.pscychresns.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pennington K, Dicker P, Hudson L, et al. Evidence for reduced neuronal somal size within the insular cortex in schizophrenia, but not in affective disorders. Schizophr. Res. 2008;106:164–171. doi: 10.1016/j.schres.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 26.Di Rosa E, Crow TJ, Walker MA, et al. Reduced neuron density, enlarged minicolumn spacing and altered ageing effects in fusiform cortex in schizophrenia. Psychiatry Res. 2009;166:102–115. doi: 10.1016/j.psychres.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Smiley JF, Konnova K, Bleiwas C. Cortical thickness, neuron density and size in the inferior parietal lobe in schizophrenia. Schizophr. Res. 2012;136:43–50. doi: 10.1016/j.schres.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Black JE, Kodish IM, Grossman AW, et al. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am. J. Psychiatry. 2004;161:742–744. doi: 10.1176/appi.ajp.161.4.742. [DOI] [PubMed] [Google Scholar]
- 29.Broadbelt K, Byne W, Jones LB. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr. Res. 2002;58:75–81. doi: 10.1016/s0920-9964(02)00201-3. [DOI] [PubMed] [Google Scholar]
- 30.Kalus P, Muller TJ, Zuschratter W, et al. The dendritic architecture of prefrontal pyramidal neurons in schizophrenic patients. Neuroreport. 2000;11:3621–3625. doi: 10.1097/00001756-200011090-00044. [DOI] [PubMed] [Google Scholar]
- 31.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 32.Kolluri N, Sun Z, Sampson AR, et al. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am. J. Psychiatry. 2005;162:1200–1202. doi: 10.1176/appi.ajp.162.6.1200. [DOI] [PubMed] [Google Scholar]
- 33.Garey LJ, Ong WY, Patel TS, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosoklija G, Toomayan G, Ellis SP, et al. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch. Gen. Psychiatry. 2000;57:349–356. doi: 10.1001/archpsyc.57.4.349. [DOI] [PubMed] [Google Scholar]
- 35.Sweet RA, Henteleff RA, Zhang W, et al. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jahn R, Schiebler W, Ouimet C, et al. A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proc. Natl. Acad. Sci. U. S. A. 1985;82:4137–4141. doi: 10.1073/pnas.82.12.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navone F, Jahn R, Di GG, et al. Protein p38: an integral membrane protein specific for small vesicles of neurons and neuroendocrine cells. J. Cell Biol. 1986;103:2511–2527. doi: 10.1083/jcb.103.6.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiedenmann B, Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. 1985;41:1017–1028. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- 39.Hamos JE, Degennaro LJ, Drachman DA. Synaptic loss in Alzheimer's disease and other dementias. Neurology. 1989;39:355–361. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- 40.Masliah E, Terry RD, Alford M, et al. Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. J. Histochem. Cytochem. 1990;38:837–844. doi: 10.1177/38.6.2110586. [DOI] [PubMed] [Google Scholar]
- 41.Masliah E, Terry RD, Alford M, et al. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am. J. Pathol. 1991;138:235–246. [PMC free article] [PubMed] [Google Scholar]
- 42.Eastwood SL, Harrison PJ. Decreased synaptophysin in the medial temporal lobe in schizophrenia demonstrated using immunoautoradiography. Neuroscience. 1995;69:339–343. doi: 10.1016/0306-4522(95)00324-c. [DOI] [PubMed] [Google Scholar]
- 43.Glantz LA, Lewis DA. Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch. Gen. Psychiatry. 1997;54:943–952. doi: 10.1001/archpsyc.1997.01830220065010. [DOI] [PubMed] [Google Scholar]
- 44.Karson CN, Mrak RE, Schluterman KO, et al. Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: a possible neurochemical basis for 'hypofrontality'. Mol. Psychiatry. 1999;4:39–45. doi: 10.1038/sj.mp.4000459. [DOI] [PubMed] [Google Scholar]
- 45.Honer WG, Falkai P, Chen C, et al. Synaptic and plasticity-associated proteins in anterior frontal cortex in severe mental illness. Neuroscience. 1999;91:1247–1255. doi: 10.1016/s0306-4522(98)00679-4. [DOI] [PubMed] [Google Scholar]
- 46.Davidsson P, Gottfries J, Bogdanovic N, et al. The synaptic-vesicle-specific proteins rab3a and synaptophysin are reduced in thalamus and related cortical brain regions in schizophrenic brains. Schizophr. Res. 1999;40:23–29. doi: 10.1016/s0920-9964(99)00037-7. [DOI] [PubMed] [Google Scholar]
- 47.Perrone-Bizzozero NI, Sower AC, Bird ED, et al. Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14182–14187. doi: 10.1073/pnas.93.24.14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao JS, Kim HW, Harry GJ, et al. Increased neuroinflammatory and arachidonic acid cascade markers, and reduced synaptic proteins, in the postmortem frontal cortex from schizophrenia patients. Schizophr. Res. 2013;147:24–31. doi: 10.1016/j.schres.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eastwood SL, Burnet PW, Harrison PJ. Altered synaptophysin expression as a marker of synaptic pathology in schizophrenia. Neuroscience. 1995;66:309–319. doi: 10.1016/0306-4522(94)00586-t. [DOI] [PubMed] [Google Scholar]
- 50.Eastwood SL, Cairns NJ, Harrison PJ. Synaptophysin gene expression in schizophrenia. Investigation of synaptic pathology in the cerebral cortex. Br. J. Psychiatry. 2000;176:236–242. doi: 10.1192/bjp.176.3.236. [DOI] [PubMed] [Google Scholar]
- 51.Eastwood SL, Harrison PJ. Synaptic pathology in the anterior cingulate cortex in schizophrenia and mood disorders. A review and a Western blot study of synaptophysin, GAP-43 and the complexins. Brain Res. Bull. 2001;55:569–578. doi: 10.1016/s0361-9230(01)00530-5. [DOI] [PubMed] [Google Scholar]
- 52.Gabriel SM, Haroutunian V, Powchik P, et al. Increased concentrations of presynaptic proteins in the cingulate cortex of subjects with schizophrenia. Arch. Gen. Psychiatry. 1997;54:559–566. doi: 10.1001/archpsyc.1997.01830180077010. [DOI] [PubMed] [Google Scholar]
- 53.Halim ND, Weickert CS, Mcclintock BW, et al. Presynaptic proteins in the prefrontal cortex of patients with schizophrenia and rats with abnormal prefrontal development. Mol. Psychiatry. 2003;8:797–810. doi: 10.1038/sj.mp.4001319. [DOI] [PubMed] [Google Scholar]
- 54.Honer WG, Falkai P, Young C, et al. Cingulate cortex synaptic terminal proteins and neural cell adhesion molecule in schizophrenia. Neuroscience. 1997;78:99–110. doi: 10.1016/s0306-4522(96)00489-7. [DOI] [PubMed] [Google Scholar]
- 55.Sweet RA, Bergen SE, Sun Z, et al. Anatomical evidence of impaired feedforward auditory processing in schizophrenia. Biol. Psychiatry. 2007;61:854–864. doi: 10.1016/j.biopsych.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 56.Moyer CE, Delevich KM, Fish KN, et al. Intracortical excitatory and thalamocortical boutons are intact in primary auditory cortex in schizophrenia. Schizophr. Res. 2013;149:127–134. doi: 10.1016/j.schres.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willard SS, Koochekpour S. Glutamate, Glutamate Receptors, and Downstream Signaling Pathways. Int. J. Biol. Sci. 2013;9:948–959. doi: 10.7150/ijbs.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collingridge GL, Olsen RW, Peters J, et al. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat. Rev. Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- 60.Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J. Pharmacol. Exp. Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- 61.Sequeira PA, Martin MV, Vawter MP. The first decade and beyond of transcriptional profiling in schizophrenia. Neurobiol. Dis. 2012;45:23–36. doi: 10.1016/j.nbd.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubio MD, Drummond JB, Meador-Woodruff JH. Glutamate Receptor Abnormalities in Schizophrenia: Implications for Innovative Treatments. Biomol. Ther. (Seoul. ) 2012;20:1–18. doi: 10.4062/biomolther.2012.20.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- 64.Sokolov BP. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998;71:2454–2464. doi: 10.1046/j.1471-4159.1998.71062454.x. [DOI] [PubMed] [Google Scholar]
- 65.Weickert CS, Fung SJ, Catts VS, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry. 2013;18:1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akbarian S, Sucher NJ, Bradley D, et al. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dracheva S, Marras SA, Elhakem SL, et al. N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am. J. Psychiatry. 2001;158:1400–1410. doi: 10.1176/appi.ajp.158.9.1400. [DOI] [PubMed] [Google Scholar]
- 68.Le Corre S, Harper CG, Lopez P, et al. Increased levels of expression of an NMDARI splice variant in the superior temporal gyrus in schizophrenia. Neuroreport. 2000;11:983–986. doi: 10.1097/00001756-200004070-00017. [DOI] [PubMed] [Google Scholar]
- 69.Rao JS, Kellom M, Reese EA, et al. Dysregulated glutamate and dopamine transporters in postmortem frontal cortex from bipolar and schizophrenic patients. J. Affect. Disord. 2012;136:63–71. doi: 10.1016/j.jad.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Mueller HT, Meador-Woodruff JH. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr. Res. 2004;71:361–370. doi: 10.1016/j.schres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 71.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60:585–598. doi: 10.1002/syn.20329. [DOI] [PubMed] [Google Scholar]
- 72.Dracheva S, Mcgurk SR, Haroutunian V. mRNA expression of AMPA receptors and AMPA receptor binding proteins in the cerebral cortex of elderly schizophrenics. J. Neurosci. Res. 2005;79:868–878. doi: 10.1002/jnr.20423. [DOI] [PubMed] [Google Scholar]
- 73.Healy DJ, Haroutunian V, Powchik P, et al. AMPA receptor binding and subunit mRNA expression in prefrontal cortex and striatum of elderly schizophrenics. Neuropsychopharmacology. 1998;19:278–286. doi: 10.1016/S0893-133X(98)00014-1. [DOI] [PubMed] [Google Scholar]
- 74.Scarr E, Beneyto M, Meador-Woodruff JH, et al. Cortical glutamatergic markers in schizophrenia. Neuropsychopharmacology. 2005;30:1521–1531. doi: 10.1038/sj.npp.1300758. [DOI] [PubMed] [Google Scholar]
- 75.Meador-Woodruff JH, Davis KL, Haroutunian V. Abnormal kainate receptor expression in prefrontal cortex in schizophrenia. Neuropsychopharmacology. 2001;24:545–552. doi: 10.1016/S0893-133X(00)00189-5. [DOI] [PubMed] [Google Scholar]
- 76.Volk DW, Eggan SM, Lewis DA. Alterations in metabotropic glutamate receptor 1alpha and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia. Am. J. Psychiatry. 2010;167:1489–1498. doi: 10.1176/appi.ajp.2010.10030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ohnuma T, Augood SJ, Arai H, et al. Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Brain Res. Mol. Brain Res. 1998;56:207–217. doi: 10.1016/s0169-328x(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 78.Gonzalez-Maeso J, Ang RL, Yuen T, et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghose S, Crook JM, Bartus CL, et al. Metabotropic glutamate receptor 2 and 3 gene expression in the human prefrontal cortex and mesencephalon in schizophrenia. Int. J. Neurosci. 2008;118:1609–1627. doi: 10.1080/00207450802330702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Humphries C, Mortimer A, Hirsch S, et al. NMDA receptor mRNA correlation with antemortem cognitive impairment in schizophrenia. Neuroreport. 1996;7:2051–2055. doi: 10.1097/00001756-199608120-00040. [DOI] [PubMed] [Google Scholar]
- 81.Eastwood SL, Mcdonald B, Burnet PW, et al. Decreased expression of mRNAs encoding non-NMDA glutamate receptors GluR1 and GluR2 in medial temporal lobe neurons in schizophrenia. Brain Res. Mol. Brain Res. 1995;29:211–223. doi: 10.1016/0169-328x(94)00247-c. [DOI] [PubMed] [Google Scholar]
- 82.Beneyto M, Kristiansen LV, Oni-Orisan A, et al. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- 83.Eastwood SL, Burnet PW, Harrison PJ. GluR2 glutamate receptor subunit flip and flop isoforms are decreased in the hippocampal formation in schizophrenia: a reverse transcriptase-polymerase chain reaction (RT-PCR) study. Brain Res. Mol. Brain Res. 1997;44:92–98. doi: 10.1016/s0169-328x(96)00195-7. [DOI] [PubMed] [Google Scholar]
- 84.Gao XM, Sakai K, Roberts RC, et al. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am. J. Psychiatry. 2000;157:1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- 85.Harrison PJ, Mclaughlin D, Kerwin RW. Decreased hippocampal expression of a glutamate receptor gene in schizophrenia. Lancet. 1991;337:450–452. doi: 10.1016/0140-6736(91)93392-m. [DOI] [PubMed] [Google Scholar]
- 86.Vrajova M, Stastny F, Horacek J, et al. Expression of the hippocampal NMDA receptor GluN1 subunit and its splicing isoforms in schizophrenia: postmortem study. Neurochem. Res. 2010;35:994–1002. doi: 10.1007/s11064-010-0145-z. [DOI] [PubMed] [Google Scholar]
- 87.Porter RH, Eastwood SL, Harrison PJ. Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 1997;751:217–231. doi: 10.1016/s0006-8993(96)01404-7. [DOI] [PubMed] [Google Scholar]
- 88.Clinton SM, Haroutunian V, Davis KL, et al. Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am. J. Psychiatry. 2003;160:1100–1109. doi: 10.1176/appi.ajp.160.6.1100. [DOI] [PubMed] [Google Scholar]
- 89.Ibrahim HM, Hogg AJ, Jr., Healy DJ, et al. Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am. J. Psychiatry. 2000;157:1811–1823. doi: 10.1176/appi.ajp.157.11.1811. [DOI] [PubMed] [Google Scholar]
- 90.Sodhi MS, Simmons M, Mccullumsmith R, et al. Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol. Psychiatry. 2011;70:646–654. doi: 10.1016/j.biopsych.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clinton SM, Meador-Woodruff JH. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2004;29:1353–1362. doi: 10.1038/sj.npp.1300451. [DOI] [PubMed] [Google Scholar]
- 92.Dracheva S, Byne W, Chin B, et al. Ionotropic glutamate receptor mRNA expression in the human thalamus: absence of change in schizophrenia. Brain Res. 2008;1214:23–34. doi: 10.1016/j.brainres.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richardson-Burns SM, Haroutunian V, Davis KL, et al. Metabotropic glutamate receptor mRNA expression in the schizophrenic thalamus. Biol. Psychiatry. 2000;47:22–28. doi: 10.1016/s0006-3223(99)00207-3. [DOI] [PubMed] [Google Scholar]
- 94.Kristiansen LV, Beneyto M, Haroutunian V, et al. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry. 2006;11:737–747. 705. doi: 10.1038/sj.mp.4001844. [DOI] [PubMed] [Google Scholar]
- 95.Corti C, Xuereb JH, Crepaldi L, et al. Altered levels of glutamatergic receptors and Na+/K+ ATPase-alpha1 in the prefrontal cortex of subjects with schizophrenia. Schizophr. Res. 2011;128:7–14. doi: 10.1016/j.schres.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 96.Crook JM, Akil M, Law BC, et al. Comparative analysis of group II metabotropic glutamate receptor immunoreactivity in Brodmann's area 46 of the dorsolateral prefrontal cortex from patients with schizophrenia and normal subjects. Mol. Psychiatry. 2002;7:157–164. doi: 10.1038/sj.mp.4000966. [DOI] [PubMed] [Google Scholar]
- 97.Gupta DS, Mccullumsmith RE, Beneyto M, et al. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57:123–131. doi: 10.1002/syn.20164. [DOI] [PubMed] [Google Scholar]
- 98.Ghose S, Gleason KA, Potts BW, et al. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? Am. J. Psychiatry. 2009;166:812–820. doi: 10.1176/appi.ajp.2009.08091445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matosin N, Frank E, Deng C, et al. Metabotropic glutamate receptor 5 binding and protein expression in schizophrenia and following antipsychotic drug treatment. Schizophr. Res. 2013;146:170–176. doi: 10.1016/j.schres.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 100.Macdonald ML, Ding Y, Newman J, et al. Altered glutamate protein co-expression network topology linked to spine loss in the auditory cortex of schizophrenia; Biol.Psychiatry; 2014. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fremeau RT, Jr., Voglmaier S, Seal RP, et al. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 102.Fremeau RT, Jr., Troyer MD, Pahner I, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 103.Fremeau RT, Jr., Kam K, Qureshi T, et al. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- 104.Wilson NR, Kang J, Hueske EV, et al. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J. Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eastwood SL, Harrison PJ. Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr. Res. 2005;73:159–172. doi: 10.1016/j.schres.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 106.Fung SJ, Sivagnanasundaram S, Weickert CS. Lack of change in markers of presynaptic terminal abundance alongside subtle reductions in markers of presynaptic terminal plasticity in prefrontal cortex of schizophrenia patients. Biol. Psychiatry. 2011;69:71–79. doi: 10.1016/j.biopsych.2010.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oni-Orisan A, Kristiansen LV, Haroutunian V, et al. Altered vesicular glutamate transporter expression in the anterior cingulate cortex in schizophrenia. Biol. Psychiatry. 2008;63:766–775. doi: 10.1016/j.biopsych.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shan D, Lucas EK, Drummond JB, et al. Abnormal expression of glutamate transporters in temporal lobe areas in elderly patients with schizophrenia. Schizophr. Res. 2013;144:1–8. doi: 10.1016/j.schres.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uezato A, Meador-Woodruff JH, Mccullumsmith RE. Vesicular glutamate transporter mRNA expression in the medial temporal lobe in major depressive disorder, bipolar disorder, and schizophrenia. Bipolar. Disord. 2009;11:711–725. doi: 10.1111/j.1399-5618.2009.00752.x. [DOI] [PubMed] [Google Scholar]
- 110.Talbot K, Eidem WL, Tinsley CL, et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Invest. 2004;113:1353–1363. doi: 10.1172/JCI20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Amara SG, Fontana AC. Excitatory amino acid transporters: keeping up with glutamate. Neurochem. Int. 2002;41:313–318. doi: 10.1016/s0197-0186(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 112.Lauriat TL, Dracheva S, Chin B, et al. Quantitative analysis of glutamate transporter mRNA expression in prefrontal and primary visual cortex in normal and schizophrenic brain. Neuroscience. 2006;137:843–851. doi: 10.1016/j.neuroscience.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 113.Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Massie A, Vandesande F, Arckens L. Expression of the high-affinity glutamate transporter EAAT4 in mammalian cerebral cortex. Neuroreport. 2001;12:393–397. doi: 10.1097/00001756-200102120-00041. [DOI] [PubMed] [Google Scholar]
- 115.Massie A, Cnops L, Smolders I, et al. High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J. Comp Neurol. 2008;511:155–172. doi: 10.1002/cne.21823. [DOI] [PubMed] [Google Scholar]
- 116.Nieoullon A, Canolle B, Masmejean F, et al. The neuronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a major actor at the brain excitatory synapse? J. Neurochem. 2006;98:1007–1018. doi: 10.1111/j.1471-4159.2006.03978.x. [DOI] [PubMed] [Google Scholar]
- 117.Bar-Peled O, Ben-Hur H, Biegon A, et al. Distribution of glutamate transporter subtypes during human brain development. J. Neurochem. 1997;69:2571–2580. doi: 10.1046/j.1471-4159.1997.69062571.x. [DOI] [PubMed] [Google Scholar]
- 118.Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem. Int. 1998;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- 119.Bauer D, Gupta D, Harotunian V, et al. Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr. Res. 2008;104:108–120. doi: 10.1016/j.schres.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bauer D, Haroutunian V, Meador-Woodruff JH, et al. Abnormal glycosylation of EAAT1 and EAAT2 in prefrontal cortex of elderly patients with schizophrenia. Schizophr. Res. 2010;117:92–98. doi: 10.1016/j.schres.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Danbolt NC. Glutamate uptake. Prog. Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 122.Rose CF, Verkhratsky A, Parpura V. Astrocyte glutamine synthetase: pivotal in health and disease. Biochem. Soc. Trans. 2013;41:1518–1524. doi: 10.1042/BST20130237. [DOI] [PubMed] [Google Scholar]
- 123.Katsel P, Byne W, Roussos P, et al. Astrocyte and glutamate markers in the superficial, deep, and white matter layers of the anterior cingulate gyrus in schizophrenia. Neuropsychopharmacology. 2011;36:1171–1177. doi: 10.1038/npp.2010.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Steffek AE, Mccullumsmith RE, Haroutunian V, et al. Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophr. Res. 2008;103:71–82. doi: 10.1016/j.schres.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Burbaeva GS, Boksha IS, Turishcheva MS, et al. Glutamine synthetase and glutamate dehydrogenase in the prefrontal cortex of patients with schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2003;27:675–680. doi: 10.1016/s0278-5846(03)00078-2. [DOI] [PubMed] [Google Scholar]
- 126.Prabakaran S, Swatton JE, Ryan MM, et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry. 2004;9:684–697. 643. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- 127.Toro CT, Hallak JE, Dunham JS, et al. Glial fibrillary acidic protein and glutamine synthetase in subregions of prefrontal cortex in schizophrenia and mood disorder. Neurosci. Lett. 2006;404:276–281. doi: 10.1016/j.neulet.2006.05.067. [DOI] [PubMed] [Google Scholar]
- 128.Martins-De-Souza D, Schmitt A, Roder R, et al. Sex-specific proteome differences in the anterior cingulate cortex of schizophrenia. J. Psychiatr. Res. 2010;44:989–991. doi: 10.1016/j.jpsychires.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 129.Gluck MR, Thomas RG, Davis KL, et al. Implications for altered glutamate and GABA metabolism in the dorsolateral prefrontal cortex of aged schizophrenic patients. Am. J. Psychiatry. 2002;159:1165–1173. doi: 10.1176/appi.ajp.159.7.1165. [DOI] [PubMed] [Google Scholar]
- 130.Bruneau EG, Mccullumsmith RE, Haroutunian V, et al. Increased expression of glutaminase and glutamine synthetase mRNA in the thalamus in schizophrenia. Schizophr. Res. 2005;75:27–34. doi: 10.1016/j.schres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 131.Mothet JP, Parent AT, Wolosker H, et al. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U. S. A. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.De Miranda J, Panizzutti R, Foltyn VN, et al. Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-D-aspartate (NMDA) receptor coagonist D-serine. Proc. Natl. Acad. Sci. U. S. A. 2002;99:14542–14547. doi: 10.1073/pnas.222421299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. U. S. A. 1999;96:13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Horiike K, TOJO H, Arai R, et al. Localization of D-amino acid oxidase in Bergmann glial cells and astrocytes of rat cerebellum. Brain Res. Bull. 1987;19:587–596. doi: 10.1016/0361-9230(87)90076-1. [DOI] [PubMed] [Google Scholar]
- 135.Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc. Natl. Acad. Sci. U. S. A. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Labrie V, Wong AH, Roder JC. Contributions of the D-serine pathway to schizophrenia. Neuropharmacology. 2012;62:1484–1503. doi: 10.1016/j.neuropharm.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 137.Bendikov I, Nadri C, Amar S, et al. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr. Res. 2007;90:41–51. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 138.Kapoor R, Lim KS, Cheng A, et al. Preliminary evidence for a link between schizophrenia and NMDA-glycine site receptor ligand metabolic enzymes, d-amino acid oxidase (DAAO) and kynurenine aminotransferase-1 (KAT-1) Brain Res. 2006;1106:205–210. doi: 10.1016/j.brainres.2006.05.082. [DOI] [PubMed] [Google Scholar]
- 139.Steffek AE, Haroutunian V, Meador-Woodruff JH. Serine racemase protein expression in cortex and hippocampus in schizophrenia. Neuroreport. 2006;17:1181–1185. doi: 10.1097/01.wnr.0000230512.01339.72. [DOI] [PubMed] [Google Scholar]
- 140.Verrall L, Walker M, Rawlings N, et al. d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. Eur. J. Neurosci. 2007;26:1657–1669. doi: 10.1111/j.1460-9568.2007.05769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Moreno S, Nardacci R, Cimini A, et al. Immunocytochemical localization of D-amino acid oxidase in rat brain. J. Neurocytol. 1999;28:169–185. doi: 10.1023/a:1007064504007. [DOI] [PubMed] [Google Scholar]
- 142.Ono K, Shishido Y, Park HK, et al. Potential pathophysiological role of D-amino acid oxidase in schizophrenia: immunohistochemical and in situ hybridization study of the expression in human and rat brain. J. Neural Transm. 2009;116:1335–1347. doi: 10.1007/s00702-009-0289-7. [DOI] [PubMed] [Google Scholar]
- 143.Madeira C, Freitas ME, Vargas-Lopes C, et al. Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr. Res. 2008;101:76–83. doi: 10.1016/j.schres.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 144.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- 145.Schwarcz R, Rassoulpour A, Wu HQ, et al. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry. 2001;50:521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- 146.Erhardt S, Blennow K, Nordin C, et al. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci. Lett. 2001;313:96–98. doi: 10.1016/s0304-3940(01)02242-x. [DOI] [PubMed] [Google Scholar]
- 147.Wonodi I, Stine OC, Sathyasaikumar KV, et al. Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch. Gen. Psychiatry. 2011;68:665–674. doi: 10.1001/archgenpsychiatry.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer's disease Cold Spring. Harb. Perspect. Biol. 2012:4. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cohen RM, Rezai-Zadeh K, Weitz TM, et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J. Neurosci. 2013;33:6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mcconnell MJ, Lindberg MR, Brennand KJ, et al. Mosaic copy number variation in human neurons. Science. 2013;342:632–637. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lewis DA. The human brain revisited: opportunities and challenges in postmortem studies of psychiatric disorders. Neuropsychopharmacology. 2002;26:143–154. doi: 10.1016/S0893-133X(01)00393-1. [DOI] [PubMed] [Google Scholar]
- 152.Chandrakala MV, Marcus SR, Nadiger HA, et al. Acute and long-term effects of chlorpromazine on glutamine synthetase and glutaminase in rat brain. J. Neurochem. 1987;49:32–34. doi: 10.1111/j.1471-4159.1987.tb03389.x. [DOI] [PubMed] [Google Scholar]
- 153.Cook JF, Hill DF, Snively BM, et al. Cognitive impairment in rapid-onset dystonia-parkinsonism. Mov Disord. 2014;29(Suppl 3):344–350. doi: 10.1002/mds.25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pietersen CY, Lim MP, Macey L, et al. Neuronal type-specific gene expression profiling and laser-capture microdissection Methods. Mol. Biol. 2011;755:327–343. doi: 10.1007/978-1-61779-163-5_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.O'Connor JA, Hemby SE. Elevated GRIA1 mRNA expression in Layer II/III and V pyramidal cells of the DLPFC in schizophrenia. Schizophr. Res. 2007;97:277–288. doi: 10.1016/j.schres.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Macdonald ML, Ciccimaro E, Prakash A, et al. Biochemical fractionation and stable isotope dilution liquid chromatography-mass spectrometry for targeted and microdomain-specific protein quantification in human postmortem brain tissue. Mol. Cell Proteomics. 2012;11:1670–1681. doi: 10.1074/mcp.M112.021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pantelis C, Yucel M, Wood SJ, et al. Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophr. Bull. 2005;31:672–696. doi: 10.1093/schbul/sbi034. [DOI] [PubMed] [Google Scholar]
- 159.Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 160.Xie Z, Huganir RL, Penzes P. Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron. 2005;48:605–618. doi: 10.1016/j.neuron.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 161.Xie Z, Srivastava DP, Photowala H, et al. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mckinney RA, Capogna M, Durr R, et al. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat. Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- 163.Cheng HW, Rafols JA, Goshgarian HG, et al. Differential spine loss and regrowth of striatal neurons following multiple forms of deafferentation: a Golgi study. Exp. Neurol. 1997;147:287–298. doi: 10.1006/exnr.1997.6618. [DOI] [PubMed] [Google Scholar]
- 164.Sa SI, Pereira PA, Paula-Barbosa MM, et al. Role of neural afferents as mediators of estrogen effects on the hypothalamic ventromedial nucleus. Brain Res. 2010;1366:60–70. doi: 10.1016/j.brainres.2010.10.043. [DOI] [PubMed] [Google Scholar]
- 165.Devito LM, Balu DT, Kanter BR, et al. Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology Genes. Brain Behav. 2011;10:210–222. doi: 10.1111/j.1601-183X.2010.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Balu DT, Basu AC, Corradi JP, et al. The NMDA receptor co-agonists, D-serine and glycine, regulate neuronal dendritic architecture in the somatosensory cortex. Neurobiol. Dis. 2012;45:671–682. doi: 10.1016/j.nbd.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liu BH, Wu GK, Arbuckle R, et al. Defining cortical frequency tuning with recurrent excitatory circuitry. Nat. Neurosci. 2007;10:1594–1600. doi: 10.1038/nn2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kaur S, Rose HJ, Lazar R, et al. Spectral integration in primary auditory cortex: laminar processing of afferent input, in vivo and in vitro. Neuroscience. 2005;134:1033–1045. doi: 10.1016/j.neuroscience.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 169.Javitt DC, Shelley A, Ritter W. Associated deficits in mismatch negativity generation and tone matching in schizophrenia. Clin. Neurophysiol. 2000;111:1733–1737. doi: 10.1016/s1388-2457(00)00377-1. [DOI] [PubMed] [Google Scholar]
- 170.Strous RD, Cowan N, Ritter W, et al. Auditory sensory (“echoic”) memory dysfunction in schizophrenia. Am. J. Psychiatry. 1995;152:1517–1519. doi: 10.1176/ajp.152.10.1517. [DOI] [PubMed] [Google Scholar]
- 171.Javitt DC, Doneshka P, Grochowski S, et al. Impaired mismatch negativity generation reflects widespread dysfunction of working memory in schizophrenia. Arch. Gen. Psychiatry. 1995;52:550–558. doi: 10.1001/archpsyc.1995.03950190032005. [DOI] [PubMed] [Google Scholar]
- 172.Javitt DC, Strous RD, Grochowski S, et al. Impaired precision, but normal retention, of auditory sensory (“echoic”) memory information in schizophrenia. J. Abnorm. Psychol. 1997;106:315–324. doi: 10.1037//0021-843x.106.2.315. [DOI] [PubMed] [Google Scholar]
- 173.Javitt DC, Steinschneider M, Schroeder CE, et al. Detection of stimulus deviance within primate primary auditory cortex: intracortical mechanisms of mismatch negativity (MMN) generation. Brain Res. 1994;667:192–200. doi: 10.1016/0006-8993(94)91496-6. [DOI] [PubMed] [Google Scholar]
- 174.Ehrlichman RS, Maxwell CR, Majumdar S, et al. Deviance-elicited changes in event-related potentials are attenuated by ketamine in mice. J. Cogn Neurosci. 2008;20:1403–1414. doi: 10.1162/jocn.2008.20097. [DOI] [PubMed] [Google Scholar]
- 175.Umbricht D, Schmid L, Koller R, et al. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch. Gen. Psychiatry. 2000;57:1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- 176.Landen M, Davidsson P, Gottfries CG, et al. Reduction of the synaptophysin level but normal levels of glycerophospholipids in the gyrus cinguli in schizophrenia. Schizophr. Res. 2002;55:83–88. doi: 10.1016/s0920-9964(01)00197-9. [DOI] [PubMed] [Google Scholar]
- 177.Breese CR, Freedman R, Leonard SS. Glutamate receptor subtype expression in human postmortem brain tissue from schizophrenics and alcohol abusers. Brain Res. 1995;674:82–90. doi: 10.1016/0006-8993(94)01384-t. [DOI] [PubMed] [Google Scholar]
- 178.Eastwood SL, Kerwin RW, Harrison PJ. Immunoautoradiographic evidence for a loss of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate-preferring non-N-methyl-D-aspartate glutamate receptors within the medial temporal lobe in schizophrenia. Biol. Psychiatry. 1997;41:636–643. doi: 10.1016/S0006-3223(96)00220-X. [DOI] [PubMed] [Google Scholar]
- 179.Benes FM, Todtenkopf MS, Kostoulakos P. GluR5,6,7 subunit immunoreactivity on apical pyramidal cell dendrites in hippocampus of schizophrenics and manic depressives. Hippocampus. 2001;11:482–491. doi: 10.1002/hipo.1065. [DOI] [PubMed] [Google Scholar]
- 180.Clinton SM, Haroutunian V, Meador-Woodruff JH. Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J. Neurochem. 2006;98:1114–1125. doi: 10.1111/j.1471-4159.2006.03954.x. [DOI] [PubMed] [Google Scholar]
- 181.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]