

Abstract
Syntheses were undertaken of derivatives of (2S, 4R)-(−)-trans-4-phenyl-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (4-phenyl-2-dimethylaminotetralin, PAT), a stereospecific agonist at the serotonin 5-HT2C G protein-coupled receptor (GPCR), with inverse agonist activity at 5-HT2A and 5-HT2B GPCRs. Molecular changes were made at the PAT C(4)-position, while preserving N, N-dimethyl substitution at the 2-position as well as trans-stereochemistry, structural features previously shown to be optimal for 5-HT2 binding. Affinities of analogs were determined at recombinant human 5-HT2 GPCRs in comparison to the phylogenetically closely-related histamine H1 GPCR, and in silico ligand docking studies were conducted at receptor molecular models to help interpret pharmacological results and guide future ligand design. In most cases, C(4)-substituted PAT analogs exhibited the same stereoselectivity ([−]-trans > [+]-trans) as the parent PAT across 5-HT2 and H1 GPCRs, albeit, with variable receptor selectivity. 4-(4′-substituted)-PAT analogs, however, demonstrated reversed stereoselectivity ([2S, 4R]-[+]-trans > [2S, 4R]-[−]-trans), with absolute configuration confirmed by single X-ray crystallographic data for the 4-(4′-Cl)-PAT analog. Pharmacological affinity results and computational results herein support further PAT drug development studies and provide a basis for predicting and interpreting translational results, including, for (+)-trans-4-(4′-Cl)-PAT and (−)-trans-4-(3′-Br)-PAT that were previously shown to be more potent and efficacious than their corresponding enantiomers in rodent models of psychoses, psychostimulant-induced behaviors, and compulsive feeding (‘binge-eating’).
Keywords: 5-HT2, Aminotetralin, Histamine H1, Molecular modeling, Serotonin receptors, X-ray crystal structure
1. Introduction
Serotonin (5-hydroxytryptamine, 5-HT) mediates its central and peripheral psychological and physiological effects through receptor subtypes grouped into the 5-HT1 – 5-HT7 families and the serotonin neurotransporter. The 5-HT2 receptor family consists of the 5-HT2A, 5-HT2B, and 5-HT2C G protein-coupled receptors (GPCRs) that signal primarily through Gαq to activate phospholipase C (PLC) and formation of inositol phosphates (IP) and diacylglycerol second messengers.1 Highly conserved transmembrane domains (~75% homology across 5-HT2 receptors) and same second messenger signaling complicate development of 5-HT2 subtype-specific ligands. Thus, despite the long-known pharmacotherapeutic potential of antagonism at 5-HT2A receptors for neuropsychiatric disorders,1,2 there are currently no approved 5-HT2-selective antagonist or inverse agonist drugs. Likewise, although activation of the 5-HT2C receptor is pharmacotherapeutic for obesity, the only approved 5-HT2C agonist drug, lorcaserin, also activates 5-HT2A and 5-HT2B receptors.3 Unfortunately, activation of 5-HT2A receptors can cause hallucinations and other psychotomimetic effects4,5 and activation of 5-HT2B receptors can lead to cardiac valvulopathy.6,7 The 5-HT2 GPCRs also share considerable transmembrane homology (~25% identical) with histamine H1 GPCRs that also signal through PLC/IP. It has been proposed that weight-gain associated with most antipsychotic drugs is due to antagonism of H1 receptors and/or antagonism of 5HT2C receptors, albeit, H1 receptor-selective antagonists are a highly successful class of anti-allergy drugs without weight-gain issues. 8–11
Recently, we reported that (2S, 4R)-(−)-trans-4-phenyl-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (4-phenyl-2-dimethylaminotetralin, PAT, Table 1)12 is a novel multifunctional 5-HT2 ligand that specifically activates 5-HT2C receptors while also having inverse agonist (and competitive antagonist) activity at 5-HT2A and 5-HT2B receptors.12 (−)-Trans-PAT functional pharmacology at 5-HT2 receptors is unique and desirable regarding translation for antipsychotic and other psychotherapeutic activities, without liability for weight gain or cardiotoxicity.13 Indeed, (−)-trans-PAT demonstrates therapeutic efficacy in rodent models of psychoses14 and in a rodent compulsive behavioral paradigm that models ‘binge-eating’.15 We determined previously that (−)-trans-PAT also is a functionally-selective histamine H1 ligand that activates H1 receptor-mediated adenylyl cyclase signaling but is an antagonist at H1-linked PLC signaling.11 It is not clear what role histamine H1 GPCRs may play in the psychotherapeutic effects of (−)-trans-PAT, however, H1 and 5-HT2 molecular modeling studies16–20 suggest it is possible to reduce or eliminate H1 affinity in PAT-type molecules, without compromising 5-HT2 activity. For example, it was observed that especially the PAT C(4)-phenyl moiety docked differently at 5-HT2A, 5-HT2B, 5-HT2C, and H1 GPCRs16–20. Accordingly, here we report the synthesis of a series of novel PAT-type analogs with molecular changes at the C(4)-position, while preserving the N, N-dimethyl substitution at the 2-position, as well as, trans-stereochemistry, structural features previously shown to be optimal for binding to 5-HT2 as well as H1 GPCRs.16,18,20,21 Pharmacological affinity of the analogs at human recombinant 5-HT2 receptor subtypes and H1 receptors also are reported, as well as, molecular docking studies of selected analogs at a homology model of the 5-HT2C receptor to help explain structure–affinity results. These studies were undertaken to facilitate design and development of drugs to treat the cognitive dysfunction common to a variety of neuropsychiatric disorders, including, psychoses, depression, dementia, as well as, substance abuse, attention-deficit hyperactivity, and other compulsive behavioral disorders.
Table 1.
Binding affinities of 4-substituted 2-dimethylaminotetralins at 5HT2A, 5HT2B and 5HT2C and H1 receptors
| Analog number | R | trans. | Affinity (Ki ± SEM; nM)
|
Ratio of affinities | ||||
|---|---|---|---|---|---|---|---|---|
| 5-HT2A | 5-HT2B | 5-HT2C | H1 | 2A/2C | H1/2C | |||
| “PAT” | C6H5 | (+) | 470(46) | 240(41) | 430(86) | 30(4) | 1.09 | 0.07 |
| (−) | 100(10) | 94(25) | 30(3) | 2(1) | 3.33 | 0.07 | ||
| 5a | 3′-F-C6H4 | (+) | 320(26) | 110(28) | 200(30) | 94(15) | 1.60 | 0.47 |
| (−) | 110(26) | 70(5) | 43(14) | 3(0.3) | 2.56 | 0.07 | ||
| 5b | 3′-Cl-C6H4 | (+) | 130(8.7) | 50(7) | 170(43) | 80(20) | 0.76 | 0.47 |
| (−) | 40(5) | 30(2) | 8(3) | 10(2) | 5.00 | 1.25 | ||
| 5c | 3′-Br-C6H4 | (+) | 260(22) | 40(8) | 200(30) | 60(10) | 1.30 | 0.30 |
| (−) | 20(3) | 8(2) | 4(1) | 30(2) | 5.00 | 7.50 | ||
| 5d | 3′-CF3-C6H4 | (+) | 1300(67) | 200(50) | 230(20) | 4100(160) | 5.65 | 17.83 |
| (−) | 80(10) | 60(20) | 10(2) | 230(27) | 8.00 | 23.00 | ||
| 5e | 3′-NO2-C6H4 | (+) | 500(20) | 20(6) | 120(13) | 1000(80) | 4.17 | 8.33 |
| (−) | 74(18) | 70(20) | 10(1) | 800(100) | 7.40 | 80.00 | ||
| 5f | 4′-F-C6H4 | (+) | 71(12) | 66(12) | 75(12) | 8(1) | 0.95 | 0.11 |
| (−) | 140(24) | 400(80) | 68(11) | 4(1) | 2.06 | 0.06 | ||
| 5g | 4′-Cl-C6H4 | (+) | 50(8) | 80(10) | 55(17) | 6(1) | 0.91 | 0.11 |
| (−) | 250(37) | 300(30) | 120(9.0) | 8(1) | 2.08 | 0.07 | ||
| 5h | 4′-Br-C6H4 | (+) | 60(10) | 30(9) | 70(8) | 530(62) | 0.86 | 7.57 |
| (−) | 220(29) | 210(15) | 100(3) | 1100(53) | 2.20 | 11.00 | ||
| 5i | 4′-CF3-C6H4 | (+) | 210(16) | 250(41) | 220(27) | 230(35) | 0.95 | 1.05 |
| (−) | 2000(200) | 540 (120) | 520(51) | 5100(360) | 3.85 | 9.81 | ||
| 5ja | 4′-NO2-C6H4 | racemic | 8100(180) | 4100 (510) | 1000 (100) | 680(31) | 8.10 | 0.68 |
| 5k | 2′-Br-C6H4 | (+) | 760(73) | 4100(510) | 78(16) | 2600(340) | 9.74 | 33.33 |
| (−) | 1100(130) | 650(72) | 70(8) | 4200(460) | 15.71 | 60.00 | ||
| 5l | cyclohexyl | (+) | 80(4) | 170(13) | 30(4) | 2(0.2) | 2.67 | 0.07 |
| (−) | 20(5) | 20(1) | 7(2) | 2(1) | 2.86 | 0.29 | ||
| 5m | cyclooctyl | (+) | 370(20) | 40(20) | 800(100) | >10000 | 0.46 | NA |
| (−) | 30(9) | 10(4) | 30(8) | 430(62) | 1.00 | 14.33 | ||
| 5n | 3′-Ph-C6H4 | (+) | 650(62) | 200(40) | 1000(10) | >10000 | 0.65 | NA |
| (−) | 44(17) | 73(13) | 5(2) | 200(20) | 8.80 | 40.00 | ||
| 5ob | 2′-Cl-C6H4 | (+) | 2800(110) | 710(64) | 50(10) | 60(20) | 56.00 | 1.20 |
| (−) | 2400(280) | 300(30) | 290(47) | 190(25) | 8.28 | 0.66 | ||
| 5pb | 2′-Me-C6H4 | (+) | 700(90) | 6000 (1000) | 2000 (200) | 120(5.1) | 0.35 | 0.06 |
| (−) | 1000(200) | 800 (100) | 1600 (150) | 400(40) | 0.63 | 0.25 | ||
NA – Not applicable.
Racemic mixture could not be resolved using chiral HPLC system;
Racemic mixture prepared by published procedure21 and resolved using chiral HPLC system.
2. Chemistry
Scheme 1 outlines the syntheses of racemic trans-4-substituted-2-dimethylaminotetralins (5a-m). The 4-(2′-[Br,])- and 4-(3′- and 4′-[Cl, Br, F, NO2, CF3])-phenyltetralones (2′a-b) or tetralen-2-ol phenylacetates (2c-k), and 4-cyclohexyl and 4-cyclooctyltetralen-2-ol phenylacetates (2l-m) were synthesized from their corresponding styrenes (1a-k) or vinyl cycloalkanes (1l-m) and trifluoroacetyl phenylacetyl anhydride, via cascade Friedel–Crafts cycli-acylalkylation, enolization, and O-acylation, following our published procedures.22 The tetralones (2′a-b) and tetralen-2-ol-phenylacetates (2c-m) were reduced with sodium borohydride to yield major cis-tetralols (3a-m). In most cases, cis-tetral-2-ol was obtained as the only product, as deduced from 1H NMR spectra and TLC, thus, the reaction could be considered as stereospecific. Assignment of cis and trans geometry was based on comparison of 1H NMR with data earlier reported22 for cis-3′-bromophenyltetralen-2-ol and the mixture of cis- and trans-3′-bromophenyltetralen-2-ol. Specifically, for the trans-isomer, the C(4) proton spectrum appeared as a triplet due to greater de-shielding in comparison to the cis-isomer, wherein, the C(4) proton spectrum appeared as a double doublet. In the case of a mixture of cis:trans isomers (90:10, determined from 1H NMR), repeated column chromatography was unsuccessful to achieve separation, thus, the diastereomeric mixture was used for subsequent steps. The tetral-2-ols (3a-m) were converted to the corresponding cis-tosylates (4a-m) in pyridine at room temperature. The cis-tosylates yielded the trans-4-substituted 2-dimethylaminotetralins (5a-m) upon reaction with aqueous dimethylamine solution in a sealed tube at 80 °C for 24 h. The 1H NMR spectra of the crude dimethylaminotetralins showed the presence of trans:cis isomers (90:10) that could not be separated by column chromatography. Success to separate the diastereomeric mixture was realized using a chiral stationary-phase (polysaccharide-based) preparative HPLC system and a combination of solvents and modifier unique for each analog (detailed in the Experimental Section) to obtain the trans-enantiomers; spectral analyses confirmed isolation of the (+)- and (−)-trans-2-dimethylaminotetralins. Previously, (−)-trans-PAT was determined by X-ray crystallography to have the (2S, 4R) absolute configuration,22,23 and new analogs here were assigned absolute stereochemistry based on chiral HPLC elution time relative to (−)-trans-PAT, as well as, to the (−)-trans-4-(4′-chlorophenyl) analog that was determined by X-ray crystallography here to also have the (2S, 4R) absolute configuration (see below). Optical rotation of all analogs was determined using a Perkin Elmer 343 series polarimeter, as reported in the Experimental Section.
Scheme 1.

Synthesis of racemic trans 4-substituted N,N dimethylaminotetralins (5a-m); Reagents and conditions: (a) Phenyl acetic acid (3 eq.), TFAA (3 eq.) 0 °C, rt for 24 h (0.5 h in case of 2′a-b); (b) NaBH4/MeOH, 50 °C,15 h; (c) TsCl/pyridine, rt, 20 h; (d) aq. NHMe2, sealed tube, 80 °C, 24 h.
Scheme 2 outlines the synthesis of racemic trans-4-(biphenyl-3-yl)-N,N-dimethyl-2-aminotetralin (5n), beginning with 3′-bromo-tetralen-2-ol-phenylacetate (2c), synthesized above, that was converted to the 3′-phenyl-tetralen-2-ol-phenylacetate (2n) using the Suzuki coupling reaction.22 Reduction of 2n with sodium borohydride at 55 °C for 15 h yielded the major cis-4-(biphenyl-3-yl)tetral-2-ol (3n), that was subsequently tosylated and treated with dimethylamine to yield 5n.
Scheme 2.

S ynthesis of racemic trans 4-(biphenyl-3-yl) N,N-dimethylaminotetralin (5n); Reagents and conditions: (a) PhB(OH)2, Pd(OAc)2, (c-Hex)2P(biphen-3-yl), K3PO4/toluene, 65 °C, 24 h; (b) NaBH4/MeOH, 50 °C,15 h; (c) TsCl/pyridine, rt, 20 h; (d) aq. NHMe2, sealed tube, 80 °C, 24 h.
Racemic trans-2′-Cl- and trans-2′-Me-phenyl-2-dimethylaminotetralins (5o,p) were prepared by our earlier procedure starting from the corresponding 1,4-diphenyl-l-buten-3-one.19–21,23
3. Pharmacological Characterization
Pharmacological experiments focused on delineating the impact of PAT analog stereochemistry and C(4)-substitution on affinity at serotonin 5-HT2A, 5-HT2B, 5-HT2C, and histamine H1 GPCRs. Results are summarized in Table 1. Consistent with previous reports,12,20,24 the parent analog, PAT, binds stereoselectively at 5-HT2 receptor subtypes and at H1 receptors, with about 3–15-fold higher affinity apparent for the (−)- over (+)-enantiomer, depending on receptor type. The (−)-trans enantiomers of the 4-(3′-[F, Cl, Br, CF3, NO2]-phenyl) analogs (5a-5e) also had higher affinity than the corresponding (+)-enantiomers at all three 5-HT2 subtypes and at H1 receptors. The 5-HT2 receptor affinity of 5c and 5g reported here is consistent with our previously published results.16,25,26 Compared to the parent trans-PAT, addition of the 3′-fluoro substituent (5a) did not result in a significant (p > 0.05) change in affinity for either the (+)- or (−)-enantiomer at 5-HT2 and H1 receptors. In general, however, the (+)- and (−)-enantiomers of trans-4-(3′-Cl-, Br-, NO2-, and CF3)-PAT analogs (5b-e) had increased affinity selectively at 5-HT2 receptors over H1 receptors, compared to the parent PAT analog. Likewise, both enantiomers of the 4-(3′-phenyl)-PAT analog (5n) had higher affinity at 5-HT2 over H1 receptors.
Analogs with substituents at the 4′-(para) position of the PAT C(4) phenyl moiety demonstrated stereoselective binding at 5-HT2 and H1 receptors that was essentially opposite of the parent PAT and 4-(3′-substituted-phenyl) analogs above, i.e., the (+)-enantiomers of trans-4-(4′-[F, Cl, Br, CF3]-phenyl) analogs (5f-5j) had about the same (p > 0.05) or significantly higher (p < 0.05) affinity than the corresponding (−)-enantiomers. Thus, the presence of the C(4) 4′-substitutent impacted stereochemical preference for binding such that higher affinity across receptors resulted when the 2-dimethylamine moiety was in the R-configuration, based on the X-ray crystal structure and chiral HPLC elution characteristics determined for the (−)-trans-4-(4′-chlorophenyl) analog (4′-Cl-PAT) (5g), as described below. These results are in contrast to previously reported results19,20 for analogs without a 4′-substitutent, wherein, higher affinity at H1 receptors is obtained when the dimethylamine moiety is in the S-configuration. The (+) and (−)-trans-4′-nitro-PAT analogs (5j) could not be resolved from the racemic mixture using the chiral HPLC system despite numerous attempts with different mobile phase conditions, hence, the affinity value given in Table 1 is for the racemate, that had relatively poor affinity across receptors.
The 4-(2′-[Br and Cl,]-phenyl) analogs (5k, 5o and 5p) displayed relatively poor affinity and stereoselectivity at 5-HT2 and H1 receptors. These results suggest there exist negative steric interactions between the 2′-position of the C(4) phenyl moiety and amino acids in the 5-HT2 and H1 receptor binding pockets. For example, modeling results suggest critical π–π interactions between the ligand C(4)-phenyl moiety and W6.48 and F6.51 residues of the 5-HT2 and H1 receptor binding pockets may be negatively impacted by a 2′-substituent (references 16–20 and Figure 2). In contrast, the robust affinity of the 4-cycloheptyl enantiomers (5l) across receptors and the high affinity of the (−)-trans-(4-cyclooctyl analog (5m) at 5-HT2 receptors confirms results from molecular modeling studies here (Figure 2) and elsewhere that suggest there exists a relatively large orthosteric binding pocket for especially the 5-HT2 subtypes.16,17,20,21
Figure 2.
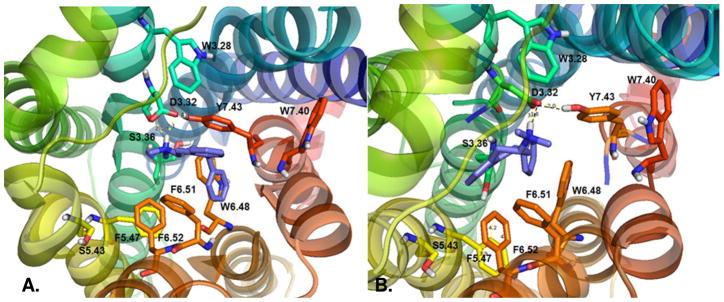
View from the extracellular domain of (2S, 4R)-(−)-trans-PAT (panel A) and (2R, 4S)-(+)-trans-PAT (panel B) docked at 5-HT2C receptor model.
Recently, the 4′-Cl-PAT analogs (5g) were reported to demonstrate 5-HT2C agonism together with 5-HT2A/2B antagonism/inverse agonism that translated to therapeutic efficacy in three different models of psychoses, as well as, efficacy to attenuate psychostimulant-induced behaviors and negatively modulate food consumption in a compulsive behavioral paradigm that models binge-eating, after oral or intraperitoneal administration in mice.25 Importantly, confirming accurate translation of the in vitro molecular pharmacology results, the (+)-trans-enantiomer of 4′-Cl-PAT (5g) had higher potency and efficacy in vivo when compared to the (−)-trans-enantiomer. The preclinical therapeutic importance of the 4′-Cl-PAT analogs justified additional studies here to characterize the molecular determinants for their stereoselective binding at 5-HT2 receptors, that was reversed compared to analogs without a 4′-phenyl substituent in Table 1.
X-ray crystallography studies were undertaken with the (−)-trans-4′-Cl-PAT analog (5g) (Figure 1) and results confirmed the absolute stereochemistry as (2S, 4R), consistent with the absolute configuration of the parent analog (−)-trans-PAT.23 In addition, molecular modeling experiments were undertaken to analyze and compare the apparently different stereochemical binding interactions of the parent PAT enantiomers in comparison to the 4′-Cl-PAT enantiomers at 5-HT2C receptors, a clinically-useful drug target for obesity.3
Figure 1.
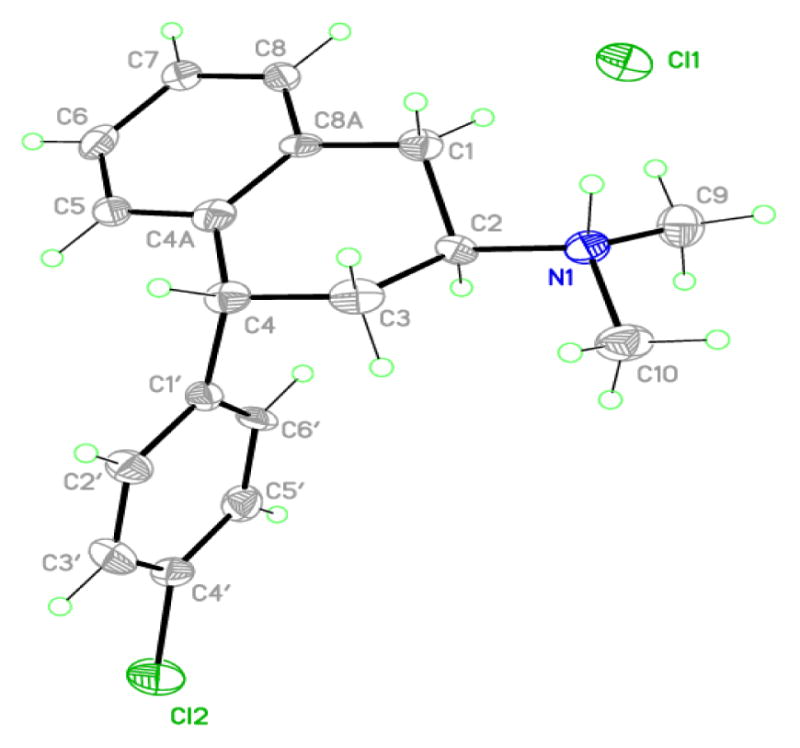
Single X-ray crystallographic structure of (2S, 4R)-(−)-trans-4′-Cl-PAT hydrochloride
4. In silico ligand docking studies
Ligand docking and molecular dynamics (MD) experiments were performed for the (2R, 4S)-(+)- and (2S, 4R)-(−)-trans-enantiomers of PAT and 4′-Cl-PAT at a molecular model of the human 5-HT2C receptor, built by homology to the β2 adrenergic GPCR (β2AR/T4-lysozyme chimera, PDB code 2rh1).30 Results indicated that the protonated amine group of (−)-trans-PAT could form a bifurcated hydrogen bond (distance 1.77–2.22 Å) with the aspartate D3.32 carboxylate oxygen atoms and a hydrogen bond (distance 1.58 Å) with the serine S3.36 hydroxyl group (Figure 2, panel A). The aromatic ring of the PAT tetrahydronaphthalene system docked nearly parallel with the aromatic ring of tyrosine Y7.43 (distance 3.7 Å), thereby able to form π-π interactions. The PAT C(4) phenyl group docked in the aromatic pocket formed by tryptophan W6.48 and phenylalanine residues F6.51 and F6.52.
The protonated amine moiety of (+)-trans-PAT could form a hydrogen bond with the D3.32 carboxylate moiety (Figure 2, panel B), but, the distance was longer (2.45 Å) in comparison to docking of (−)-trans-PAT. Also different for the (+)-trans-enantiomer is that the tetrahydronaphthalene aromatic ring cannot interact with the aromatic ring of Y7.43, and the C(4) phenyl substituent is far (6.6–7 Å) from the residues in the aromatic pocket formed by W6.48, F6.51 and F6.52. Also, interactions between 5-HT2C receptor amino acids were different after docking of the PAT enantiomers—specifically, the distance for interaction between D3.32 and Y7.43 was closer after docking of (−)-trans-PAT (1.92 Å) as compared to (+)-trans-PAT (2.0 Å).
Figure 3 shows the (−)-trans- and (+)-trans-enantiomers of 4′-Cl-PAT (5g) docked at the 5-HT2C receptor model. For both enantiomers, the presence of the 4′-Cl substituent modified positioning of the ligand in the binding pocket compared to the PAT enantiomers, particularly, for (−)-trans-4′-Cl-PAT, that docked close to transmembrane helices VI and VII (panel A), whereas, (+)-trans-4′-Cl-PAT was positioned closer to transmembrane helix V (panel B). The protonated amine moiety of (−) and (+)-trans-4′-Cl-PAT could form a hydrogen bond with the D3.32 carboxylate moiety. Only for the (+)-trans enantiomer, however, was the 4′-Cl moiety able to interact with the S5.43 hydroxyl group in transmembrane helix V, presumably, explaining the higher affinity of (+)-trans-4′-Cl-PAT in comparison to (−)-trans-4′-Cl-PAT at 5-HT2C receptors.
Figure 3.
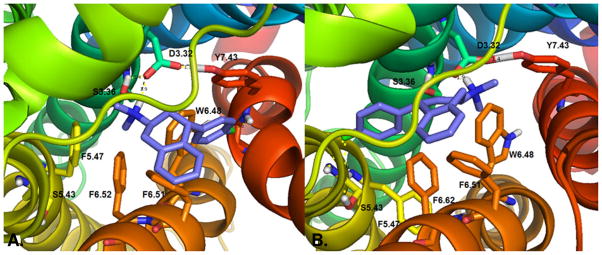
View from the extracellular domain of (2S, 4R)-(−)-trans-4′-Cl-PAT (panel A) and (2R, 4S)-(+)-trans-4′-Cl-PAT (panel B) docked at the 5-HT2C receptor model.
5. Discussion
The novel PAT-type analogs and receptor affinity results reported here helped to develop a structure–affinity relationship for these and related compounds at 5-HT2 receptor subtypes and H1 receptors, as well as, characterize differences in the ligand interactions at the orthosteric binding pockets of closely-related aminergic neurotransmitter GPCRs that may be exploited for drug development purposes. Despite the high homology (~75%) between 5-HT2 receptor subtypes and with H1 receptors (~25%), and same predominant PLC/IP signaling pathway shared by all the receptors, preferential binding of certain analogs at specific receptors was observed and information about binding pocket structure can be gleaned from the results to inform drug design. For example, the orthosteric binding pocket of 5-HT2 as well as H1 receptors apparently is large enough to accommodate the C(4)-cyclohexyl substituent of 5l with high affinity binding interactions (especially for the [−]-trans-enantiomer). Molecular interactions between 5l and the 5-HT2C (in comparison to 5-HT2A) receptor previously have been analyzed in computational studies35 and those results are consistent with the high-affinity binding results (for the [−]-enantiomer) obtained experimentally, as reported in Table 1. While the (−)- and especially (+)-trans enantiomers of the C(4)-cyclooctyl analog 5m had relatively low affinity at H1 receptors, both enantiomers and especially the (−)-enantiomer had relatively high affinity at 5-HT2 receptors, similar to the cyclohexyl analog (−)-trans-5l. Thus, even a relatively simple change in the analog steric parameter resulted in at least 10-fold selectivity for 5-HT2 over H1 receptors. Molecular modeling studies here and elsewhere corroborate structure–affinity relationship results in Table 1 that suggest the orthosteric binding pocket of 5-HT2 receptors is larger compared to H1 receptors.16–18,20,21 Likewise, both enantiomers of the 4-(3′-Cl-, Br-, NO2-, CF3, and phenyl)-PAT analogs (5b-e and 5n) had higher affinity at 5-HT2 receptors over H1 receptors, making them more selective for 5-HT2 receptors than the parent PAT analog. For compounds in Table 1, in general, analogs with substituents in the 3′ (meta) position of the parent PAT C(4) phenyl moiety had highest affinity and stereoselectivity associated with the (2S, 4R)-(−)-trans-enantiomer, favoring binding at 5-HT2 receptor subtypes over H1 receptors. Thus, it appears there are differences in binding pocket structures that can be exploited to achieve selective high affinity binding of trans-PAT-type analogs at 5-HT2 subtypes over H1 receptors.
Substitutions at the 4′-(para) position of the PAT C(4) phenyl ring resulted in a reversed stereoselectivity (compared to other analogs in Table 1) regarding high affinity binding at all three 5-HT2 receptor subtypes and at H1 receptors. X-ray crystallography results confirmed the absolute stereochemistry of trans-4′-Cl-PAT as (2S, 4R) for the (−)-enantiomer, thus, an accurate ligand stereochemical conformation was established for use in the computational docking studies at the 5-HT2C molecular model. For both 4′-Cl-PAT enantiomers, the presence of the 4′-Cl substituent modified positioning of the ligand relative to the parent PAT analog in the binding pocket, particularly, with regard to (−)-trans-4′-Cl-PAT, that docked close to transmembrane helices VI and VII. In contrast, (+)-trans-4′-Cl-PAT docked closer to transmembrane helix V, where the 4′-Cl moiety interacted with the 5-HT2C S5.43 hydroxyl group, an interaction not observed for the (−)-enantiomer, which, likely explains, in part, the higher affinity of (+)-trans-4′-Cl-PAT in comparison to (−)-trans-4′-Cl-PAT at 5-HT2C receptors.
For the 4′-Cl-PAT analogs (5g), stereoselective high affinity binding that favored the (+)-trans-enantiomer across receptors translated in functional studies reported elsewhere,25 e.g., (+)-trans-4′-Cl-PAT had higher potency and efficacy than (−)-trans-4′-Cl-PAT regarding agonist activity at 5-HT2C receptors. Importantly, too, the in vitro molecular pharmacology of (+)-trans-4′-Cl-PAT translated in vivo to higher potency and efficacy compared to the (−)-enantiomer after oral and intraperitoneal administration in three different mouse models of psychoses, as well as, efficacy to attenuate psychostimulant-induced behaviors and negatively modulate food consumption in a compulsive behavioral paradigm that models binge-eating.25 The apparently successful translation of the 5-HT2/H1 in vitro pharmacology of the 4′-Cl-PAT analogs using in vivo neurobehavioral paradigms is further corroborated by translational results reported elsewhere14,15,27 for the parent PAT analog, i.e., the (−)-trans-enantiomer that demonstrates higher potency and efficacy with regard to affinity and function in vitro also is the more active enantiomer regarding neurochemical and neurobehavioral activity in vivo (neurochemical and neurobehavioral activity).11,12
In addition to the parent and 4′-Cl-PAT analogs, results of translational studies for the 3′-Br-PAT analogs (5c) recently were reported.26 (−)-Trans-3′-Br-PAT was determined to be a novel potent and efficacious human 5-HT2C receptor agonist that is a competitive antagonist and inverse agonist at human 5-HT2A and 5-HT2B receptors, respectively. Moreover, in three different mouse models of drug-induced psychoses, (−)-trans-3′-Br-PAT had efficacy comparable to the prototypical “second-generation” antipsychotic drug, clozapine, after intraperitoneal administration. Unlike clozapine which suppressed locomotor activity, (−)-trans-3′-Br-PAT did not alter locomotor activity when administered alone. (−)-Trans-3′-Br-PAT also decreased consumption of a highly palatable food in a mouse model of compulsive “binge” eating. (+)-Trans-3′-Br-PAT was less active across in vitro and in vivo pharmacological assays. The current structure-affinity results will help guide design of analogs with improved potency especially at 5-HT2A/2C receptors, as it appears that PAT-type 5-HT2C-specific agonists with 5-HT2A inverse agonist/antagonist activity, such as (−)-trans-PAT, (+)-trans-4′-Cl-PAT, and (−)-trans-3′-Br-PAT, are promising novel pharmacotherapeutics for psychoses, without liability for weight gain, hallucinations, cardiac valvulopathy, sedation, or bradykinesia.
6. Conclusion
Brain 5-HT2 GPCRs are pragmatic drug development targets. For example, strategies involving inactivation of 5-HT2A signaling and/or activation of 5-HT2C signaling are proposed for a variety of neuropsychiatric disorders. Moreover, activation of 5-HT2C receptors is clinically effective for obesity, albeit, the only approved 5-HT2C agonist (lorcaserin) also activates 5-HT2A and 5-HT2B signaling that is associated with untoward psychiatric and cardiac side effects, respectively, and should obviously be avoided. Activation of histamine H1 GPCRs, closely related to the 5-HT2 family, also is untenable due to their association with the immunological response. The current work suggests that drug candidates targeting specific 5-HT2 receptor subtypes can be designed using homology-based molecular modeling protocols where the target structure is unknown (i.e., 5-HT2A and 5-HT2C), as well as, structure-based (i.e., for H1) molecular modeling. In addition to corroborating structure–affinity relationship data, for example, regarding results here that suggest the orthosteric binding pocket of 5-HT2 receptors is larger compared to H1 receptors, molecular modeling results helped to explain unexpected affinity results regarding the impact of substitution to the PAT molecular scaffold on stereochemical preference for binding. To expand on structure–affinity information learned here, the synthetic methodology involving trifluoroacetyl phenylacetyl anhydride and Friedel–Crafts reaction22 used in Scheme 2 appears to be an efficient strategy to provide a wide variety of PAT-type analogs.
7. Experimental
7.1. Chemistry
All starting materials, reagents and solvents were purchased as the highest-grade available and used without further purification. Reactions sensitive to moisture and/or oxygen were carried out under an inert atmosphere of anhydrous nitrogen. Analytical thin-layer chromatography (TLC) was performed on Merck pre-coated 60 F254 plates, and spots were visualized with UV light (254 nm), I2, 10% phosphomolybdic acid solution in ethanol. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck). 1H and 13C NMR were collected at a Varian instrument at resonance frequencies 400 and 100 MHz correspondingly, in CDCl3. The chemical shifts are reported in parts per million (ppm) using trimethylsilane (TMS) as the standard, with multiplicity defined as follows: s = singlet, d = doublet, t = triplet, q = quartet, bs = broad singlet, m = multiplet. Coupling constants (J) are expressed in hertz (Hz). Optical rotations were determined using a Perkin Elmer 343 series polarimeter. High resolution mass spectrometry (HRMS) was performed with an Agilent 6210 instrument using time-of-flight (TOF-MS) with electro spray ionization (ESI). Purity of targeted compounds was ≥95% (determined by HPLC) unless otherwise noted. Elemental analyses were performed on a Carlo Erba EA-1108 instrument. Melting points were determined on a Mel-Temp apparatus equipped with a mercury thermometer and are uncorrected. HPLC retention times of (+) and (−)-trans-enantiomers were determined by UV Trace 220/254 nm on an Shimadzu™ instrument equipped with a semi-preparative (s-prep)-RegisCell™ (5 μm, 25 cm × 10 mm i.d.) or preparative (prep)-RegisCell™ (5 μm, 25 cm × 30 mm i.d.) chiral (polysaccharide-based) column.
7.1.1. General procedure for preparation of tetralones (2′a-b) and enol-esters (2c-m)
Phenylacetic acid (3 eq.) was dissolved in TFAA (3 eq.), ≤5 min, at rt under N2 atm to generate the mixed anhydride, that was transferred to 1a–m (1 eq.) using a double ended needle, with stirring at 0 °C. The reaction mixture was stirred at r.t. for 24 h and then quenched with deionized water and extracted with ethyl acetate (3x). Organic layers were combined, washed with saturated aqueous sodium bicarbonate (4x), dried over anhydrous sodium sulfate, filtered, and concentrated. Crude products were purified by flash chromatography (Si-gel, hexanes:toluene 3:2) to yield the tetralones (2′a-b) or enol-esters (2c-m) in pure form.
7.1.1.1. 4-(3′-(Trifluoromethyl)phenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2d)
2d was obtained from 1d (5.81 mmol) as a yellow oil, yield: 55% (1.30 g); 1H NMR (400 MHz, CDCl3): δH 2.78 (d, J = 8.4, 2H), 3.73 (s, 2H), 4.38 (t, J = 8.8 Hz, 1H), 6.33 (s, 1H), 6.73 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.27–7.44 (m, 7H), 7.51 (bs, 2H); 13C NMR (100 MHz, CDCl3): δC 34.5, 41.2, 45.0, 114.9, 123.8, 125.1, 126.7, 127.3, 127.3, 127.4, 128.7, 129.0, 129.1, 129.2, 131.8, 133.0, 133.2, 134.9, 144.4, 149.1, 169.5; HRMS: Calcd. for C25H19F3O2Na [M+Na]+ : 431.1229. Found: 431.1249.
7.1.1.2. 4-(3′-Nitrophenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2e)
2e was obtained from 1e (6.71 mmol) as a yellow oil, yield: 41% (1.07 g); 1H NMR (400 MHz, CDCl3): δH 2.74 (dd, J = 17.2, 8.8 Hz, 1H), 2.91 (dd, J = 16.8, 7.6 Hz, 1H) 3.72 (s, 2H), 4.42 (t, J = 8.4 Hz, 1H), 6.34 (s, 1H), 6.79 (d, J = 7.2 Hz, 1H), 7.17 (d, J = 8.4 Hz, 2H), 7.21 (t, J = 7.6 Hz, 1H), 7.29–7.34 (m, 5H), 7.47 (t, J = 8.0 Hz, 1H), 7.54 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 34.3, 41.2, 45.6, 115.0, 122.0, 123.1, 127.0, 127.4, 127.5, 127.5, 127.6, 128.7, 129.2, 129.5, 133.0, 133.1, 134.1, 134.5, 145.6, 148.4, 148.6, 169.5; HRMS : Calcd. for C24H19NO4K [M+K]+ : 424.0946. Found: 424.0934.
7.1.1.3. 4-(4′-Fluorophenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2f)
2f was obtained from 1f (25.0 mmol) as a brown oil, yield: 15% (1.30 g); 1H NMR (400 MHz, CDCl3): δH 2.75 (dd, J = 8.8, 1.0 Hz, 2H), 3.72 (s, 2H), 4.30 (t, J = 8.8 Hz, 1H), 6.30 (s, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.94–7.40 (m, 12H); 13C NMR (100 MHz, CDCl3): δC 34.7, 41.2, 44.4, 114.8, 115.4 (JCF = 20.4 Hz), 125.3, 126.6, 127.1, 127.2, 127.3, 127.5, 128.2, 128.7, 129.0, 129.8 (JCF = 8.1 Hz), 133.0, 133.3, 135.7, 137.9, 139.1, 149.3, 169.5; HRMS : Calcd. for C24H23FNO2 [M+NH4]+: 376.1713. Found: 376.1715.
7.1.1.4. 4-(4′-Chlorophenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2g)
2g was obtained from 1g (25.0 mmol) as brown oil, yield: 21% (2.01 g); 1H NMR (400 MHz, CDCl3): δH 2.71–2.82 (m, 2H), 3.73 (s, 2H), 4.29 (t, J = 8.8 Hz, 1H), 6.31 (s, 1H), 6.76 (d, J = 7.6 Hz, 1H), 7.01–7.42 (m, 12H); 13C NMR (100 MHz, CDCl3): δC 34.5, 41.3, 44.5, 114.8, 126.6, 127.1, 127.2, 127.3, 127.5, 128.7, 129.2, 129.7, 132.5, 133.0, 133.2, 135.3, 141.9, 149.2, 169.1; HRMS : Calcd. for C24H23ClNO2 [M+NH4]+: 392.1417. Found: 392.1418.
7.1.1.5. 4-(4′-Bromophenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2h)
2h was obtained from 1h (25.0 mmol) as a brown oil, yield: 45% (4.72 g); 1H NMR (400 MHz, CDCl3): δH 2.64–2.82 (m, 2H), 3.72 (s, 2H), 4.27 (t, J = 7.8 Hz, 1H), 6.31 (s, 1H), 6.76 (d, J = 7.6 Hz, 1H), 7.05–7.43 (m, 12H); 13C NMR (100 MHz, CDCl3): δC 34.4, 41.2, 44.6, 114.8, 120.6, 125.3, 126.6, 127.1, 127.2, 127.3, 127.4, 128.2, 128.7, 129.0, 129.2, 130.0, 131.6, 133.0, 133.2, 135.2, 137.8, 142.4, 149.1, 169.5; HRMS : Calcd. for C24H20BrO2 [M+H]+ : 419.0647. Found: 419.0641.
7.1.1.6. 4-(4′-Trifluoromethyl)phenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2i)
2i was obtained from 1i (5.81 mmol) as brown oil, yield: 46% (1.08 g); 1H NMR (400 MHz, CDCl3): δH 2.75 (dd, J = 16.8, 10.1 Hz, 1H), 2.83 (dd, J = 16.8, 7.6 Hz, 1H), 3.72 (s, 2H), 4.37 (t, J = 8.6 Hz, 1H), 6.32 (s, 1H), 6.75 (d, J = 7.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 2H), 7.18 (t, J = 7.4 Hz, 1H), 7.25–7.38 (m, 7H), 7.56 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 34.4, 41.2, 44.9, 114.9, 122.5, 126.8, 127.3, 127.5, 128.7 (JCF = 2.2 Hz), 129.2, 133.0, 133.2, 134.8, 147.6, 149.0, 169.5; HRMS : Calcd. for C25H20F3O2 [M+H]+ : 409.1415. Found: 409.1427.
7.1.1.7. 4-(4′-Nitrophenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2j)
2j was obtained from 1j (13.4 mmol) as a brown oil, yield: 48% (2.5 g); 1H NMR (400 MHz, CDCl3): δH 2.71 (dd, J = 16.9, 8.4 Hz, 1H), 2.90 (dd, J = 16.9, 7.4 Hz, 1H), 3.72 (s, 2H), 4.41 (t, J = 8.2 Hz, 1H), 6.33 (s, 1H), 6.78 (d, J = 7.2 Hz, 1H), 7.11 (d, J = 6.0 Hz, 2H), 7.20 (t, J = 7.8 Hz, 1H), 7.26–7.41 (m, 7H), 8.15 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 34.2, 41.2, 44.8, 115.0, 123.8, 126.9, 127.4, 127.5, 127.6, 128.7, 129.1, 129.2, 132.9, 133.1, 134.0, 146.9, 148.6, 151.2, 169.5; HRMS : Calcd. for C24H19NNaO4 [M+Na]+ : 408.1212. Found: 408.1217.
7.1.1.8. 4-(2′-Bromophenyl)-3,4-dihydronaphthalen-2-yl 2-phenylacetate (2k)
2k was obtained from 1k (5.49 mmol) as orange oil, yield: 38% (0.873 g); 1H NMR (400 MHz, CDCl3): δH 2.73 (dd, J = 17.0, 8.8 Hz, 1H), 2.84 (dd, J = 17.0, 8.0 Hz, 1H), 3.66 (s, 2H), 4.86 (t, J = 8.4 Hz, 1H), 6.31 (s, 1H), 6.77 (d, J = 7.4 Hz, 1H), 7.01–7.06 (m, 4H), 7.12–7.16 (m, 2H) 7.23–7.31 (m, 5H), 7.54–7.57 (m, 1H); 13C NMR (100 MHz,CDCl3): δC 33.2, 41.1, 43.5, 114.6, 124.4, 126.6, 127.1, 127.2, 127.3, 127.5, 127.6, 128.2, 128.6, 129.1, 130.0, 132.9, 133.2, 133.3, 134.4, 142.3, 148.9, 169.3.
7.1.1.9. 4-(3′-Fluorophenyl)-tetralon-2-one (2′a) and 4-(3′-(chlorophenyl)-tetralon-2-one (2′b)
2′a and 2′b were prepared from 1a and 1b respectively by our earlier reported procedure.22
7.1.1.10. 4-(Biphenyl-3-yl)tetralen-2-ol phenylacetate (2n)
2n was prepared from 2c by our earlier reported procedure22 using the Suzuki coupling method.
7.1.1.11. 4-Cyclohexyltetralen-2-ol Phenylacetate (2l) and 4-Cyclooctyltetralen-2-ol Phenylacetate (2m)
2l and 2m were prepared from 1l and 1m respectively by our earlier reported procedure.22
7.1.2. General procedure for preparation of 2-tetralols (3a-m)
Sodium borohydride (2.5 eq.) was added to methanol at 0 °C. Tetralones (2′a-b) or enol-esters (2c-n) (1 eq.) dissolved in toluene was added to methanol solution at 0 °C and the mixture stirred at 50 °C for 15 h. The reaction mixture was quenched by brine, extracted with ethyl acetate (3x), and the organic layer was dried over anhydrous magnesium sulfate. Solvent was evaporated under reduced pressure and the residue was purified by column chromatography (100% dichloromethane).
7.1.2.1. cis-4-(3′-Fluorophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3a)
3a was obtained from tetralone 2′a (3.4 mmol) as colorless oil, yield: 38% (0.312 g); 1H NMR (400 MHz, CDCl3): δH 1.84 (dd, J = 23.8, 12.0 Hz, 1H, H-3), 2.16–2.27 (m, 1H, H-3′), 2.33–2.40 (m, 1H, H-1), 2.79–2.91 (m, 1H, H-1′), 3.13–3.23 (m, 1H, H-2), 4.10–4.18 (m, 1H, H-4), 6.74 (d, J = 7.6 Hz, 1H), 6.75–6.96 (m, 3H), 7.00–7.31 (m, 4H); 13C NMR (100 MHz, CDCl3): δC 39.4, 42.9, 45.9, 67.4, 113.3 (JCF = 21.8 Hz), 115.4 (JCF = 20.6 Hz), 124.4, 126.2, 126.4, 129.0, 129.3, 129.9 (JCF = 8.1 Hz), 134.9, 138.1, 148.5 (JCF = 7.4 Hz), 162.9 (JCF = 245.5 Hz); Anal. Calcd for C16H15FO + H: C, 78.99; H, 6.63. Found: C, 78.19; H, 7.17.
7.1.2.2. cis-4-(3′-Chlorophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3b)
3b was obtained from tetralone 2′b (2.5 mmol) as colorless oil, yield: 68% (0.438 g); 1H NMR (400 MHz, CDCl3): δH 1.85 (dd, J = 23.8, 12.0 Hz, 1H), 2.01–2.06 (m, 1H), 2.33–2.39 (m, 1H), 2.80–2.92 (m, 1H), 3.13–3.23 (m, 1H, H-2), 4.08–4.18 (m, 1H, H-4), 6.73 (d, J = 7.6 Hz, 1H), 7.03–7.28 (m, 7H); 13C NMR (100 MHz, CDCl3): δC 39.4, 43.0, 46.0, 67.5, 126.3, 126.5, 126.7, 127.0, 128.7, 129.1, 129.3, 129.8, 134.3, 135.0, 138.0, 148.0; Anal. Calcd for C16H15ClO + H: C, 73.98; H, 6.21. Found: C, 74.15; H, 6.48; HRMS : Calcd for C16H14ClO [M]−: 257.0733, C16H19ClNO [M+NH4]+ : 276.1150. Found: 257.0709, 276.1143.
7.1.2.3. cis-4-(3′-Bromophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3c)
3c was prepared from 2c by our earlier reported procedure.22
7.1.2.4. cis-4-(3′-Trifluoromethylphenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3d)
3d was obtained from 2d (3.16 mmol) as a pale yellow solid, mp 96–98 °C, yield: 62% (0.570 g); 1H NMR (400 MHz, CDCl3): δH 1.89 (dd, J = 23.6, 12.1 Hz, 1H), 2.30–2.43 (m, 1H), 2.93 (dd, J = 15.2, 10.8 Hz, 1H), 3.19–3.25 (m, 1H), 4.21–4.26 (m, 2H, H-2 & H-4), 6.70 (d, J = 7.6 Hz, 1H), 7.03–7.07 (m, 1H), 7.16 (d, J = 3.2 Hz, 2H), 7.35 (d, J = 7.2 Hz, 1H), 7.41–7.47 (m, 2H), 7.51 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 39.5, 43.1, 46.2, 67.5, 125.4, 126.4, 126.6, 128.6, 129.0, 129.1, 129.4, 132.1, 135.1, 137.8, 146.9.
7.1.2.5. cis-4-(3′-Nitrophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3e)
3e was obtained from 2e (2.59 mmol) as a white solid, mp 137–139 °C, yield: 55% (0.384 g); 1H NMR (400 MHz, CDCl3): δH 1.89 (dd, J = 23.2, 11.2 Hz, 1H), 2.41–2.45 (m, 1H), 2.94 (dd, J = 15.6, 10.4 Hz, 1H), 3.22 (dd, J = 16.0, 4.8 Hz, 1H), 4.20–4.23 (m, 1H), 4.31 (dd, J = 12.0, 5.2 Hz, 1H), 6.68 (d, J = 7.6 Hz, 1H), 7.03–7.07 (m, 1H), 7.17 (d, J = 4.4 Hz, 2H), 7.47–7.53 (m, 2H), 8.07 (s, 1H), 8.12 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 39.4, 42.9, 45.9, 67.3, 121.7, 123.6, 126.5, 126.8, 128.9, 129.5, 129.6, 134.9, 135.1, 137.2, 148.2; HRMS : Calcd. for C16H15NO3Na [M+Na]+ : 292.0944. Found: 292.0936.
7.1.2.6. cis-4-(4′-Fluorophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3f)
3f was obtained from 2f (4.4 mmol) as a pale yellow solid, mp 85–87 °C, yield: 74% (0.784 g); 1H NMR (400 MHz, CDCl3): δH 1.85 (dd, J = 23.4, 12.2 Hz, 1H), 2.32–2.40 (m, 1H), 2.82–2.93 (m, 1H), 3.18 (dd, J = 15.7, 5.1, 2.1 Hz, 1H), 4.09–4.21 (m, 2H), 6.73 (d, J = 7.6 Hz, 1H), 6.93–7.37 (m, 7H); 13C NMR (100 MHz, CDCl3): δC 36.5, 43.3, 45.5, 67.6, 115.4 (JCF = 21.1 Hz), 126.2, 126.4, 126.5, 128.6, 129.0 (JCF = 5.8 Hz), 129.3, 130.0 (JCF = 7.3 Hz), 135.0, 138.6, 141.7. 161.5 (JCF = 243.3 Hz); HRMS : Calcd. for C16H15FO [M]+ : 242.1107. Found: 242.1106.
7.1.2.7. cis-4-(4′-Chlorophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3g)
3g was obtained from 2g (5.0 mmol) as a pale yellow solid, mp: 100–105 °C, yield: 85% (1.10 g); 1H NMR (400 MHz, CDCl3): δH 1.84 (dd, J = 23.6, 12.0 Hz, 1H), 2.32–2.41 (m, 1H), 2.89 (dd, J = 15.8, 10.2 Hz, 1H), 3.17 (dd, J = 15.4, 3.4 Hz, 1H), 4.10–4.20 (m, 2H), 6.72 (d, J = 7.6 Hz, 1H), 6.98–7.34 (m, 7H); 13C NMR (100 MHz, CDCl3): δC 39.4, 43.1, 45.6, 67.5, 126.2, 126.4, 128.7, 129.1, 129.3, 130.0, 132.1, 135.0, 138.3, 144.5; HRMS : Calcd. for C16H15ClO [M]+ : 258.0811. Found: 258.0822.
7.1.2.8. cis-4-(4′-Bromophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3h)
3h was obtained from 2h (5.0 mmol) as a pale yellow semisolid, yield: 91% (1.37 g); 1H NMR (400 MHz, CDCl3): δH 1.84 (dd, J = 23.6, 12.0 Hz, 1H), 2.34–2.41 (m, 1H), 2.89 (dd, J = 15.8, 10.2 Hz, 1H), 3.13–3.24 (m, 1H), 4.08–4.20 (m, 2H, H-2 & H-4), 6.72 (d, J = 8.0 Hz, 1H), 6.84–7.44 (m, 7H); 13C NMR (100 MHz, CDCl3): δC 39.5, 43.1, 45.7, 67.5, 126.3, 126.5, 128.6, 129.0, 129.1, 130.0, 130.4, 131.7, 135.0, 138.2, 145.0; HRMS : Calcd. for C16H14BrO [M-H]− : 301.0228. Found: 301.0230.
7.1.2.9. cis-4-(4′-Trifluoromethylphenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3i)
3i was obtained from 2i (5.0 mmol) as a white solid, mp 98–100 °C, yield: 81% (1.18 g); 1H NMR (400 MHz, CDCl3): δH 1.87 (dd, J = 23.6, 12.0 Hz, 1H), 2.36–2.44 (m, 1H), 2.92 (dd, J = 15.6, 10.4 Hz, 1H), 3.20 (dd, J = 16.2, 3.8 Hz, 1H), 4.14–4.31 (m, 2H, H-2 & H-4), 6.70 (d, J = 7.6 Hz, 1H), 7.01–7.08 (m, 1H), 7.16 (d, J = 4.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) : δC 39.5, 43.0, 46.1, 67.5, 125.6 (JCF = 3.7 Hz), 126.4, 126.6, 129.0, 129.1, 129.4, 135.1, 137.8, 150.1; HRMS : Calcd. for C17H14F3O [M-H]− : 291.0997. Found: 291.1002.
7.1.2.10. cis 4-(4′-Nitrophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3j)
3j was obtained from 2j (5.0 mmol) as a pale yellow solid, mp 143–145 °C, yield: 94% (1.26 g); 1H NMR (400 MHz, CDCl3): δH 1.87 (dd, J = 23.4, 11.8 Hz, 1H), 2.38–2.46 (m, 1H), 2.85–2.99 (m, 1H), 3.20 (dd, J = 16.0, 3.2 Hz, 1H), 4.16–4.27 (m, 1H, H-2), 4.31 (dd, J = 12.0, 5.6 Hz, 1H, H-4), 6.67 (d, J = 8.0 Hz, 1H), 7.02–7.09 (m, 1H), 7.17 (d, J = 4.0 Hz, 2H), 7.35 (d, J = 8.8 Hz, 2H), 8.18 (d, J = 8.8 Hz, 2H); 13C NMR (CDCl3): δC 39.4, 42.7, 46.1, 67.3, 124.0, 126.5, 126.9, 129.0, 129.5, 129.6, 135.1, 137.1, 146.7, 153.9; HRMS : Calcd. for C16H14NO3 [M-H]- : 268.0974. Found: 268.0979.
7.1.2.11. cis-4-(2′-Bromophenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (3k)
3k was obtained from 2k (1.24 mmol) as a pale yellow semi-solid, yield: 68% (0.250 g). The compound was used immediately for the next step.
7.1.2.12. cis-4-Cyclohexyl-1,2,3,4-tetrahydronaphthalen-2-ol (3l)
3l was obtained from 2l (0.43 mmol) as white needles, mp 93–95 °C, yield: 96% (0.95 g); 1H NMR (400 MHz, CDCl3): δH 0.85–1.42 (m, 7H), 1.51 (dd, J = 23.4, 11.1 Hz, 1H), 1.66–1.86 (m, 4H), 1.99–2.14 (m, 2H), 2.65–2.74 (m, 1H), 2.95–3.10 (m, 2H), 3.93–4.08 (m, 1H, H-2), 7.05–7.18 (m, 3H), 7.25 (d, J = 7.6 Hz, 1H); 13C NMR (CDCl3): δC 26.1, 26.6, 26.7, 27.2, 31.5, 33.8, 40.0, 42.2, 43.1, 68.3, 125.6, 126.3, 126.8, 129.2, 135.7, 138.6; Anal. Calcd for C16H22O: C, 83.43; H, 9.63. Found: C, 83.28; H, 9.93. HRMS : m/z Calcd for C16H21 [M+H–H2O]+ : 213.1643, C16H26NO [M+NH4]+ : 248.2014. Found: 213.1636, 248.1997.
7.1.2.13. cis-4-Cyclooctyl-1,2,3,4-tetrahydronaphthalen-2-ol (3m)
3m was obtained from 2m (0.73 mmol) as colorless oil, yield: 85% (0.160 g); 1H NMR (400 MHz, CDCl3): δH 1.19–1.83 (m, 16H), 2.12–2.17 (m, 1H), 2.30–2.33 (m, 1H), 2.66–2.73 (m, 1H), 2.95–3.01 (m, 2H), 3.91–3.97 (m, 1H, H-2), 7.05–7.18 (m, 3H), 7.29 (d, J = 7.6 Hz 1H); 13C NMR (CDCl3):δC 25.7, 26.0, 26.7, 26.9, 27.4, 27.6, 33.6, 33.7, 40.1, 40.5, 45.6, 68.1 125.6, 126.3, 126.9, 129.3, 135.9, 138.9.
7.1.2.14. cis-4-(Biphenyl-3-yl)tetral-2-ol (3n)
3n was obtained from 2n (1.0 mmol) as colorless oil, yield: 86% (0.260 g); 1H NMR (400 MHz, CDCl3): δH 1.90 (dd, J = 23.6, 12.1 Hz, 1H), 2.34–2.40 (m, 1H), 2.63 (bs, 1H), 2.84–2.91 (m, 1H), 3.10–3.18 (m, 1H), 4.07–4.17 (m, 2H), 6.79 (d, J = 7.6 Hz, 1H), 6.97–7.54 (m, 12H); 13C NMR (CDCl3): δC 39.4, 43.1, 46.3, 67.5, 125.2, 126.1, 126.2, 126.3, 127.1, 127.2, 127.5, 127.6, 128.4, 128.6, 128.9, 128.9, 129.1, 129.2, 134.9,137.8, 138.7, 140.9, 141.4, 146.4; HRMS : Calcd for C22H24NO [M+NH4]+: 318.1858. Found: 318.1857.
7.1.3. General procedure for tosylation of alcohols (3a-m)
p-Toluenesulfonyl chloride (1.1 eq.) was added to a solution of tetralols 3a-m (1 eq.) in pyridine and the reaction mixture was stirred at rt for 24 h under nitrogen atmosphere. Pyridine was removed under reduced pressure at 40–50 °C and the residue was purified by column chromatography (n-hexanes: dichloromethane = 1: 1), and used immediately due to thermal instability of the tosylates 4a-m.
7.1.3.1. cis-4-(3′-Fluorophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4a)
4a was obtained from 3a (1.23 mmol) as colorless oil, yield: 25% (0.122 g); 1H NMR (400 MHz, CDCl3): δH 2.00–2.09 (m, 1H), 2.43–2.49 (m, 4H), 3.12 (d, J = 7.8 Hz, 2H), 4.12 (dd, J = 11.7, 5.7 Hz, 1H), 4.86–4.92 (m, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.78–6.82 (m, 1H), 6.90–7.29 (m, 6H), 7.34 (d, J = 8.0 Hz, 2H), 7.79–7.82 (m, 2H); 13C NMR (100 MHz, CDCl3) : δC 21.7, 36.4, 39.4, 45.4, 77.5, 113.7 (JCF = 21.4 Hz), 115.4 (JCF = 21.4 Hz), 124.3, 126.6, 126.7, 126.8, 127.7, 129.1 (JCF = 12.5 Hz),129.8, 129.9, 130.0, 130.1, 133.3, 134.1, 137.3, 144.8, 147.4 (JCF = 6.6 Hz), 162.9 (JCF = 246.2 Hz).
7.1.3.2. cis-4-(3′-Chlorophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4b)
4b was obtained from 3b (1.66 mmol) as colorless oil, yield: 38% (0.260 g); 1H NMR (400 MHz, CDCl3): δH 2.04 (dd, J = 24.2, 11.7 Hz, 1H), 2.42–2.48 (m, 4H), 3.12 (d, J = 7.8 Hz, 2H), 4.09 (dd, J = 11.7, 5.7 Hz, 1H), 4.85–4.93 (m, 1H), 6.71 (d, J = 7.8 Hz, 1H), 6.99–7.25 (m, 7H), 7.34 (d, J = 8.0 Hz, 2H), 7.80 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 21.6, 36.4, 39.4, 45.3, 77.4, 126.6, 126.7, 126.8, 126.9, 127.0, 127.7, 128.7, 129.1, 129.2, 129.8, 129.9, 130.0, 133.3, 134.0, 134.4, 137.2, 144.7, 147.0.
7.1.3.3. cis-4-(3′-Bromophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4c)
4c was obtained from 3c (2.15 mmol) as colorless oil, yield: 44% (0.431 g); 1H NMR (400 MHz, CDCl3): δH 2.06 (dd, J = 23.8, 12.0 Hz, 1H), 2.42–2.50 (m, 4H), 3.16 (d, J = 7.6 Hz, 2H), 4.20 (dd, J = 11.6, 6.0 Hz, 1H), 4.90–4.95 (m, 1H), 6.68 (d, J = 7.6 Hz, 1H), 7.03–7.10 (m, 2H), 7.13–1.17 (m, 1H), 7.29–7.32 (m, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.40–7.45 (m, 2H), 7.52 (d, J = 7.6 Hz, 1H), 7.80 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 21.6, 36.3, 39.5, 45.4, 77.3, 122.5, 126.6, 126.7, 127.3, 127.4, 127.5, 128.9, 129.1, 129.7, 129.7, 129.8, 130.1, 131.5, 133.2, 133.9, 137.1, 144.7,147.2; HRMS : Calcd for C23H25BrNO3S [M+NH4]+ : 474.0739, 476.0718. Found: 474.0721, 476.0700.
7.1.3.4. cis-4-(3′-(Trifluoromethyl)phenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4d)
4d was obtained from 3d (1.84 mmol) as pale-yellow semi-solid, yield: 13% (0.107 g); 1H NMR (400 MHz, CDCl3): δH 2.06 (dd, J = 24.0, 11.6 Hz, 1H), 2.44–2.53 (m, 4H), 3.14 (d, J = 7.6 Hz, 2H), 4.21 (dd, J = 11.6, 6.0 Hz, 1H), 4.84–4.98 (m, 1H), 6.67 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 7.4 Hz, 2H), 7.15 (t, J = 7.2 Hz, 1H), 7.24 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 7.6 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 21.6, 36.3, 39.3, 45.4, 104.9, 123.7 (JCF = 3.7 Hz), 125.3, 126.8, 126.9, 127.6, 127.7, 129.1, 129.2, 129.4, 129.9, 132.0, 133.4, 134.0, 137.0, 144.8, 145.9; HRMS : Calcd for C24H21F3O3SNa [M+Na]+ : 467.1056. Found: 469.1074.
7.1.3.5. cis-4-(3′-Nitrophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4e)
4e was obtained from 3e (1.37 mmol) as white semi-solid, yield: 36% (0.210 g); 1H NMR (400 MHz, CDCl3): δH 2.09 (dd, J = 23.6, 12.0 Hz, 1H), 2.43–2.53 (m, 4H), 3.16 (d, J = 7.6 Hz, 2H), 4.28 (dd, J = 10.8, 5.6 Hz, 1H), 4.92–4.95 (m, 1H), 6.67 (d, J = 7.6 Hz, 1H), 7.04–7.19 (m, 3H), 7.34 (d, J = 8.0 Hz, 2H), 7.46–7.49 (m, 2H), 7.78 (d, J = 7.6 Hz, 2H), 7.99 (s, 1H), 8.11 (d, J = 6.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 21.7, 36.2, 39.3, 45.1, 121.9, 123.5, 127.0, 127.2, 127.7, 129.0, 129.6, 129.6, 129.9, 133.4, 134.9, 136.3, 144.9, 147.2, 148.4; HRMS : Calcd for C23H21NO5SNa [M+Na]+: 446.1033. Found: 4461034.
7.1.3.6. cis-4-(4′-Fluorophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4f)
4f was obtained from 3f (3.09 mmol) as colorless oil, and was immediately used for the next step, yield: 21% (0.257 g).
7.1.3.7. cis-4-(4′-Chlorophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4g)
4g was obtained from 3g (3.87 mmol) as colorless oil, and was immediately used for the next step, yield: 64% (1.02 g).
7.1.3.8. cis-4-(4′-Bromophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4h)
4h was obtained from 3h (1.0 mmol) as colorless oil, yield: 61% (0.279 g). 1H NMR (400 MHz, CDCl3): δH 2.03 (dd, J = 23.8, 11.8 Hz, 1H), 2.29–2.51 (m, 4H), 3.12 (d, J = 7.6 Hz, 2H), 4.08 (dd, J = 11.8, 5.8 Hz, 1H), 4.82–4.96 (m, 1H), 6.68 (d, J = 7.6 Hz, 1H), 6.95–7.15 (m, 5H), 7.28–7.35 (m, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.0, 2H); 13C NMR (100 MHz, CDCl3): δC 21.6, 36.3, 39.3, 44.9, 77.4, 126.5, 126.6, 127.6, 127.7, 129.0, 129.1, 129.6, 129.7, 129.8, 130.3, 131.6, 133.2, 134.0, 137.3, 144.0,144.7.
7.1.3.9. cis-4-(4′-(Trifluoromethyl)phenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4i)
4i was obtained from 3i (2.11 mmol) as pale-yellow semi-solid, yield: 81% (0.762 g). 1H NMR (400 MHz, CDCl3): δH 2.06 (dd, J = 24.0, 11.6 Hz, 1H), 2.44–2.53 (m, 4H), 3.14 (d, J = 7.6 Hz, 2H), 4.21 (dd, J = 11.6, 6.0 Hz, 1H), 4.84–4.98 (m, 1H), 6.67 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 7.4 Hz, 2H), 7.15 (t, J = 7.2 Hz, 1H), 7.24 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 7.6 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) : δC 21.6, 36.3, 39.3, 45.4, 125.6 (JCF = 3.6 Hz), 126.8, 126.9, 128.9, 129.0, 129.1, 129.3, 129.7, 129.8, 133.4, 134.0, 137.0, 144.8, 149.1.
7.1.3.10. cis-4-(4′-Nitrophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4j)
4j was obtained from 3j (2.11 mmol) as pale-yellow semi-solid, yield: 55% (1.37 g); 1H NMR (400 MHz, CDCl3): δH 2.09 (dd, J = 23.4, 11.0 Hz, 1H), 2.41–2.55 (m, 4H), 3.15 (d, J = 7.6 Hz, 2H), 4.21 (dd, J = 10.6, 4.8 Hz, 1H), 4.88–5.02 (m, 1H), 6.66 (d, J = 7.6 Hz, 1H), 7.09 (t, J = 7.8 Hz, 2H), 7.17 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 8.14 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 21.6, 36.2, 39.0, 45.1, 123.9, 126.9, 127.2, 127.6, 129.1, 129.5, 129.9, 133.4, 134.0, 136.3, 144.9, 146.8, 152.8.
7.1.3.11. cis-4-(2′-Bromophenyl)-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4k)
4k was obtained from 3k (0.824 mmol) as colorless oil, and was immediately used for the next step, yield: 78% (0.290 g).
7.1.3.12. cis-4-Cyclohexyl-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4l)
4l was obtained from 3l (1.63 mmol) as colorless oil, yield: 81% (0.465 g); 1H NMR (400 MHz, CDCl3): δH 0.87–1.32 (m, 6H), 1.55–1.79 (m, 6H), 1.89–1.97 (m, 1H), 2.11–2.15 (m, 1H), 2.44 (s, 3H), 2.85–2.97 (m, 2H), 4.66–4.75 (m, 1H), 6.95 (d, J = 7.4 Hz, 1H), 7.05–7.20 (m, 3H), 7.33 (d, J = 8.2 Hz, 2H), 7.78–7.83 (m, 2H); 13C NMR (100 MHz, CDCl3): δC 21.5, 26.1, 26.4, 26.5, 26.9, 30.7, 31.2, 36.6, 42.0, 42.8, 79.3, 125.8, 126.6, 126.9, 127.6, 129.1, 129.7, 133.8, 134.2, 137.8, 144.5; HRMS : Calcd. for C23H28NaO3S [M+Na]+ : 407.1657. Found: 407.1631.
7.1.3.13. cis-4-Cyclooctyl-1,2,3,4-tetrahydronaphthalen-2-yl 4-methylbenzenesulfonate (4m)
4m was obtained from 3m (0.619 mmol) as colorless oil, and was immediately used for the next step, yield: 44% (0.112 g).
7.1.3.14. cis-4-(Biphenyl-3-yl)tetral-2-yl 4-methylbenzenesulfonate (4n)
4n was obtained from 3n (0.75 mmol) as a colorless oil, yield: 59% (0.200 g); 1H NMR (400 MHz, CDCl3): δH 2.13 (dd, J = 24.0, 11.9 Hz, 1H), 2.47 (s, 3H), 2.48–2.53 (m, 1H), 3.03–3.28 (m, 2H), 4.14–4.22 (m, 1H), 4.89–5.10 (m, 1H), 6.78 (d, J = 7.6 Hz, 1H), 7.00–7.69 (m, 14H), 7.80 (d, J = 8.4, 2H); 13C NMR (CDCl3): δC 21.6, 36.5, 39.6, 45.8, 77.8, 125.6, 126.6, 126.7, 127.0, 127.3, 127.4, 127.5, 127.6, 127.8, 128.5, 128.7, 128.8,129.0, 129.1, 129.7, 129.8, 133.3, 134.1, 137.9, 140.9, 141.6, 144.7,145.4.
7.1.4. General procedure for synthesis of 4-substituted-2-N,N-dimethylaminotetralins (5a-m)
cis-Tosylates 4a-m (1 eq.) were dissolved in a minimum amount of dichloromethane and then added to an aqueous solution of dimethylamine (40% in water, 100 eq.) in a sealed tube. The reaction mixture was heated at 80 °C for 24 h. The crude material was extracted with ethyl acetate (3x), dried over anhydrous magnesium sulfate, and solvent was evaporated under reduced pressure. The crude mixture was purified by column chromatography (dichloromethane: methanol = 95: 5) to afford racemic 5a-m.
The racemic mixtures of trans-analogs (and cis-analogs in the case of 5q) were separated by semi-preparative chiral HPLC using conditions and solvents specific to each analog to elute the trans- (+)-(2R,4S) and (−)-(2S,4R) enantiomers (and cis- [+]-[2R, 4R] and [−]-[2S, 4S] in the case of 5q) at retention time t1 at t2, respectively, with absolute stereochemistry assigned according to retention time of the the parent analog, PAT.23 The (+) and (−) free base amines were converted to HCl salts for use in pharmacological assays by passing HCl through an ethereal solution of the free base
7.1.4.1. trans-4-(3′-Fluorophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5a)
5a was obtained from 4a (0.30 mmol) as colorless oil, yield: 70% (0.057 g); 1H NMR (400 MHz, CDCl3): δH 2.39–2.42 (m, 2H, H-3 & H-3′), 2.70 (s, 6H), 3.16–3.43 (m, 3H, H-1, H-1′ & H-2), 4.52 (bs, 1H, H-4), 6.60 (d, J = 10.0 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 6.89–6.97 (m, 1H), 7.00 (d, J = 7.2 Hz, 1H), 7.18–7.31 (m, 4H); 13C NMR (100 MHz, CDCl3): δC 30.5, 31.7, 40.1, 43.3, 58.3, 113.7 (JCF = 21.4 Hz), 115.3 (JCF = 22.1 Hz), 123.9, 127.2, 127.5, 129.5, 130.0, 130.2 (JCF = 8.8 Hz), 132.9, 135.0, 147.3, 162.8 (JCF= 246.9 Hz); HRMS : Calcd for C18H21FN [M+H]+ : 270.1658. Found: 270.1656.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.1% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 10.9, t2 = 12.5.
7.1.4.2. trans-4-(3′-Chlorophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5b)
5b was obtained from 4b (0.60 mmol) as colorless oil, yield: 30% (0.051 g); 1H NMR (400 MHz, CDCl3): δH 2.25–2.32 (m, 2H), 2.63 (s, 6H), 3.02–3.37 (m, 3H), 4.41 (bs, 1H, H-4), 6.77–6.92 (m, 3H), 7.11–7.20 (m, 5H); 13C NMR (100 MHz, CDCl3): δC 30.6, 32.0, 40.3, 43.4, 58.2, 126.6, 127.1, 127.3, 127.5, 128.4, 129.5, 129.9, 130.3, 133.1, 134.6, 135.0, 146.8; HRMS : Calcd for C18H21ClN [M+H]+ : 286.1363, 288.1333. Found 286.1357, 288.1332.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.1% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 11.0, t2 = 12.5.
mp (HCl salt): 218–220 °C. (+)-trans-5b HCl: [α]25D = +51.6 (c 1.0, CH2Cl2), (−)-trans-5b HCl: [α]25D = −52.0(c 1.0, CH2Cl2).
7.1.4.3. trans-4-(3′-Bromophenyl)-N,N-dimethyl-1,2,3,4- tetrahydronaphthalen-2-amine (5c)
5c was obtained from 4c (0.92 mmol) as colorless oil, yield: 35% (0.106 g); 1H NMR (400 MHz, CDCl3): δH 2.05–2.09 (m, 2H), 2.26 (s, 6H), 2.58–2.63 (m, 1H), 2.78–2.87 (m, 1H), 2.96–3.01 (m, 1H), 4.28 (t, J = 5.1 Hz, 1H, H-4), 6.84–6.87 (m, 2H), 7.03–7.13 (m, 5H), 7.25 (dd, J = 7.8, 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 30.6, 32.0, 40.2, 43.2, 58.0, 122.7, 126.9, 127.0, 127.1, 127.3, 129.4, 129.8, 129.9, 130.1, 131.2, 133.2, 135.0, 147.2; HRMS : Calcd for C18H21BrN [M+H]+ : 330.0852, 332.0837, 331.0890, 333.0869. Found 330.0852, 332.0831, 331.0848, 333.0828.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.1% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 11.0, t2 = 12.7.
mp (HCl salt): 208–210 °C. (+)-trans-5c HCl: [α]25D = +54.3 (c 1.0, CH2Cl2), (−)-trans-5c HCl: [α]25D = −54.0 (c 1.0, CH2Cl2).
7.1.4.4. trans-4-(3′-Trifluoromethylphenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5d)
5d was obtained from 4d (0.22 mmol) as colorless oil, yield: 46% (0.032 g); 1H NMR (400 MHz, CDCl3): δH 2.18–2.25 (m, 2H), 2.38 (s, 6H), 2.74–2.78 (m, 1H), 2.95 (dd, J = 16.4, 10.0 Hz, 1H), 3.13 (dd, J = 15.6, 4.4 Hz, 1H), 4.47 (t, J = 4.4 Hz, 1H, H-4), 6.91 (d, J = 7.6 Hz, 1H), 7.14 (d, J = 7.6 Hz, 2H), 7.21 (d, J = 4.0 Hz, 2H), 7.32 (s, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 31.6, 34.2, 41.4, 43.8, 56.6, 123.1, 125.1 (JCF = 3.7 Hz), 126.5, 126.9, 128.7, 129.5, 129.9, 135.5, 136.4, 147.3; HRMS : Calcd. for C19H21F3N [M+H]+ : 320.1621. Found: 320.1631.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.2% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 10.6, t2 = 12.1.
mp (HCl salt): 235–237 °C. (+)-trans-5d HCl: [α]25D = +26.8 (c 1.0, CH2Cl2), (−)-trans-5d HCl: [α]25D = -27.0 (c 1.0, CH2Cl2).
7.1.4.5. trans-4-(3′-Nitrophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5e)
5e was obtained from 4e (0.472 mmol) as colorless oil, yield: 31% (0.044 g); 1H NMR (400 MHz, CDCl3): δH 2.11–2.22 (m, 2H), 2.29 (s, 6H), 2.58–2.61 (m, 1H), 2.89 (dd, J = 16.0, 10.2 Hz, 1H), 3.06 (dd, J = 16.4, 4.4 Hz, 1H), 4.49 (t, J = 4.4 Hz, 1H, H-4), 6.88 (d, J = 7.6 Hz, 1H), 7.11–7.15 (m, 1H), 7.21 (d, J = 3.6 Hz, 1H), 7.35 (d, J = 7.6 Hz, 2H), 7.43 (t, J = 8.0 Hz, 1H), 7.92 (s, 1H), 8.05 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 31.9, 35.0, 41.8, 43.8, 56.0, 121.3, 123.4, 126.4, 126.9, 129.1, 129.7, 134.9, 136.2, 136.3, 148.3, 149.0; HRMS : Calcd. for C18H21N2O2 [M+H]+ : 297.1598. Found: 297.1612.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.2% of TEA (modifier); flow rate = 1.5 mL/min; t1 =12.8, t2 =14.3.
mp (HCl salt): 195–197 °C. (+)-trans-5e: [α]25D = +24.3 (c 1.0, CH2Cl2), (−)-trans-5e : [α]25D = −25.3 (c 1.0, CH2Cl2).
7.1.4.6. trans-4-(4′-Fluorophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5f)
5f was obtained from 4f (0.64 mmol) as yellow semisolid, yield: 42% (0.072 g); 1H NMR (400 MHz, CDCl3): δH 2.28–2.37 (m, 2H), 2.65 (s, 6H), 3.06–3.24 (m, 2H), 3.34 (dd, J = 15.0, 4.6 Hz, 1H), 4.48 (t, J = 4.0 Hz, 1H, H-4), 6.89–7.01 (m, 4H), 7.11–7.25 (m, 4H); 13C NMR (100 MHz, CDCl3): δC 30.6, 32.3, 40.2, 42.9, 57.9, 115.4 (JCF = 21.2 Hz), 127.1 (JCF = 21.2 Hz), 129.4, 129.8 (JCF = 8.1 Hz), 130.0, 133.2, 135.8, 140.6, 160.2; HRMS : Calcd. for C18H21FN [M+H]+ : 270.1658. Found: 270.1649.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.2% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 11.9, t2 = 13.5.
mp (HCl salt): 198–200 °C. (+)-trans-5f HCl: [α]25D = +28.7 (c 1.0, CH2Cl2), (−)-trans-5f HCl : [α]25D = -29.0 (c 1.0, CH2Cl2).
7.1.4.7. trans-4-(4′-Chlorophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5g)
5g was obtained from 4g (2.40 mmol) as yellow oil, yield: 37% (0.244 g); 1H NMR (400 MHz, CDCl3): δH 2.42 (s, 2H), 2.72 (s, 6H), 3.17–3.38 (m, 2H), 3.49 (d, J = 15.2 Hz, 1H), 4.52 (bs, 1H, H-4), 6.87 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 7.6 Hz, 1H), 7.15–7.30 (m, 5H), 12.70 (s, 1H); 13C NMR (100 MHz, CDCl3): δC 30.4, 31.4, 38.9, 40.6, 43.0, 58.4, 127.3, 127.6, 128.8, 129.5, 129.6, 130.0, 132.5, 132.8, 134.9, 142.9; HRMS : Calcd. for C18H21ClN [M+H]+ : 286.1363. Found: 286.1364.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.2% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 10.9, t2 =12.8.
mp (HCl salt): 243–245 °C. (+)-trans-5g HCl: [α]25D = +45.5 (c 1.0, CH2Cl2), (−)-trans-5g HCl : [α]25D = −46.0 (c 1.0, CH2Cl2).
7.1.4.8. trans-4-(4′-Bromophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5h)
5h was obtained from 4h (0.5 mmol) as colorless oil, yield: 70% (0.115 g); 1H NMR (400 MHz, CDCl3): δH 2.14–2.17 (m, 2H), 2.38 (s, 6H), 2.69–2.80 (m, 1H), 2.88–2.95 (m, 1H), 3.07–3.11 (m, 1H), 4.36 (t, J = 4.9 Hz, 1H, H-4), 6.87 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 7.6 Hz, 1H), 7.06–7.21 (m, 3H), 7.37 (d, J = 8.4 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 31.9, 34.1, 41.5, 43.5, 56.6, 126.4, 126.7, 128.9, 129.1, 129.4, 129.9, 130.3, 130.4, 131.3, 131.7, 136.8, 145.4; HRMS : Calcd. for C18H21BrN [M+H]+ : 330.0857. Found: 330.0849.
HPLC (s-prep): solv. sys = MeOH:EtOH:2-PrOH:Hexanes (5:5:5:85) + 0.1% of DEA (modifier); flow rate = 10 mL/min; t1 = 16.06, t2 = 19.3.
mp (HCl salt): = 240–243 °C. (+)-trans-5h HCl: [α]25D = +66.1 (c 1.0, CH2Cl2), (−)-trans-5h HCl : [α]25D = −65.8 (c 1.0, CH2Cl2).
7.1.4.9. trans-4-(4′-Trifluoromethylphenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5i)
5i was obtained from 4i (1.7 mmol) as colorless oil, yield: 60% (0.326 g); 1H NMR (400 MHz, CDCl3): δH 2.19–2.28 (m, 2H), 2.41 (s, 6H), 2.74–2.84 (m, 1H), 2.97 (dd, J = 16.2, 9.8 Hz, 1H), 3.15 (dd, J = 16.2, 4.6 Hz, 1H), 4.48 (t, J = 4.4 Hz, 1H, H-4), 6.92 (d, J = 7.6 Hz, 1H), 7.08–7.17 (m, 3H), 7.22 (d, J = 3.6 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 31.6, 33.9, 41.3, 43.8, 56.7, 125.3 (JCF = 3.7 Hz), 126.5, 126.9, 128.9, 129.5, 129.9, 135.3, 136.3, 150.2; HRMS : Calcd. for C19H21F3N [M+H]+ : 320.1626. Found: 320.1619.
HPLC (s-prep): solv. sys = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.2% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 10.8, t2 = 13.7.
mp (HCl salt): 260–263 °C. (+)-trans-5i HCl: [α]25D = +28.3 (c 1.2, CH2Cl2), (−)-trans-5i HCl: [α]25D = −28.0 (c 1.2, CH2Cl2).
7.1.4.10. trans-4-(4′-Nitrophenyl)-N,N-dimethyl-1,2,3,4- tetrahydronaphthalen-2-amine (5j)
5j was obtained from 4j (2.28 mmol) as yellow oil, yield: 70% (0.473 g); 1H NMR (400 MHz, CDCl3): δH 2.13–2.21 (m, 2H), 2.30 (s, 6H), 2.57–2.66 (m, 1H), 2.90 (dd, J = 16.2, 9.4 Hz, 1H), 3.07 (dd, J = 16.0, 4.8 Hz, 1H), 4.49 (t, J = 5.0 Hz, 1H, H-4), 6.88 (d, J = 7.6 Hz, 1H), 7.09–7.15 (m, 1H), 7.18 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 4.0 Hz, 2H), 8.12 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δC 29.7, 32.0, 34.6, 41.6, 44.1, 56.2, 123.5, 126.4, 127.0, 129.5, 129.7, 129.8, 136.1, 136.1, 154.5; HRMS : Calcd. for C18H21N2O2 [M+H]+ : 297.1603. Found: 2907.1601. The racemic mixture 5j could not be resolved to its individual enantiomers using the chiral HPLC system. The free base racemate was converted to the hydrochloride salt and used in pharmacological assays.
7.1.4.11. trans-4-(2′-Bromophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5k)
5k was obtained from 4k (0.21 mmol) as pale yellow oil, yield: 46% (0.032 g); 1H NMR (400 MHz, CDCl3): δH 2.25–2.47 (m, 2H), 2.70 (s, 6H), 3.21–3.24 (m, 1H), 3.36 (bs, 1H), 3.61–3.65 (m, 1H), 4.90 (bs, 1H, H-4), 6.44–6.47 (m, 1H), 6.95 (d, J = 7.6 Hz, 1H), 7.05–7.20 (m, 5H), 7.63 (d, J = 7.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 28.1, 31.2, 43.2, 58.7, 123.9, 127.4, 127.5, 127.6, 128.7, 129.6, 130.1, 131.1, 133.1, 133.4, 133.2, 135.0, 143.2; HRMS : Calcd for C18H21BrN [M+H]+ : 330.0852, 332.0837, 331.0890, 333.0869. Found: 330.0852, 332.0831, 331.0848, 333.0828.
HPLC (s-prep): solv. sys = MeOH:EtOH:2-PrOH:Hexanes (5:5:5:85), 0.1% of DEA (modifier); flow rate = 10 mL/min; t1 = 15.3, t2 = 17.2.
mp (HCl salt): = 165–167 °C. (+)-trans-5h HCl: [α]25D = +34.3 (c 1.0, CH2Cl2), (−)-trans-5h HCl : [α]25D = -34.8 (c 1.0, CH2Cl2).
7.1.4.12. trans-4-Cyclohexyl-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5l)
5l was obtained from 4l (0.12 mmol) as white microcrystals, mp: 137–139 °C, yield: 71% (0.220 g); 1H NMR (400 MHz, CDCl3): δH 1.00–1.31 (m, 5H), 1.39–1.47 (m, 1H), 1.57–1.85 (m, 6H), 2.48–2.52 (m, 1H), 2.72–2.77 (m, 1H), 2.80 (s, 6H), 3.03 (dd, J = 9.0, 16.6 Hz, 1H), 3.34 (dd, J = 6.8, 16.0 Hz, 1H), 3.72–3.75 (m, 1H, H-4), 7.09–7.29 (m, 4H); 13C NMR (100 MHz, CDCl3): δC 26.5, 26.8, 27.3, 30.2, 31.8, 32.3, 41.6, 41.9, 43.5, 56.8, 124.9, 125.7, 129.1, 129.2, 136.0, 139.7. Anal. Calcd for C18H27N + HCl + H2O: C, 69.32; H, 9.70; N, 4.49. Found: C, 69.48; H, 9.98, N, 4.34. HRMS : Calcd for C18H28N [M+H]+: 258.2222. Found: 258.2220.
HPLC (s-prep): solv. sys = MeOH:EtOH:2-PrOH:Hexanes (5:5:5:85), 0.1% of DEA (modifier); flow rate = 10 mL/min; t1 = 15.3, t2 = 17.2.
mp (HCl salt): = 174–176 °C. (+)-trans-5l HCl [α]25D = +34.3 (c 0.37, CH2Cl2), (−)-trans-5l HCl: [α]25D = −34.4 (c 0.37, CH2Cl2).
7.1.4.13. trans-4-Cyclooctyl-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5m)
5m was obtained from 4m (0.271 mmol) as colorless oil Yield: 58% (0.044 g); 1H NMR (400 MHz, CDCl3): δH 1.21–1.64 (m, 13H), 1.72–1.81 (m, 2H), 1.89–1.96 (m, 1H), 2.00–2.07 (m, 1H), 2.31 (s, 6H), 2.65–2.95 (m, 4H), 7.05–7.21 (m, 4H); 13C NMR (100 MHz, CDCl3): δC 25.9, 26.4, 26.6, 26.7, 27.1, 27.2, 28.8, 32.6, 32.7, 40.7, 42.2, 43.2, 58.0, 125.4, 125.5, 128.1, 129.0, 136.3, 140.1. HRMS : m/z Calcd for C20H32N; C20H31NNa 286.2535; 308.2354 [M+H]+; [M+Na]+, Found: 286.2555; 308.2366.
HPLC (s-prep): solv. sys = MeOH:EtOH:2-PrOH:Hexanes (5:5:5:85), 0.1% of DEA (modifier); flow rate = 10 mL/min; t1 = 15.3, t2 = 17.2.
7.1.4.14. trans-4-(Biphenyl-3-yl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5n)
5n was obtained from 4n (0.4 mmol) as colorless oil, yield: 70% (0.091 g); 1H NMR (400 MHz, CDCl3): δH 2.38–2.48 (m, 2H), 2.66 (s, 6H), 3.17 (dd, J = 15.1, 11.0 Hz, 1H), 3.27–3.34 (m, 1H), 3.42 (dd, J = 15.2, 4.9 Hz, 1H), 4.56 (t, J = 4.5 Hz, 1H, H-4), 6.87 (d, J = 7.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 7.16–7.25 (m, 4H), 7.31–7.52 (m, 7H); 13C NMR (100 MHz, CDCl3): δC 30.7, 31.9, 40.1, 43.6, 58.3, 125.6, 126.9, 127.0, 127.1, 127.2, 127.3, 127.4, 128.6, 128.7, 128.9, 129.3, 130.0, 133.3, 135.7, 140.7, 141.4, 145.2; HRMS : m/z Calcd for C24H26N 328.2065 [M+H]+, Found: 328.2074.
HPLC (s-prep): solv. sys = MeOH:EtOH:2-PrOH:Hexanes (5:5:5:85), 0.1% of TEA (modifier); flow rate = 1.5 mL/min; t1 = 11.2, t2 = 12.3.
7.1.4.15. trans-4-(2′-Chlorophenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5o)
Racemic 5o was prepared starting from 2-cholorobenzaldehyde and phenyl acetone following a six-step procedure earlier published by us2 as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δH 2.08–2.14 (m, 2H), 2.28 (s, 6H), 2.58–2.66 (m, 1H), 2.84–2.91 (m, 1H), 3.01–3.06 (m, 1H), 4.83 (t, J = 5.1 Hz, 1H, H-4), 6.64 (dd, J = 7.6, 1.7 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 7.04–7.20 (m, 5H), 7.38 (dd, J = 7.8, 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 32.1, 32.3, 40.9, 41.8, 56.2, 126.2, 126.3, 126.4, 127.3, 129.3, 129.4, 129.9, 131.0, 133.7, 136.8, 137.5, 144.2.
HPLC (s-prep): solv. sys. = EtOH:Hexanes (15:85) + 0.1% of TEA; flow rate = 1.0 mL/min; t1 = 15.9, t2 = 16.8.
mp (HCl salt): 207–209 °C (lit21 mp [racemic]: 209–210 °C).
7.1.4.16. trans-4-(2′-Methylphenyl)-N,N-dimethyl-1,2,3,4-tetrahydronaphthalen-2-amine (5p)
Racemic 5p was prepared starting from 2-methylbenzaldehyde and phenyl acetone following a six-step procedure earlier published by us2 as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δH 1.97–2.13 (m, 2H), 2.28 (s, 6H), 2.44 (s, 3H), 2.66–2.73 (m, 1H), 2.85–2.91 (m, 1H), 3.01–3.07 (m, 1H), 4.57 (t, J = 5.7 Hz, 1H, H-4), 6.62 (d, J = 7.6 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 6.99–7.20 (m, 6H); 13C NMR (100 MHz, CDCl3): δC 19.5, 32.0, 32.9, 40.1, 41.9, 56.4, 125.6, 125.8, 126.0, 126.1, 129.1, 129.4, 129.8, 130.3, 135.5, 136.5, 138.9, 144.9.
HPLC (s-prep): solv. sys. = MeOH:EtOH:1-PrOH:2-PrOH:Hexanes (5:5:5:5:80) + 0.1% of TEA; flow rate = 1.5 mL/min; t1 = 11.1, t2 = 11.5.
mp (HCl salt): 220–221 °C (lit21 mp [racemic]: 220–222 °C).
7.2. Crystallography
X-Ray intensity data were collected at 100 K on a Bruker DUO diffractometer using CuKα radiation (λ = 1.54178 Å), from an IMU power source, and an APEXII CCD area detector. Raw data frames were read by program SAINT and integrated using 3D profiling algorithms. The resulting data were reduced to produce hkl reflections and their intensities and estimated standard deviations. The data were corrected for Lorentz and polarization effects and numerical absorption corrections were applied based on indexed and measured faces. The structure was solved and refined in SHELXTL6.1, using full-matrix least-squares refinement. The non-H atoms were refined with anisotropic thermal parameters and all of the H atoms were calculated in idealized positions and refined riding on their parent atoms. The amine proton was obtained from a Difference Fourier map and refined without any constraints. In the final cycle of refinement, 1194 reflections (of which 1193 are observed with I > 2σ (I)) were used to refine 196 parameters and the resulting R1, wR2 and S (goodness of fit) were 1.75%, 5.46% and 1.434, respectively. The refinement was carried out by minimizing the wR2 function using F2 rather than F values. R1 is calculated to provide a reference to the conventional R value and its function is not minimized.
7.3. Radioreceptor competitive binding assays11,12,14,25
The radioligands [3H]mesulergine, [3H]ketanserin, and [3H]mepyramine at the highest specific activity available were purchased from Perkin-Elmer Life Science (Boston, MA). The cDNA encoding the human serotonin 5-HT2A, 5-HT2B, 5-HT2C-INI, and histamine H1 wild type receptors were obtained from UMR cDNA Resource Center (Rolla, MO). HEK-293 cells were grown to 90% confluency in 10 cm plates using Dulbecco’s modified Eagle’s medium (DMEM; 10-013-CV, Mediatech, Manassas, VA) supplemented with 5% dialyzed fetal bovine serum and 1% penicillin-streptomycin (30-0020-CI, Mediatech). Cells were washed 1x with phosphate-buffered saline, and then transfected with 24 μg of cDNA mixed with 40 μL Lipofectamine 2000 reagent in Opti-MEM media and placed in an incubator at 37°C, 5% CO2, 95% humidity for 24 to 48 hr. Membranes then were collected in 50 mM Tris, 10 mM MgCl2-6H2O, 0.1 mM EDTA (assay buffer), as described previously,12 and stored at −80 °C until binding assays were performed.
Radioligand competitive displacement binding assays were performed in 96-well plates, using 3–5 μg of protein from membrane samples per well, similar to laboratory methods used previously.12 Each concentration point in the binding experiments was performed in triplicates of samples, and each experiment was performed a minimum of three times. Final concentration of radioligands in the assay mixtures was ~KD concentration, i.e., 2.0 nM [3H]ketanserin (5-HT2A receptors), 1.95 nM [3H]mesulergine (5-HT2B receptors), 1.4 nM [3H]mesulergine (5-HT2C receptors), or 1.0 nM [3H]mepyramine. Non-specific binding was determined in the presence of or 10 μM mianserin for all three 5-HT2 receptors or 10 μM triprolidine for H1 receptors. Radioreceptor binding assay mixtures were incubated for 1.0 h at 37 °C, with termination by rapid filtration through Whatman GF/B filters using a 96-well cell harvester (Tomtec, Hamden, CT). and subsequently washed five times with 50 mM Tris-HCl at rt. Filters containing bound [3H]radioligand were dried, placed in vials containing 2 mL scintillation cocktail (ScintiVerse), allowed to equilibrate overnight, and then were counted for 3H-induced scintillation using a Beckman-Coulter LS6500 counter.
7.1.1. Data analysis
Competition binding data were analyzed using nonlinear regression curve-fitting algorithms in GraphPad Prism, 5.03 for Windows (San Diego, CA). Hill slopes were not calculated as only eight data points were used to plot the graphs,28 thus, the algorithm for one-site fit Ki determination was used wherein the Hill slope was set to 1.0; the two-site curve-fit algorithm did not result in an improved fit (data not shown). Ligand affinity is expressed as an approximation of Ki values by conversion of the IC50 value using the Cheng-Prusoff equation where L is the concentration of radioligand.29 Comparisons of Ki values were performed using one-way ANOVAs and Student’s t-tests. The Ki values were considered to be statistically significant when the p-value was less than 0.05.
7.1.2. Receptor Model Building
A homology model of the human serotonin 5-HT2C receptor was built based on the crystal structure of the β2AR/T4-lysozyme chimera, PDB code 2rh1.30 Validation of the models and other details are described elsewhere.16,17,21 Briefly, the native sequences of the receptors were aligned to the β2AR sequence using ClustalW multiple sequence alignment.31,32 Point mutations were performed as needed and the gaps were analyzed, followed by the appropriate sequence additions and deletions. The transmembrane domains were built using the Biopolymer module of Sybyl-X 1.2 (Tripos International, St. Louis, MO) and the resulting bundle was optimized using Tripos force field.33 The resulting model was inserted into a rectangular box containing a pre-equilibrated 1-palmitoyl-2-oleyl-sn-glycero phosphatidyl choline (POPC) bilayer.33 The system containing the receptor model and POPC was relaxed using the Tripos force field34 to a gradient 0.05 Kcal/Å mol, and subjected to a 5 ns MD simulation using NVT canonical ensemble, 300 K temperature, Boltzmann initial velocities, and non-bonded cutoff set at 8 Å.
During performance of the research reported in this manuscript, the 5HT2B crystal structure was released (pdb code = 4IB4). Accordingly, we compared the β2AR-based 5HT2C homology model to a new 5HT2C homology model based generated as described above but based on the reported 5HT2B crystal structure. The alpha carbon atoms of both models were superimposed. The RMSD value obtained was 2.05 Å, which is smaller than the X-ray resolution of the template structures pdb codes 2RH1 and 4IB4 (2.40 Å and 2.70 Å, respectively), indicating, the two models are very similar, and consequently, ligand docking poses and ligand–receptor interactions are analogous.35
7.4. Ligand Docking
The ligand structures were built as monocations (protonated amines) using HyperChem 8.0 (Hypercube, Inc., Gainesville, FL) and structures were optimized using PM3 model Hamiltonian to a gradient of 0.01 Kcal/ Å mol. Ligands were pre-positioned in the receptor binding pocket, ensuring the critical interaction between ligand protonated amine group and the carboxylate side chain of residue D3.32.16–18,35 Flexible docking runs were carried out using Flexidock module in Sybyl-X 1.2. Optimal poses were selected and subject to molecular dynamics simulations as previously described,16–17 to obtain the most representative pose of the ligand in the binding pocket for analysis. Figures of the ligands in the receptor’s binding site were generated using Pymol version 1.3 (Schrödinger, LLC).36
Acknowledgments
These studies were funded by grants from the U.S. National Institutes of Health (R01 DA023928, DA030989, MH081193). X-ray equipment was purchased with funding from the National Science Foundation and the University of Florida.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Roth BL, Willins DL, Kristiansen K, Kroeze WK. Pharmacol Ther. 1998;79:231. doi: 10.1016/s0163-7258(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 2.Eggers AE. Med Hypotheses. 2013;80:791. doi: 10.1016/j.mehy.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Thomsen WJ, Grottick AJ, Menzaghi F, Reyes-Saldana H, Espitia S, Yuskin D, Whelan K, Martin M, Morgan M, Chen Al-Shamma H, Smith B, Chalmers D, Behan D. J Pharmacol Exp Ther. 2008;325:577. doi: 10.1124/jpet.107.133348. [DOI] [PubMed] [Google Scholar]; Smith SR, Weissman NJ, Anderson CM, Sanchez M, Chuang E, Stubbe S, Bays H, Shanahan WR Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. . N Engl J Med. 2010;363:245. doi: 10.1056/NEJMoa0909809. [DOI] [PubMed] [Google Scholar]
- 4.Glennon RA, Titeler M, McKenney JD. Life Sci. 1984;198435:2505. doi: 10.1016/0024-3205(84)90436-3. [DOI] [PubMed] [Google Scholar]
- 5.Nichols DE. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Roth BL. N Engl J Med. 2007;356:6. [Google Scholar]
- 7.Elangbam CS. Toxicol Pathol. 2010;38:837. doi: 10.1177/0192623310378027. [DOI] [PubMed] [Google Scholar]
- 8.Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL. Neuropsychopharmacology. 2003;28:519. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- 9.Kirk SL, Glazebrook J, Grayson B, Neill JC, Reynolds GP. Psychopharmacology (Berl) 2009;207:119. doi: 10.1007/s00213-009-1639-8. [DOI] [PubMed] [Google Scholar]
- 10.Roerig JL, Steffen KJ, Mitchell JE. CNS Drugs. 2011;25:1035. doi: 10.2165/11596300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Moniri NH, Covington-Strachan D, Booth RG. J Pharmacol Exp Ther. 2004;311:274. doi: 10.1124/jpet.104.070086. [DOI] [PubMed] [Google Scholar]
- 12.Booth RG, Fang L, Huang Y, Wilczynski A, Sivendran S. Eur J Pharmacol. 2009;615:1. doi: 10.1016/j.ejphar.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marquis KL, Sabb AL, Logue SF, Brennan JA, Piesla MJ, Comery TA, Grauer SM, Ashby CR, Nguyen HQ, Jr, Dawson LA, Barrett JE, Stack G, Meltzer HY, Harrison BL, Rosenzweig-Lipson S. J Pharmacol Exp Ther. 2007;320:486. doi: 10.1124/jpet.106.106989. [DOI] [PubMed] [Google Scholar]
- 14.Canal CE, Booth RG, Morgan D. Neuropharmacology. 2013;70:112. doi: 10.1016/j.neuropharm.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowland NE, Crump EM, Nguyen N, Robertson K, Sun Z, Booth RG. Pharmacol Biochem Behav. 2008;91:176. doi: 10.1016/j.pbb.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Córdova-Sintjago T, Sakhuja R, Kondabolu K, Canal CE, Booth RG. Int J Quantum Chem. 2012;112:3807. doi: 10.1002/qua.24237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Córdova-Sintjago T, Villa N, Canal C, Booth RG. Int J Quantum Chem. 2012;112:140. doi: 10.1002/qua.24237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Córdova-Sintjago T, Fang L, Bruysters M, Leurs R, Booth RG. J Chem Pharm Res. 2012;4:2937. [PMC free article] [PubMed] [Google Scholar]
- 19.Bucholtz EC, Wyrick SD, Owens CE, Booth RG. Med Chem Res. 1998;8:322. [Google Scholar]
- 20.Bucholtz EC, Brown RL, Tropsha A, Booth RG, Wyrick SD. J Med Chem. 1999;42:3041. doi: 10.1021/jm980428x. [DOI] [PubMed] [Google Scholar]
- 21.Canal CE, Córdova-Sintjago T, Villa N, Fang LJ, Booth RG. Eur J Pharmacol. 2011;673:1. doi: 10.1016/j.ejphar.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincek AS, Booth RG. Tetrahedron Lett. 2009;50:5107. doi: 10.1016/j.tetlet.2009.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyrick SD, Booth RG, Myers AM, Owens CE, Kula NS, Baldessarini RJ, McPhail AT, Mailman RB. J Med Chem. 1993;36:2542. doi: 10.1021/jm00069a013. [DOI] [PubMed] [Google Scholar]
- 24.Booth RG, Fang L, Wilczynski A, Sivendren S, Sun Z, Travers S, Bruysters M, Sansuk K, Leurs R. Inflamm Res. 2008;57(Suppl 1):S43. doi: 10.1007/s00011-007-0621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan D, Kondabolu K, Kuipers A, Sakhuja R, Robertson KL, Rowland NE, Booth RG. Neuropharmacology. 2013;72:274. doi: 10.1016/j.neuropharm.2013.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canal CE, Morgan D, Felsing D, Kondabolu K, Rowland NE, Robertson KL, Sakhuja R, Booth RG. J Pharmacol Exp Ther. 2014;349:310. doi: 10.1124/jpet.113.212373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choksi NY, Nix WB, Wyrick SD, Booth RG. Brain Res. 2000;852:151. doi: 10.1016/s0006-8993(99)02228-3. [DOI] [PubMed] [Google Scholar]
- 28.Motulsky HJ, Christopoulos A. GraphPad Software, Inc; San Diego, CA: 2003. [Google Scholar]
- 29.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 30.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG, Clustal W Clustal X version 20 . Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 32.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heller H, Schaefer M, Schulten K. The J Phys Chem. 1993;97:8343. [Google Scholar]
- 34.Clark M, Cramer RD, Opdenbosch NV. J Comput Chem. 1989;10:982. [Google Scholar]
- 35.Córdova-Sintjago T, Villa N, Fang L, Booth RG. Molecular Physics. 2014;112:398. doi: 10.1080/00268976.2013.833656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283. [Google Scholar]