

Abstract
Antigen-mediated crosslinking of Immunoglobulin E (IgE) bound to mast cells/basophils via FcεRI, the high affinity IgE Fc-receptor, is a well-known trigger of allergy. In humans, but not mice, dendritic cells (DCs) also express FcεRI that is constitutively occupied with IgE. In contrast to mast cells/basophils, the consequences of IgE/FcεRI signals for DC function remain poorly understood. We show that humanized mice that express FcεRI on DCs carry IgE like non-allergic humans and do not develop spontaneous allergies. Antigen-specific IgE/FcεRI crosslinking fails to induce maturation or production of inflammatory mediators in human DCs and FcεRI-humanized DCs. Furthermore, conferring expression of FcεRI to DCs decreases the severity of food allergy and asthma in disease-relevant models suggesting anti-inflammatory IgE/FcεRI signals. Consistent with the improved clinical parameters in vivo, antigen-specific IgE/FcεRI crosslinking on papain or LPS-stimulated DCs inhibits the production of pro-inflammatory cytokines and chemokines. Migration assays confirm that the IgE-dependent decrease in cytokine production results in diminished recruitment of mast cell progenitors; providing a mechanistic explanation for the reduced mast cell-dependent allergic phenotype observed in FcεRI-humanized mice. Our study demonstrates a novel immune regulatory function of IgE and proposes that DC-intrinsic IgE signals serve as a feedback mechanism to restrain allergic tissue inflammation.
INTRODUCTION
Latest reports from the World Allergy Organization state that approximately 30% of the population worldwide suffers from chronic allergic diseases such as asthma or food allergy.1 In all allergic disorders, large varieties of commonly innocuous antigens become allergens and induce detrimental immune responses. Immunoglobulin E (IgE) is well-appreciated for its central role in allergy.2 Induced by an antigen recognition event and facilitated by T cell help, B cells start to produce antigen-specific IgE. The body's IgE pool consists of a short-lived serum IgE fraction and a cell-bound IgE fraction, which forms when monovalent IgE binds to FcεRI, the high affinity IgE Fc receptor. IgE interactions with FcεRI significantly prolong the half-life of the immunoglobulin and stabilize the engaged Fc receptors on the surface of mast cells and basophils. Then, minor amounts of antigen are sufficient to elicit acute allergic reactions via instant release of preformed intracellular mediators, such as histamine, from these innate effector cells. Additionally, inflammatory cytokines induced by the signaling cascade downstream of IgE/FcεRI participate in the induction and dissemination of chronic allergic symptoms.3, 4
FcεRI is the multimeric immune recognition receptor that conveys the high efficiency of the antigen/IgE-mediated signals to innate IgE effector cells. On human and murine mast cells and basophils, FcεRI is expressed in its classical tetrameric isoform. This isoform comprises the IgE-binding α-chain, which is associated with ITAM-bearing signaling subunits (i.e. the FcεRI β-chain and the FcεRI γ-chain dimer). Additionally, a splice variant of the β-chain (FcεRI βt) was recently described as a regulatory subunit that modulates mast cell degranulation and IL-8 production in humans.5 While pro-inflammatory signals downstream of tetrameric FcεRI on mast cells are thoroughly studied, the functions of the second receptor isoform, trimeric FcεRI, remain poorly understood. This is likely because trimeric FcεRI is constitutively expressed in humans, but not in mice. This trimeric FcεRI isoform, which can be found on the cell surface of human DCs, lacks the FcεRI-β chain and thus has one ITAM-signaling module less than the tetrameric FcεRI isoform on mast cells and basophils3, 6. In addition to the constitutive expression on human DCs in the absence of any obvious pathology, inducible forms of trimeric FcεRI have been described in allergic humans and in murine models of allergy on several cell types including monocytes, neutrophils, and inflammatory DCs.7-9 Additionally, inducible expression of trimeric FcεRI on neutrophils has been found in murine experimental cerebral malaria10 implying that IgE signals via inducible FcεRI might have functions in diseases other than allergies. How cells respond to IgE-mediated signals following induction of trimeric FcεRI and whether differences in the signaling of constitutively expressed and inducible receptors exist is presently unknown.
As a consequence of FcεRI expression, the surface of human DCs is loaded with monomeric IgE comparable to mast cells and basophils at all times independently of the allergic status of an individual.11-13 Importantly, this element of the human IgE pool is not reflected in murine models. As sentinel gatekeepers of innate and adaptive immunity, DCs are thought to contribute to the initiation as well as the chronicity of allergic responses.14-20 For our understanding of immunological functions of IgE in humans, it is important to address the gaps in our understanding of how antigen/allergen-specific activation via IgE/FcεRI influences key immunoregulatory functions of DCs.
DCs actively secrete inflammatory cytokines and chemokines, which impact the magnitude of the allergic tissue response directly and indirectly via the recruitment of inflammatory cells and/or their progenitors. The secretion profile of DCs during inflammation thus contributes to chronic allergies independently of their antigen presentation function. The latter mechanism equips DCs with the unique ability to activate primary T cells and induce T helper type (Th)1, Th2, Th17, and regulatory T cell responses,21 implying that DCs are the central antigen presenting cells for the induction of allergy. Although the detailed mechanisms still remain controversial and unclear,14, 22 most recent evidence points to an IRF4-dependent DC subpopulation as Th2-inducing antigen presenting cells.23 The ensuing Th2-type T cells produce signature cytokines of allergic responses, such as IL-4, IL-5, and IL-13, and are thought to initiate and perpetuate the chronicity of allergy.24 The current hypothesis for the function of IgE-mediated antigen presentation by DCs is that FcεRI-mediated antigen sampling educates the adaptive immune system to elicit Th2-type inflammation de novo.25 This model was mainly proposed based on studies reporting higher FcεRI expression on DCs from individuals suffering from atopic diseases such as allergic rhinitis, atopic dermatitis, and asthma.8, 26-29 While these correlations argue in favor of an allergy-promoting role of IgE/FcεRI-mediated antigen presentation, they do not provide direct evidence. A recent publication demonstrated a contribution of DC-specific FcεRI to serum IgE clearance; further challenging the concept that the DC-bound IgE pool is a major contributor for promoting chronic allergic inflammation.30
During allergic responses in humans, antigen-specific IgE/FcεRI signals likely influence all immune regulatory aspects of DC biology. In this study, we addressed how the DC-bound IgE pool impacts allergic responses using FcεRI-humanized mice that were engineered to constitutively express a trimeric isoform of FcεRI on DCs like humans while preserving expression of endogenous tetrameric FcεRI on mast cells and basophils.31
RESULTS
FcεRI-bound IgE on murine DCs does not result in the development of a spontaneous allergic phenotype in FcεRI-humanized animals
The surface of humans DCs is coated with IgE bound to FcεRI.11-13 To study the consequences of IgE/FcεRI-mediated DC activation for the regulation of IgE-mediated allergic responses in vivo, we capitalized on the availability of a mouse strain that is humanized for FcεRI expression on DCs (referred to as IgER-TG;31). IgE is a constituent of the baseline polyclonal humoral response and is found in non-sensitized animals that are reared under specific pathogen-free (SPF) conditions. We first analyzed whether FcεRI expression on murine DCs (Supplementary Figure S1) alters the distribution of the baseline IgE pool in SPF-housed mice. Similar to healthy non-allergic humans (Figure 1a and 11, 12, 32), a fraction of DCs from non-sensitized IgER-TG mice bears IgE at the cell surface (Figure 1b and 1c). Importantly, the FcεRI-humanized mice do not display a gross-morphological allergic phenotype despite the presence of the DC-specific IgE pool. These cells are further comparable to human DCs (Figure 1a) in that the cells with the highest FcεRI expression and IgE binding capacity in the IgER-TG animals are CD8− DCs (Platzer et al manuscript under review and 31), which have been recently identified to be the homologous DC subset to human CD1c+ DCs.33, 34 This expression profile demonstrates that IgER-TG mice phenocopy the FcεRI- and IgE-binding pattern of human DCs. In contrast to IgER-TG DCs, DCs from wild type (WT) mice do not carry any surface IgE (Figure 1b). In BALB/c IgER-TG mice, a strain with higher baseline IgE levels than C57BL/6, the DC-bound IgE fraction was readily detectable at ≤8 weeks, and no age-dependent increase was found (Supplementary Figure S2a). IgER-TG mice on the C57BL/6 background showed a significant increase in the DC-bound IgE pool with age (Supplemental Supplementary Figure S2b). The age-dependent induction of IgG1 was comparable in IgER-TG and WT animals (Supplementary Figure S2c). In the absence of inflammation, serum IgE levels of IgER-TG mice were actually reduced when compared to WT controls (Figure 1c), likely due to a shift from the serum to the cell-bound IgE fraction. We further confirmed that comparable levels of CD23, the low affinity IgE receptor, are found on B cells in WT and IgER-TG animals but that CD23 is not expressed on DCs (Supplementary Figure S3). Thus, CD23 cannot contribute to the DC-bound IgE pool at steady state in mice.
Figure 1.

Expression of FcεRI on DCs results in a DC-specific IgE pool as seen in non-allergic humans. (a) Human peripheral blood DCs of a non-allergic individual carry surface IgE. DCs (in blue) were identified by gating on CD1c+ CD19− cells to distinguish them from CD1c+CD19+ B cells (in green) and analyzed for cell surface-bound IgE. DCs carry significant amounts of IgE but less than CD203+ basophils and mast cells (in purple). (b) CD11c+ DCs of IgER-TG mice carry IgE at steady state. Representative FACS plots of WT- and IgER-TG DCs isolated from spleens and quantification of the DC-bound IgE pool using mean fluorescence intensities (MFI) determined by flow cytometric analysis. (c) Baseline serum IgE levels are lower in non-sensitized IgER-TG animals. Symbols in are representative of individual mice of ≥ 2 independent experiments (*p < 0.05; ***p< 0.001).
In summary, these results show that IgER-TG mice accurately mimic the cell-bound fraction of the human IgE pool in the absence of inflammation and that FcεRI-bound IgE on DCs does not induce a spontaneous allergic phenotype.
Antigen-specific IgE/FcεRI crosslinking on DCs does not result in DC maturation or production of pro-inflammatory cytokines
Aiming at characterizing the consequences of IgE/FcεRI-crosslinking for DC activation, we next modeled antigen-specific signals via IgE/FcεRI. Splenic DCs from IgER-TG mice were loaded with monomeric hapten-specific IgE (NP-IgE), and haptenized antigen (NP-OVA) was used to engage surface FcεRI (Figure 2a). It has been previously described that DCs from IgER-TG animals express the trimeric receptor as a chimera of the human α-chain and the rodent γ-chains.31 We found that crosslinking of IgE/FcεRI induced rapid phosphorylation of spleen tyrosine kinase (Syk) and extracellular signal-regulated kinases (Erk1 and Erk2), which was not seen in identically treated WT DCs or after stimulation of DC with CpG DNA (Figure 2a). These results demonstrate that the signaling cascade down-stream of FcεRI on DCs involves signaling molecules that also have been described downstream of the tetrameric FcεRI in human and mouse mast cells.35
Figure 2.

IgE/FcεRI-crosslinking does not induce phenotypic maturation or production of inflammatory cytokines in DCs. (a) Antigen-mediated IgE/FcεRI activation induces phosphorylation of Syk and Erk1/2 in DCs. Splenic DCs were loaded with NP-specific IgE prior to incubation with NP-OVA (see schematic). IgER-TG DCs were also stimulated with CpG DNA, or antigen-crosslinking was omitted (NT = not treated). As a control, WT DCs were treated identically. Immunoblots for phospho-Syk, total Syk, phospho-Erk1/2 or total Erk1/2 are shown. (b) IgE/FcεRI-crosslinking fails to upregulate expression of maturation marker molecules in DCs from IgER-TG mice and (c) human monocyte-derived DCs. (d) Absence of cytokine secretion by splenic DCs upon antigen-specific IgE/FcεRI-crosslinking. Mean of triplicates +/− SEM, representative experiment (n=2); below detection level (bd) (e) TNF-α secretion from bone-marrow derived mast cells upon antigen-specific IgE/FcεRI-crosslinking. (f) Absence of transcriptional responses in murine DCs after antigen-specific IgE/FcεRI-crosslinking. mRNA expression was determined after 8 h. OVA uptake in the presence of CpG-DNA or papain was compared to IgE/FcεRI-mediated OVA uptake. Fold change compared to DCs that received OVA was calculated, and the mean of triplicates +/− SEM is shown, representative experiment (n=2).
After having confirmed that antigen-specific IgE/FcεRI-crosslinking induces a functional signaling cascade downstream of common γ-chain phosphorylation in DCs, we studied phenotypic maturation and cytokine production. Humanized DCs did not responded to antigen-specific IgE/FcεRI-crosslinking with upregulation of co-stimulatory molecules (Figure 2b), indicating that IgE signals do not provide a maturation stimulus. To exclude that the lack of DC maturation was an artifact of humanized FcεRI expression, we confirmed the absence of maturation signals in human monocyte-derived DCs after IgE/FcεRI-activation (Figure 2c). Analysis of culture supernatants from splenic DCs further demonstrated that neither TNF-α, IL-6, nor IL-10 were induced by IgE-mediated DC activation, although these mediators were readily detectable when DCs had been stimulated with CpG DNA or papain (Figure 2d). In contrast, identical IgE-mediated stimulation of mast cells from humanized FcεRI mice36 induced production of TNF-α (Figure 2e) as described for mast cells of WT animals.37 Microarray analysis of IgE/FcεRI-activated DCs confirmed the lack of induction of TNF-α or any other inflammatory mediator on the mRNA level (Supplementary Figure S4). To rule out that relative changes induced downstream of IgE/FcεRI were too subtle for detection by microarray, we additionally confirmed that no inflammatory cytokines are transcribed (Figure 2f and Supplementary Table S1), using digital mRNA profiling with sensitivity comparable to qRT-PCR.38 As a positive control for the functionality of the humanized DCs, we show that stimulation with CpG DNA, which is well-known to induce Th1-type immune responses, induced robust transcription of IL-12p40, TNF-α, and IL-1α, while papain induced the chemokine CCL-5 (RANTES), which is associated with Th2-type immune responses14 (Figure 2f). This set of experiments unequivocally demonstrates that antigen-specific IgE/FcεRI signals fail to induce a proinflammatory signature in DCs.
Mice with FcεRI-expressing DCs display diminished food allergic responses in vivo
To address how IgE/FcεRI-activation of DCs affects the severity of allergic responses in vivo, we turned to a well-characterized model of experimental food allergy that uses Th2-type sensitization with OVA/alum injections followed by repeated gavages of the model antigen to mimic physiological allergen exposure.39 First, we confirmed that in FcεRI-humanized mice DCs of the small intestine express FcεRI, an expression pattern likewise found in humans (Figure 3a and 3b, respectively). Comparable levels of sensitization were observed in WT and IgER-TG animals as determined by serum levels of antigen-specific IgE and IgG1 (Figure 3c and 3d) arguing against a pronounced role for DC-mediated FcεRI-facilitating serum IgE clearance30 during the sensitization phase in our model. As described for allergic humans,29 sensitization and challenge significantly increased the DC-bound IgE pool systemically in the spleen as well as locally in the mesenteric lymph nodes (Figure 3e and 3f, respectively). Even though OVA-specific IgE was readily detected in serum of WT mice (Figure 3e), no IgE was found on the DC surface in these animals (Figure 3e and 3f), indicating that the inducible murine form of FcεRI on DCs, which was described after house dust mite (HDM) exposure and in models of viral infection,22, 40 was not present in this model.
Figure 3.

Intestinal DCs express FcεRI and allergic sensitization increases the DC-bound IgE pool of IgER-TG animals. (a) FcεRI expression (in red) in the small intestine of IgER-TG mice is found on CD11c+ DCs (in blue). CD11c+ DCs from IgER-TG are GFP positive because the transgenic construct contains an IRES-GPF reporter element. (b) FcεRI expression on CD11c+ DCs in the human small intestine. Human FcεRIα (green, first panel), CD11c (red, second panel). Mast cells as depicted by c-kit are not found in non-inflamed human tissue using immunofluorescence (row two, red). DAPI-stain for nuclei (blue). (c) and (d) Analysis of OVA-specific IgE and IgG1 levels in serum of sensitized mice before antigen challenge. (e) and (f) Sensitization and antigen challenge increases the amount of surface-bound IgE on DCs of IgER-TG mice but not WT animals. DCs were isolated from the spleens and mesenteric lymph nodes (MLNs) of OVA-challenged WT and IgER-TG mice, respectively. Sensitized mice are indicated by (+), non-sensitized mice by (−). Symbols are representative of individual mice (n=2).
IgE-mediated food allergy presents with extensive Th2-type tissue inflammation in the small intestine, and intestinal mucosal mast cell numbers are an established quantitative measure of disease severity in mice and humans.39 Comparative quantitative mRNA profiling was performed to assess the extent of tissue inflammation in the small intestine of food-allergic animals. We found that the lamina propria of IgER-TG animals was overall substantially less inflamed (Figure 4a and Supplementary Table S2). mRNA transcripts of mast cell-specific proteases (MCPT-1 and MCPT-2, and carboxypeptidase A3), Th2-type cytokines (IL-4 and IL-13), and DC-derived inflammatory mediators such as CCL-2 and IL-6 were significantly less abundant in the tissue of IgER-TGs than in WT controls (Figure 4a). Expression of IFN-γ was low and did not change upon challenge (Supplementary Figure S5a). Additionally, significantly lower transcript levels of the Th2-specific cytokines IL-33 and IL-13 were found in the MLNs of IgER-TGs. In contrast, transcript levels of IL-2 and IL-12p40 and were comparable in both strains demonstrating a specific inhibition of cytokines associated with mucosal Th2 immune responses (Supplementary Figure S5b). We did not detect significant changes between challenged WT and IgER-TG animals in regard to percentages of Foxp3+CD25+ T cells in the MLNs (Supplementary Figure S5c). Furthermore, IL-10 production was similar (Supplementary Figure S5d), suggesting that the regulatory T cell compartment was not responsible for the restrained tissue inflammation in the IgER-TG mice.
Figure 4.

Food allergic responses are significantly reduced in IgER-TG animals. (a) Decreased mast cell-specific transcripts (i.e. mast cell proteases MCPT1, MCPT2, and carboxypeptidase A3 (CPA3)) and lower IL-4, IL-13, CCL-2, and IL-6 expression were detected in IgER-TGs. Sensitized mice are indicated by (+), non-sensitized mice by (−). Normalized mRNA counts or fold change in mRNA levels +/− SEM of small intestinal tissue of 4 independent experiments (per experiment tissue of ≥2 mice was pooled) are shown. (b) and (c) Small intestinal tissue sections were stained, and infiltrating mast cells were counted. (d) Assessment of mast cell protease MCPT1 in serum as a systemic readout for IgE-mediated mast cell activation. Serum was collected 1h after oral challenge. Symbols are representative of individual mice (n=2).
Chloroacetate esterase staining for mucosal mast cells on intestinal tissue sections confirmed that mast cell numbers were likewise profoundly decreased in antigen challenged IgER-TG animals (Figure 4b and 4c). To assess IgE-dependent mast cell activation systemically,41 we quantified serum MCPT-1 levels 1 h after antigen challenge. MCPT-1 was readily detectable after oral challenge and MCPT-1 levels in IgER-TG mice were significantly lower compared to WT animals (Figure 4d), demonstrating that systemic consequences of IgE-mediated mast cell activation are also dampened in FcεRI-humanized mice. These data demonstrate that DC-bound IgE pool has an inhibitory function that reduces mucosal inflammation as well as systemic responses during food allergy. This effect was only seen after oral challenge. When mice were systemically challenged intravenously, similar amounts of MCPT-1 were found in the serum of both strains (Supplementary Figure S5e). This finding further argues against enhanced IgE clearance as a mechanism of mitigated allergic inflammation in IgER-TG animals and suggests a tissue-specific anti-inflammatory function of DC-bound IgE.
Chronic asthmatic inflammation is diminished in mice with FcεRI-expressing DCs
We next aimed at analyzing whether the dampening of inflammation by the DC-specific IgE pool is a general regulatory principle for allergic responses or is, alternatively, specific for intestinal allergies. We here decided to study allergic asthma as the most common allergic disease worldwide.1 An earlier observation from our collaborators demonstrated an exacerbation of airway inflammation in FcεRI-humanized animals.31 Therefore, it was also important to revisit the effects of the DC-bound IgE pool on allergic asthma. Since our current study found a pronounced effect on the mast cell-dependent IgE-effector axis, we chose a chronic asthma model, which induces the disease by repeated intranasal antigen exposure without systemic sensitization. This model is currently considered closest to the induction of human asthma and is also characterized by increased tissue mast cell numbers.42, 43 First, we confirmed that, in contrast to CD11c+ DCs, CD11c+ macrophages in the lung do not express FcεRI or bind IgE (data not shown and 31). Applying this model, we found that IgER-TG mice exhibited less severe disease symptoms based on measurements of airway resistance (Figure 5a). Consistent with the food allergy model, a significant decrease in mast cell/basophil specific mRNA transcripts was detected in the lung (Figure 5b). Importantly, the protective features of the DC-bound IgE were also apparent at the cytokine/chemokine level. Transcripts of proinflammatory cytokines CCL-2, TNF-α, and IL-6 were overall decreased in lung tissue of IgER-TG mice (Figure 5c and Supplementary Figure S6a). The bronchoalveolar lavage fluid of IgER-TG mice contained fewer cells (Supplementary Figure S6b), including decreased numbers of neutrophils (Figure 5d), while similar numbers of eosinophils were detected (Figure 5d). Interestingly, an increase in CD11c+ cells was observed (Supplementary Figure S6b). No increase in IL-13 was detectable in the bronchoalveolar lavage fluid of IgER-TG mice (Supplementary Figure S6c). These data suggest that IgE/FcεRI-mediated DC activation predominantly affects mast cell-associated inflammation and might not affect eosinophils. Thus our results are not necessarily contradicting the study of Sallman et al. which focused on a model that mimics the early eosinophil-dependent phase of allergic asthma but not the chronic allergic lung inflammation, which involves mast cells. Importantly, our results show improvement of a clinical parameter of another chronic allergic disease in IgER-TG mice.
Figure 5.

Chronic allergic asthma presents with a less severe clinical phenotype in IgER-TG animals. (a) Airway hyper-responsiveness (airway resistance, Rn) without and with methacholine challenge. (b) Infiltration of mast cell/basophils in lung tissue quantified by using mRNA levels of the cell-type specific marker FcεRI β-chain. Symbols are representative of individual mice ≥ 2 independent experiments. (c) mRNA profiling of lung tissue after induction of allergic asthma. (d) Counts of neutrophils and eosinophils in the bronchoalveolar lavage fluid of sensitized (+) WT and IgER-TG mice. Symbols represent individual mice of 2 independent experiments. Due to the high biological variation of the tissue response no statistical significance was reached.
We next studied HDM-induced allergic airway inflammation to address whether high protease content of antigens combined with pattern recognition receptor (PRR)-activation can overcome the immune regulatory effect the DC-bound IgE pool. Unlike the food allergy model and the chronic airway inflammation model induced with OVA, the allergic pathology in the HDM model is not predominantly driven by mast cells. 44 As described for the HDM model, we observed more pronounced eosinophil infiltration than in the OVA sensitization model. Comparable to the OVA model, no significant difference of eosinophil infiltration in BALF or lung tissue was found in WT and IgER-TG mice (Supplementary Figure S7a). No significant infiltration of mast cells was detected in the HDM model, however, we found a trend for lower expression of CCL-2, IL-6 and TNF-α (Supplementary Figure S7b and S7c). Although tissue expression levels of IL-4, IL-13 and IL-5 were not lower in the lungs of IgER-TG mice, the CD4+T cells expressed significantly less IL-13 receptor (Supplementary Figure S7d and S7e). Importantly, a significant reduction of IL-33 was found (Supplementary Figure S7f), which is indicative for ameliorated lung inflammation in the IgER-TG mice. This set of experiments confirms in a third independent model of allergy that the production of pro-inflammatory Th2-type cytokines is not enhanced in IgER-TG mice and further demonstrates that even in the presence of strong protease and PRR signals allergic Th2-type inflammation is not amplified by the DC-bound IgE pool. Interestingly, the anti-inflammatory effects of the DC-bound IgE pool were more pronounced in models of allergy that depended heavily on mast cell infiltration for disease severity.
The DC-specific IgE pool restrains food allergy in mice that lack FcεRI expression on mast cells and basophils
To further study the immune regulatory features of IgE/FcεRI-mediated DC activation, we generated mice that lack expression of FcεRI on mast cells and basophils and express the IgE receptor only on DCs (IgER-TG x muα-KO, Figure 6a). DCs from IgER-TG x muα-KO mice were more densely loaded with IgE than cells from their parental humanized strain (Figure 6b), and OVA-specific serum IgE after challenge was significantly higher compared to IgER-TG and WT animals (Figure 6c). Notably, in mice that completely lack murine FcεRI (muα-KO), IgG1-mediated immune activation compensates for the lack of IgE-derived inflammatory signals, and muα-KO animals display all common signs of allergic inflammation.45 In line with this previous report, we found a severe allergic response in muα-KO and WT animals as determined by a profound increase of intestinal mast cell transcripts (Figure 6d). In contrast, IgER-TG x muα-KO mice displayed a significantly less severe allergic phenotype that was comparable to IgER-TG animals (Figure 6d). This data set confirms that the immune regulatory function of IgE is uniquely executed by the cell-bound DC-specific IgE fraction and does not involve IgE-mediated crosstalk with mast cells. Furthermore, the experiments show that DC-specific IgE signals restrain the allergic response beyond the inflammation that results from IgE-dependent mast cell and basophil activation.
Figure 6.
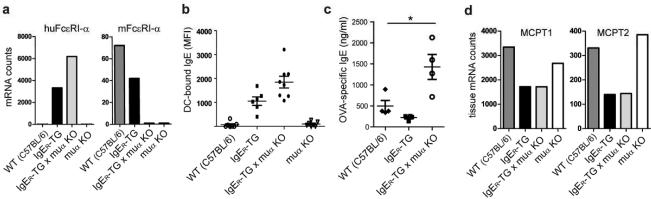
Reduction of allergic inflammation is regulated by the DC-bound IgE pool. (a) Expression patterns of murine and/or human FcεRIα-chains in WT mice, IgER-TG mice, and IgER-TG mice backcrossed onto the murine α-chain knock strain (muα KO). Backcrossed animals are referred to as IgER-TG mice x muα KO (b) DC-bound IgE is found exclusively on strains that express FcεRI on this cell type after OVA/alum sensitization. (c) Analysis of OVA-specific serum IgE after sensitization. (d) Comparison of intestinal mast cell-specific transcripts of MCPT1 and MCPT2 as readout for the severity of food allergy. Representative experiment (n=3); All animals used for experiments (a-d) were on the C57BL/6 background.
IgE-mediated antigen presentation does not promote generation of Th2-type effector T cells
The inhibitory activity of IgE/FcεRI-bearing DCs described above is in potential disagreement with the Th2-promoting function of IgE/FcεRI-mediated antigen presentation noted previously.31, 46 We therefore revisited the role of the IgE presentation pathway in T cell proliferation and Th2-type T cell priming. First, we confirmed the extraordinary sensitivity of this antigen presentation pathway31, 47 with DCs from IgER-TG animals. We isolated splenic DCs, pre-loaded them with NP-IgE and pulsed with NP-OVA for 1 h. We then injected these DCs into WT recipient mice to assess induction of T cell proliferation in vivo. Analysis of the draining lymph nodes revealed that only IgE-loaded DCs induced a strong proliferative response of adoptively transferred CFSE-labeled OVA-specific T cells in the low antigen concentration range (Figure 7a, second antigen concentration: Supplementary Figure S8a). In contrast to the Th2-promoting stimulus papain,14, 48 IgE/FcεRI mediated-antigen uptake by DCs failed to induce an inflammatory Th2 profile in CD4+T cells in vivo (Supplementary Figure S8b). To rule out that the Th2 response was simply under the detection limit in this assay, we confirmed these data with in vitro antigen presentation assays. Here, we altered the antigen loading conditions to allow for a more physiological setting. In contrast to previous studies where DCs were pulsed with antigen in the cold, our loading conditions allowed for IgE-dependent and IgE-independent antigen uptake in parallel over prolonged periods of time at 37°C. As noted previously, when IgE-loaded DCs from IgER-TG animals were used, T cell proliferation was induced in a low antigen concentration range that otherwise failed to initiate T cell responses. Titration experiments over a broader antigen concentration range demonstrated that IgE/FcεRI-mediated antigen uptake by DCs increased T cell proliferation most effectively at low concentrations (≤0.5 μg/ml), whereas no significant impact on the proliferative responses was observed at higher concentrations (Figure 7c). In line with previously published data,31 day 3 supernatants of the DC/T cell co-cultures contained more IL-4 and IL-13 when IgE/FcεRI-mediated uptake was used for antigen sampling (Figure 7d). However, antigen titration experiments showed that this early IL-4 production by T cells was a response to increased antigen uptake by the DCs at a low antigen concentration rather than a specific consequence of IgE/FcεRI signaling (Figure 7e). At the higher antigen concentration, we actually observed less IL-4 after IgE/FcεRI-mediated uptake when compared to fluid phase uptake (Figure 7e). Since priming of naïve T cells into fully differentiated T effector cells requires more than 3-4 days, we next analyzed the effector T cell phenotype at day 7 by staining for intracellular cytokines. Notably, T cells that were induced after IgE-mediated antigen presentation failed to differentiate into IL-4+ Th2 cells (Figure 7f). Since in vivo basophils and innate lymphoid cells have been demonstrated to provide additional IL-4, which is essential for Th2 cell priming via DCs 48, we added recombinant IL-4 to the DC/T cell co-cultures. Importantly, even in the presence of exogenous IL-4, IgE/FcεRI-mediated antigen presentation failed to generate more efficient Th2 effector responses than seen in the controls (Figure 7e and Supplementary Figure S8c). Addition of LPS to the DC/T cell co-cultures results in a Th1-type response as evident by the presence of a high percentage of IFN-γ+ T cells (Figure 7f). IgE/FcεRI-mediated antigen presentation did not diminish the induction of Th1 cells by LPS and rather reduced the numbers of IL-4+ T cells in LPS cultures (Figure 7f and Supplementary Figure S8c). To test if increased antigen uptake through IgE/FcεRI can promote Th1 responses, we added IL-12 or CpG DNA to the DC/T cell co-cultures. We detected more IFN-γ+ T cells in the presence of IL-12 and IgE (Figure 8g). Similar to IL-12, we found more IFN-γ and less IL-13 production in the presence of CpG DNA and IgE (Supplementary Figure S8d). This data suggest that IgE/FcεRI-mediated antigen uptake can, in fact, increase Th1 immune responses when the DCs receive Th1-supporting stimuli during antigen presentation. We also tested whether IgE/FcεRI-mediated antigen uptake could promote the priming of inducible regulatory T cells (iTreg). Addition of TGF-β1 to DC/T cell co-cultures resulted in the de novo generation of CD25+Foxp3+ iTregs and IL-10 expressing CD4+ T cells from purified naïve CD25− OT-II T cells. No significant difference in iTreg priming or IL-10+ CD4+ T cells was detectable in the presence or absence of IgE-mediated antigen uptake through DCs (Supplementary Figure S8e and S8f).
Figure 7.

IgE/FcεRI-mediated antigen presentation by DCs does not result in efficient generation of Th2-type effector T cells. (a) Induction of in vivo T cell proliferation. Splenic DCs were pulsed with NP-OVA in vitro (0.5 μg/ml antigen) in the presence (plus) or absence (no) of NP-specific IgE. Representative experiment (n=3). (b) Induction of in vitro T cell proliferation by DCs via IgE-mediated antigen uptake. Soluble OVA was present continuously; OVA-peptide323-339 was used as control. Mean of triplicates +/− SEM, representative experiment (n≥5). (c) In vitro DC/T cells proliferation assay in the presence of increasing antigen concentrations. Triplicates of representative experiment (n=2). (d) IL-4 and IL13 in DC/T cell co-culture supernatant at day 3. (e) IL-4 and IFN-γ increase in culture supernatants when DC-bound IgE is used for antigen uptake. Expression levels of IL-4 mRNA are compared in T cells that were induced IgE-independently (no IgE) or via the IgE/ FcεRI-mediated uptake (plus IgE). (f) Intracellular cytokine staining for IL-4 and IFN-γ in T cells. Recombinant IL-4 or LPS was added to the antigen presentation assays as indicated. Bar diagram shows percentages of IL-4+ and IFN-γ CD4+ T cells in DC/T cell co-cultures. Data represent triplicates of biological replicates +/− SEM of a representative experiment (n=3). Representative FACS blots depict gated CD4+ T cells at day 7. (g) Recombinant IL-12 was added to the co-cultures and priming of Th1 cells was determined by intracellular staining for IFN-γ at day 7. Data represent triplicates of biological replicates +/− SEM of a representative experiment (n=2).
Figure 8.

Antigen-specific IgE/FcεRI-crosslinking on activated DCs inhibits the production of proinflammatory cytokines and chemokines resulting in impaired migration of inflammatory cells. (a) IgE-crosslinking inhibits the papain-induced production of CCL-2, IL-6, and TNF-α by murine DCs. (b) Inhibition of CCL-2 production in human DCs by IgE/FcεRI-crosslinking. (c) The chemokine CCL-2 shows strong chemotactic activity for c-Kit+ mast cell progenitors and CD11c+ DC. Indicated concentrations of recombinant CCL-2 were added to the medium in the lower wells of transwell plates and the migration of cells from mast cell progenitor cultures towards the chemokine was determined. (d) Antigen-specific IgE/FcεRI-mediated DC activation (OVA plus IgE) inhibits migration of bone-marrow derived mast cell progenitors and DC. DCs were activated as indicated and DC-conditioned supernatants were used for chemotaxis assays. Migratory response of cKit+ cells and CD11c+ cells towards DC-conditioned supernatants was calculated from 3 independent experiments. Migratory response of DC stimulated with LPS or papain in the absence of IgE/FcεRI-mediated DC activation (OVA alone) was set at 100 %.
In conclusion, we confirmed that IgE-mediated antigen presentation enables DCs to initiate T cell proliferation in response to extraordinarily low doses of soluble antigen. The ensuing T cells, however, failed to fully differentiate into a Th2 effector type. This result is in line with our in vivo experiments in IgER-TGs mice, where no evidence of increased Th2-type T cell responses was detected (Figure 4a, Supplementary Figures S5b and S7d).
Cross-linking of IgE/FcεRI inhibits production of pro-inflammatory mediators from activated DCs and thereby interferes with recruitment of mast cell progenitors
So far we showed that antigen-specific IgE/FcεRI-crosslinking does not induce phenotypic DC maturation, that the DC-specific IgE pool does not promote Th2-type inflammation in vivo, and that IgE-mediated antigen presentation fails to efficiently induce Th2 effector cells. In summary, these findings imply an immune regulatory function of DC-specific IgE signals. We next turned our attention to identifying the cellular basis of the inhibitory function of IgE/FcεRI-mediated DC activation observed in the in vivo studies describe above. To this end we determined the responses of DCs stimulated with papain and low dose LPS, i.e., stimuli that mimic DC activation occurring during allergic responses in vitro and then measured the cytokine response to such stimulation. 48, 49 Based on the suppression of cytokine patterns in our in vivo experiments (Figure 4 and 5), we selected CCL-2, IL-6, and TNF-α as likely candidates for being subjected to regulatory IgE signals in activated DCs. In vitro stimulation with papain strongly induced CCL-2, IL-6, and TNF-α production by DCs (Figure 8a). Importantly, in the presence of antigen-specific IgE/FcεRI signals, significantly lower amounts of all three inflammatory mediators were detected (Figure 8a). IgE/FcεRI cross-linking also inhibited inflammatory cytokine production in response to low dose LPS (Supplementary Figure S9a). We detected no significant difference whether FcεRI cross-linking occurred simultaneously or consecutively to LPS stimulation. In a time course experiment, we saw that the inhibitory effect of IgE activation on inflammatory cytokine production was not detectable after 6h, already measurable after 24h, but most evident after 48h (Supplemental Figure S9b). This experiment indicates that IgE-derived inhibitory signals are cumulative over time as evident by the strongest inhibition at later time points.
To further investigate the different outcomes on cytokine production in mast cells and DCs, we compared side-by-side the signaling cascades after IgE/FcεRI cross-linking (Supplementary Figure S9c). We found similar levels of total ERK1/2 in both cell types, but stronger ERK1/2 phosphorylation upon IgE/FcεRI crosslinking was observed in mast cells compared to DCs. No inhibition of ERK1/2 phosphorylation upon IgE/FcεRI crosslinking in the presence of LPS was detected in the DCs (Supplementary Figure S9d). When analyzing SHP-1 and SHIP-1, two phosphatases that have been described to regulate ITAM signaling, we found higher expression of both proteins in DCs compared to mast cells (Supplementary Figure S9c). We did not detect an increase in phosphorylation of SHIP-1 in DCs upon receptor crosslinking with 0.5 μg/ml OVA, the concentration commonly used in our experiments. Since SHP-1-dependent inhibitory ITAM signal have been demonstrated for FcγRIII,50 we also tested whether FcεRI crosslinking in the presence of LPS would lead to SHP-1 phosphorylation. We did not detect an increase in pSHP-1 (Supplementary Figure S9d). Thus, the pathway operative downstream of trimeric FcεRI in DCs seems different from those described so far for mast cells and other inhibitory Fc receptors.
We next validated the presence of inhibitory IgE-mediated signals for its relevance to humans by demonstrating that activated human DCs also produce significantly lower amounts of CCL-2 upon IgE-mediated activation (Figure 8b). High donor variability with regards to responses to papain stimulation was found in humans. Papain responsive DCs showed the same inhibitory pattern of CCL-2 production upon antigen-specific IgE crosslinking as LPS-stimulated cells (Figure 8b). This set of experiments demonstrates that antigen-specific IgE/FcεRI-crosslinking on activated DCs reduces, rather than promotes, the production of pro-inflammatory mediators. Since human DCs show the same restrained cytokine/chemokine release after IgE/FcεRI activation, we provide evidence that our findings directly translate to human DC function.
CCL-2, also known as monocyte chemotactic protein 1 (MCP-1), attracted our attention particularly because it exhibits strong chemotactic activity for CD117+ (KIT+) mast cell progenitor cells and DCs, in addition to its function in the recruitment of monocytes and T cells to sites of inflammation 51-54. Since repeated antigen challenges increase allergic tissue inflammation over time through the influx of inflammatory progenitor cells, we hypothesized that inhibition of CCL-2 production by antigen-specific IgE signals should impair trafficking of mast cell progenitors. To test this hypothesis, we first confirmed the chemotactic properties of CCL-2 for murine mast cell progenitors and CD11c+ DCs (Figure 8c). Next, we performed chemotaxis assays using conditioned supernatants from IgE-preloaded DCs, which we activated with papain or LPS in the presence or absence of antigen-mediated crosslinking. Papain and LPS activation induced the production of cell supernatants with strong chemotactic activity for mast cell progenitors as well as CD11c+ DCs (Supplementary Figure S10). In line with our hypothesis we found that migration of mast cell progenitors and CD11c+ DCs from bone marrow progenitor cultures was reduced in average by over 30% to 50%, respectively, when supernatants from IgE/FcεRI-activated DCs were used (Figure 8d). Antigen-specific IgE/FcεRI-crosslinking, however, did not interfere with LPS-induced upregulation of co-stimulatory molecules, such as CD86, by neither mouse nor human DCs (Supplementary Figure S11 and data not shown).
In summary, these experiments demonstrate that IgE-mediated signals interfere with the production of inflammatory, Th2-promoting mediators by activated DCs. In an allergic setting in vivo, inhibited production of these mediators explains why the allergic tissue is overall less inflamed in IgER-TG mice (see model, Supplementary Figure S12). Additionally, the decreased production of the mast cell-tropic chemokine CCL-2 implies that the dampening of the IgE-effector cell axis is due to the diminished inflammatory progenitor cell recruitment to sites of allergic tissue inflammation.
DISCUSSION
This study on the functions of DC-bound IgE reveals to our knowledge an unidentified immune regulatory mechanism mediated by antigen-specific crosslinking of IgE/FcεRI on DCs. We found that IgER-TG animals, which mimic the constitutive IgE receptor expression pattern of humans and therefore bind IgE at the cell surface of DCs, suffer from significantly less severe clinical symptoms of allergies than WT animals. This protective feature of IgE-mediated immune activation was likely not detected to date because the DC-specific IgE pool is not found in laboratory WT mice. Our study thus adds allergic Th2-type diseases to the list of human conditions that are not fully modeled by the murine immune system in their entire complexity.55
Comparable sensitization levels were detected in FcεRI-humanized and WT mice arguing against a redistribution of the available IgE pool from serum to the DC-bound fraction as a simple explanation for impeded allergic tissue responses. Additionally, the availability of similar amounts of antigen-specific IgE for mast cell activation was confirmed by demonstrating that mast cell degranulation after systemic antigen exposure is similar in IgER-TG and WT mice. Our findings do not exclude that FcεRI on DCs also contributes to serum IgE clearance as described recently.56 We show, however, that DC-bound IgE has an additional active role in regulating mucosal allergic responses on the tissue level.
A mechanistic explanation for the observation that the DC-bound IgE pool executes regulatory rather than pro-allergic signals derives from our demonstration that IgE/FcεRI-crosslinking on activated DCs inhibits production of allergy-promoting inflammatory cytokines. Furthermore, decreased generation of chemokines indicates that lower mast cell numbers in allergic tissues of IgER-TG animals result from less efficient recruitment of inflammatory cells to the affected tissue. Our experiments clearly identify CCL-2 as one of the key chemokines that regulates trafficking of mast cell precursors under the direct control of DC-specific IgE signals. The observation that decreased levels of CCL-2 result in ameliorated allergy is also supported by the literature, demonstrating that CCL-2 serves as an amplifier of pro-inflammatory effector functions of mast cells and basophils during allergic airway inflammation.57-60
The anti-inflammatory role of IgE-mediated DC activation directly opposes the pro-inflammatory role of IgE/FcεRI on mast cells/basophils. How can these obviously cell type-specific outcomes be explained? Although the signaling cascade in DCs appears comparable to the one induced in mast cells by IgE/FcεRI crosslinking, the lack of the signal-amplifying β-chain in the DC-specific trimeric FcεRI isoform is an obvious difference between the two cell types. Thus, differences in signal strength and/or activation of alternative signaling cascades might explain the divergent functional outcomes. Importantly, our finding that Syk phosphorylation via IgE/FcεRI crosslinking in DCs inhibits cytokine production is not the first demonstration of an inhibitory signal by this kinase. Distinct immunoregulatory roles for Syk signaling through several types of ITAM-associated receptors have been described, including CD89 (the high affinity IgA Fc receptor), β2-integrins, NKp44, Siglec-H, immunoglobulin like transcript 7 (ILT7), and CD303.61-67 For example, Syk activation downstream of the integrin CD11b has been shown to inhibit inflammatory responses triggered by TLR signaling in innate immune responses.67, 68 Additionally, it has been demonstrated that Syk signaling can fine-tune innate immune responses to lessen inflammation.69 Mechanistically, the inhibitory signals mediated by the diverse receptors have been demonstrated to include indirect ITAM cross-regulatory mechanisms, ITAM-mediated sequestration of effector molecules as well as recruitment of inhibitory effectors, like the Scr homology2 domain-containing phosphatase (SHP-1).61, 70 Since the IgE/FcεRI interaction is of a very high affinity and we cross-linked the receptor with haptenized antigen, we speculate that the inhibitory mechanism of IgE/FcεRI signaling in DCs probably differs from the inhibitory signals induced by the monomeric low-avidity ligation of CD89 by IgA, which functions through recruitment of SHP-1.66, 71 How Syk activation after IgE/FcεRI crosslinking exactly interferes with DC activation and results in the downregulation of cytokine production will be an important question to address in the future.
We further demonstrated that antigen presentation via the DC-bound IgE pool does not promote expansion of Th2-type effector T cells. Thus, our data suggest that this pathway of antigen recognition does not fuel the Th2-perpetuating immune machinery in allergy. Our findings are in line with several epidemiologic observations, which also challenge the previous interpretation that IgE/FcεRI signals provide DCs with Th2-promoting stimuli. First, although human DCs are preloaded with IgE independent of the age or the allergic status of an individual, not every human develops allergy. Secondly, while IgE/FcεRI expression on DCs appears to correlate with serum IgE,29 high IgE levels are frequently found in the absence of an allergic phenotype.11, 12, 32
It is important to discuss why IgE/FcεRI-mediated antigen presentation by DCs has traditionally been believed to foster generation of allergic T cell responses. The IgE-mediated presentation pathway was originally described with birch pollen-specific T cell clones and HLA-matched DCs from allergic donors.6, 47 The use of established human T cell clones is a shortcoming in this setting because obtained data do not allow for conclusions with regards to priming of naïve T cells or their differentiation towards specific T effector cells. Only the availability of DCs from FcεRI-humanized mice, which can be analyzed in combination with primary antigen-specific T cells, allows for an evaluation of the IgE/FcεRI presentation pathway for the generation of Th2 effector cells. The intracellular cytokine staining of T cells generated with this strategy in our study demonstrated convincingly that the IgE/FcεRI pathway is not superior in its ability to induce Th2-type effector T cells. However, we confirmed earlier observations that IgE/FcεRI-mediated antigen uptake induces T cell proliferation to otherwise sub-stimulatory antigen concentrations.31, 47 With regards to this question, in contrast to previous studies where the antigen binding to IgE/FcεRI of was performed at 4°C to restrict fluid phase antigen uptake,6, 31 our experimental conditions of continuous antigen availability at 37°C allowed for the first straightforward comparison of IgE-dependent and independent uptake pathways. Thus, these experiments better replicate the in vivo situation where DCs use multiple antigen sampling pathways simultaneously. We cannot formally exclude that the increased T cell proliferation and early IL-4 production initiated through IgE-mediated antigen uptake may contribute to Th2 responses under certain circumstances. However, the in depth characterization of IgE/FcεRI-mediated antigen presentation by DCs as presented in our study ranks this event very low as a causative candidate for initiating allergen-specific Th2 cells.
Targeting FcεRI on DCs with antigen-IgE fusion proteins was recently demonstrated as a pathway to induce antigen-specific T cell tolerance in FcεRI-humanized mice.72 It is important to stress that the major difference of this study to ours was the use of monomeric IgE fused with antigen. Such an IgE fusion binds to FcεRI but does not allow for crosslinking as seen with antigen in vivo. The intracellular pathway used by monomerically-engaged FcεRI is unlikely to be operative in our models where internalization is induced by multimeric ligation. Irrespectively, targeting an FcεRI-mediated antigen uptake pathway to specifically induce T cell deletion is an interesting approach to make use of non-inflammatory IgE-mediated antigen presentation events.
At first glance, the immunoprotective characteristics of the DC-specific IgE pool stand in contrast to a previous report that demonstrated exacerbation of early eosinophil-dependent airway inflammation in IgER-TGs mice.31 Note that the study of Sallman et al. used a different, less physiological sensitization approach than we did in our asthma models. Additionally, this study did not evaluate airway resistance as a clinical parameter of allergic asthma. Even though our experiments support the protective nature of DC-specific IgE in allergic lungs by additionally demonstrating an overall decrease in inflammatory cytokines/chemokines and the absence of increased eosinophil numbers, our data do not formally exclude that early eosinophil-dependent inflammation is enhanced in the humanized animals. Irrespectively, we demonstrate that the consequences of DC-intrinsic IgE signals for chronic allergic inflammation are an improved clinical phenotype and not more severe asthma symptoms. We also argue that the immunoregulatory mechanism of IgE is supported by the description of an inducible form of FcεRI on murine inflammatory DCs in the lung.22, 40 The authors originally interpreted induction of FcεRI on DCs as a mechanism to promote allergic inflammation. At the time of these studies, such interpretations were the most plausible based on the current paradigms of IgE-mediated immune activation. Based on our findings, we propose as an alternative explanation that FcεRI is induced in mice as part of a counter-regulatory mechanism aimed at dampening allergic inflammation. The relevance of this new interpretation for human allergy is supported by our finding that human monocyte-derived DCs, which are generally considered to be an inflammation-associated DC subset, show the same immune-suppressive pathways of IgE-mediated cell activation.
However, during the development of human allergy, regulatory signals from the DC-bound IgE pool are obviously not sufficient to prevent or resolve overshooting inflammatory Th2-type response. It is here important to keep in mind that the consequences of IgE-triggering via constitutively expressed FcεRI on DCs cannot automatically be translated to functions of the inducible form of trimeric FcεRI on innate cells such as neutrophils or monocytes as described in allergic humans.7, 11 It is conceivable that IgE signals via inducible trimeric FcεRI contribute to the exacerbation of allergy.
We propose that IgE-mediated DC activation acts as a checkpoint and serves as an inhibitory feedback mechanism to down-regulate inflammation during allergic responses. It is important to keep in mind that overshooting inflammatory Th2-type immune responses are not only seen in allergy, but can occur during encounters with helminth parasites.73 Amongst the detrimental consequences of the failure to restrain Th2 responses is tissue fibrosis.74 The existence of a counter-regulatory feedback mechanism that controls the magnitude of these immune responses via antigen-specific IgE seems thus highly beneficial and evolutionarily justified.
In summary, we suggest that re-examining our view of IgE is warranted because the anti-inflammatory DC-specific functions demonstrated here challenge the current notion of this immunoglobulin as a dominantly pro-inflammatory mediator. Additionally, our findings may have the potential to serve as the basis for novel therapeutic strategies for the treatment of allergy by exploiting the DC-based immunoregulatory features of IgE.
MATERIALS AND METHODS
Mice
All animals were bred under SPF conditions at Boston Children's Hospital (Boston, MA). In vivo experiments were performed in littermate-controlled settings. Age-matched WT mice that had been co-housed after weaning were used in addition to littermates. OT-II TCR transgenic animals were purchased from The Jackson Laboratory. All animal studies were approved by the Boston Children's Hospital or the Brigham and Women's Hospital Institutional Animal Care and Use Committees.
Antibodies and Reagents
Hapten-specific chimeric human IgE anti- 4-hydroxy-3-nitrophenylacetyl (NP) was derived from Jw 8/5/13 cells (clone JW8/1, AbD Serotech). NP(7)-PE and NP-OVA (~19 NP-moieties per OVA) were purchased from Biosearch Technologies Inc. NP-OVA was size purified (10kDa > NP-OVA < 100 kDa) to remove OVA peptide and higher MW OVA aggregates in the soluble antigen solution with Amicon Ultra Ultracel 10k and 100k centrifugal filters (EMD Millipore).
Flow cytometry
Single cell suspensions were pretreated with mouse Fc-Block™ (BD Biosciences/ Pharmingen) and incubated with fluorophore-labeled antibodies or appropriate isotype controls. Acquisitions were performed using a BD FACSCanto™ II (BD Bioscience). For intracellular staining of cytokines BD Cytofix/Cytoperm (BD Biosciences) was used. Analysis was done using BD FACSDiva™ and FlowJo (Treestar INC.) software. Following antibodies were used: Phycoerythrin-(PE), allophycocyanin-(APC) and PE/Cy7 anti-human FcεRIα mAb CRA1 (clone AER-37, eBioscience or BioLegend), PE-anti-mouse IgE (RME-1, Biolegend), APC-anti-CD11c, Alexa Fluor® 647 anti-mouse CD8α and Alexa Fluor® 647 anti-mouse CD4 from Biolegend (San Diego, CA), PE-anti-mouse CD86 (BD Pharmingen), APC-anti- CD117 (Biolegend), and PE- anti-mouse FcεRIα mAb (Mar1, Biolegend). Intracellular cytokine stain included PE anti-mouse IFN-γ (clone XMG1.2, Biolegend) and APC anti-mouse IL-4 (clone 11B11, eBioscience). Anti-human CD1c (BDCA-1, clone AD5-8E7, Miltenyi Biotech), APC-anti-human CD19 (clone HIB19, BioLegend), PE/Cy7 anti- human IgE (clone MHE-18, BioLegend), PE anti-human CD203 (Biolegend), Alexa Fluor® anti human CD83 (clone HB15e), PE anti-human CD80 (Biolegend) and APC anti-human CD1a (BD Pharmingen).
Preparation of single cell suspensions from spleens and mesenteric lymph nodes
Tissue was digested with either Collagenase Type VIII or Collagenase D (Sigma) for 30 minutes and homogenized using 70 μm cell strainers (BD Biosciences). Red blood cells were lysed using 1x RBC lysis buffer (eBioscience).
Measurement of immunoglobulins
Serum levels of total and OVA-specific IgE and IgG1 and were determined by ELISA. Total IgG1 was determined using the mouse IgG1 ELISA Ready-SET-Go! from eBioscience. For total IgE, 96-well plates were coated overnight at 4°C with goat isotype-specific antibodies to IgE (1110-01, Southern Biotech) diluted 1:1000 in coating buffer (eBioscience). After six washes with 0.05% Tween 20 (Sigma) in PBS, plates were blocked with 2% (w/v) bovine serum albumin (BSA, Sigma) in PBS (Life Technologies) for 4 h. Serum samples were diluted 25-200 fold and incubated overnight at 4°C. HRP-conjugated anti-mouse IgE (1110-05, Southern Biotech) was added for 1 h prior to development with tetramethylenbenzidine (KPL). After addition of 2 M H2SO4, spectrophotometric analysis at 450 nm (Spectramax 250, Molecular Devices) was obtained and serum Ig concentrations were calculated from a standard curves with a commercially available IgE antibody (557079, BD Pharmingen). For OVA-specific IgE and IgG1 the above described protocol was adjusted as follows: Plates were coated with goat α-mouse IgE (d1:1000, Southern Biotech) or goat anti-mouse IgG1 (Southern Biotech), respectively. For the standard curves mouse anti-OVA specific IgE (AbD Serotech) or mouse anti-OVA IgG1 (Sigma Aldrich) were used. Detection was performed by incubating with biotinylated OVA (1 μg/ml) for 1h, followed by Streptavidin-HRP (BD Pharmingen) for 30min.
Measurement of cytokine production
Cytokine production was determined using commercially available Ready SET Go ELISAs!® (eBioscience) or was assessed using the BD™ Cytometric Bead Array and analyzed using FCAP Array™ Software v1.0.1 (BD Bioscience).
In vitro stimulation of murine DCs
Bone marrow-derived or splenic DCs were stimulated with 25 μg/ml papain (Calbiochem), 500 ng/ml CpG DNA (ODN 1668), and/or NP-OVA (0.5 μg/ml) in the presence or absence of NP-specific IgE.
Cell culture and generation of bone marrow (bm)-derived DC
DCs were cultured in RPMI-1640, 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. T cell cultures were supplemented with 55 μM β-mercaptoethanol. Bone marrow was collected and cultured as described.75, 76 For generation of DCs, bone marrow cells were cultured at 1.5 × 106 cells/ml with 200 ng/ml human Flt3-ligand (Peprotech) for 8 – 10 days.
Human monocyte-derived DCs
Monocytes were isolated from healthy blood donors (IRB approved and provided by the Kraft Family Blood Donor Center of the Dana-Farber Cancer Institute and Brigham and Women's Hospital) using the MACS CD14+ positive selection kit (Miltenyi Biotech). Cells were cultured with recombinant human GM-CSF (1000 U/ml) and IL-4 (500 U/ml) for 5 days. FcεRI expression was confirmed by FACS. To obtain highly pure DC populations, CD1a+ cells preloaded with NP-specific IgE were purified using MACS microbeads (Miltenyi Biotech). 5×105 DCs were cultured in 1ml RPMI in 48 well plates. DCs were activated as indicated with 0.05 μg/ml NP-OVA, 100 ng/ml LPS, or 25 μg/ml papain. NP-OVA was added 5 to 10 min prior to LPS or papain. Cells and culture supernatants were collected after 16h. CCL-2 was measured using the human CCL-2 Ready SET Go ELISAs!® (eBioscience).
Food allergy model
Intestinal allergic reactions were induced as previously described (Ahrens et al., 2012). Briefly, mice were sensitized with 100 μg of OVA (grade V, Sigma-Aldrich) in a 1:1 alum adjuvant suspension (Imject Alum Adjuvant, Thermo Scientific) by intraperitonial (i.p.) injection on days 0 and 14. Control mice received alum alone. Briefly, commencing on day 28, all mice were challenged with 50 mg of OVA on alternating days for a total of 3 to 6 administrations. Mice were deprived of food for 3 hours prior to each challenge. Tissue was harvested 24h after the last challenge. Intestinal mast cell quantification jejunum tissue was collected 10–12 cm distal to the stomach and processed by standard histological techniques. Tissue sections were stained for mucosal mast cells with chloroacetate esterase activity.77 15 high power fields of tissue from 3 IgER-TG and 3 WT mice were counted by two blinded investigators.
Asthma model
Mice were sensitized to OVA by repeated intranasal applications 3 times a week for a total of 10 applications as previously described (Williams and Galli, 2000; Yu et al., 2006). Sensitization was confirmed by the presence of OVA-specific IgE in serum. Mice were subjected to analysis 24h after the last OVA application. Measurement of airway resistance: Methacholine challenge test: Mice were anesthetized with sodium pentobarbital (100mg/Kg, i.p., Oak Pharmaceuticals), tracheotomized, and connected to the Flexivent rodent ventilator (Scireq) via a tracheal cannula. Methacholine (Sigma Chemical Company) was aerosolized trough the inhalation port for 10 seconds, and the airway resistance (Rn) was measured at a positive end-expiratory pressure (PEEP) of 2.5 cm H2O, as described.78 For unprovoked lung physiology studies, mice were anesthetized and connected to the Flexivent ventilator as above. After unprovoked lung physiology studies, lungs were lavaged trough the tracheal cannula to collect bronchoalveolar lavage (BAL) fluid. Total cells in the BAL were determined; neutrophils and eosinophil numbers were obtained after staining a cytospin with Hema 3 Staining kit (Fisher Scientific, MI). For the collection of lung tissue, mice were exanguinated through right ventricular puncture and pulmonary circulation was perfused via the right ventricle. For HDM induced airway inflammation, mice were sensitized once intranasal with 5 μg house dust mite (HDM) extract (Greer Labs) per mouse. Commencing 1 week after the initial sensitization, mice were challenged with 25μg HDM per mouse, 3 times a week for a total of 8 intranasal applications. Single cell suspensions of lung tissue for flow cytometric analysis were prepared using the Lung Dissociation Kit from Miltenyi Biotech.
Chemotaxis assay with mast cell progenitors
DCs were activated with papain or LPS in the presence or absence of antigen-specific IgE/FcεRI crosslinking (NP-OVA was added 5 min prior to LPS or papain and supernatants of these DC cultures were harvested after 30h. DC-derived supernatants were diluted 1:2 with fresh medium and placed into the lower wells of 24 well transwell plates with polycarbonate membrane filters (5 μm pore size; Corning). Mast cell progenitor cultures were set up as described (Collington et al., 2010). Briefly, bone marrow cells were isolated and cultured for 4 to 5 days with 10 ng/ml IL-3 and 50 ng/ml SCF (kindly provided by Dr. M. Gurish), and 5×105 cells (in 100 μl, average cell size 10 μm) were placed into the upper chambers of the transwell plates. Migrated cells were collected after 2 h, stained for cell type specific marker molecules, and analyzed by flow cytometry. To quantify cell migration, CountBright Counting Beads (Invitrogen) were used.
mRNA expression analysis
Digital mRNA profiling was performed using multiplexed mRNA expression analysis applying the nCounter™ assay system (NanoString Technologies). Tissue was harvested and rapidly transferred into RNAlater solution (Ambion) and stored at −80°C. Tissue was homogenized in RLT Buffer (Qiagen) with β-mercaptoethanol (Sigma) using the GentleMACS™ Dissociator with GentleMACS M tubes (Miltenyi Biotech). Tissue lysates of small intestine, lungs or purified RNA (100 ng) from DCs were hybridized with customized nCounter gene expression code sets and direct mRNA counts were determined by the nCounter® Digital Analyzer System according to the manufacturer's protocol. mRNA counts were first normalized to internal positive and negative controls before normalization to 5 house-keeping genes (i.e. β-actin, HPRT-1, HSP90ab1, Shfm-1, and Tomm7). Data are presented as direct mRNA counts or as fold difference compared to control treated samples.
Immunoblotting
Cells were lysed in RIPA buffer (Santa Cruz Biotechnologies) containing protease inhibitors (Complete, Roche) and 2mM sodium ortho-vanadate. Samples were run on 10% SDS-PAGE gels, transferred to PVDF membranes (Pierce) and probed with following antibodies: anti-Syk and anti-phospho SYK, anti-ERK1/2 and anti-phospho ERK1/2 (all from Cell Signaling Technology Inc.) followed by peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG. Peroxidase activity was detected using SuperSignal chemiluminescent substrate reagents (Pierce).
Microscopy
Frozen small intestinal tissue sections of IgER-TG mice were stained with PE-labeled mouse anti-human FcεRI (Cra1; eBioscience) and Alexa Fluor® 647labeled- anti mouse CD11c. Specificity of the staining was confirmed with appropriate isotype control antibodies and tissue from WT mice. Immunofluorescence histochemistry of human GI tissue was performed as described using discarded tissue of a published cohort 79. All microscopy was performed at the Harvard Digestive Disease Center Core Facility.
T cell proliferation assays
DCs were isolated using CD11c MicroBeads (Miltenyi Biotec). Purity (>90%) was assessed by FACS. To preload DCs with IgE, cells were cultured over night at 37°C with NP-specific IgE (500 ng/ml). For T cell isolation OT-II mice, the CD4+ T Cell Isolation Kit II (Miltenyi Biotec) was used. Purified T cells were labeled using the CellTrace™ CFSE Cell Proliferation Kit (Molecular Probes). Prior to loading with antigen, unbound IgE was removed. In vitro assays were performed in the continuous presence of indicated antigen concentrations. Antigen presenting cells were plated with CFSE-labeled CD4+ T cells at 1:2 to 1:4 ratios. For in vivo experiments, DCs (1-2 × 106 cells/ml) were pulsed for 1 h with NP-OVA at concentrations of 0.05μg/ml or 0.5μg/ml. 2.5 ×105 DCs loaded with IgE and NP-OVA or equal DC numbers loaded only with NP-OVA were injected into opposite hind footpads of WT mice adoptively transferred with CFSE-labeled OT-II T cells. Popliteal lymph nodes were harvested after 72h. For determining intracellular cytokine production, OT-II T cells were cultured with DCs at 1:5 ratios in 48-well plates with or without recombinant IL-4 (15 ng/ml, Peprotech) or LPS (1 μg/ml). On day 3 and 5, half of the culture medium was exchanged, and IL-4 and/or IL-2 (20 ng/ml) were added. On day 6 or 7, cells were restimulated with PMA and ionomycin for 4 h. Brefeldin A (BFA) was added for the last 3 h. For the generation of Th1 cells IL-12 (25ng/ml) or CpG DNA (500ng/ml) were added to the co-cultures. For the iTreg assays, purified CD4+ OT-II cells were depleted for CD25+ cells using the CD25 MicroBead Kit (Miltenyi Biotec). T cells were co-cultured in 96 well plates at 1:3 ratios with DCs in the presence of 5ng/ml TGF-β1. On day 3 half of the culture medium was exchanged, and fresh TGF-β1 and IL-2 (20 ng/ml) were added. On day 6, cells were analyzed by intracellular staining for Foxp3 and IL-10 using the Foxp3 staining Kit from eBiosciences.
Statistical Analysis
Data are presented as mean +/− SEM of ≥ 3 independent experiments unless stated otherwise. Statistical analysis was performed using PRISM software (GraphPad Software Inc). Significance was assessed using unpaired two-tailed Student's t tests for all comparisons between two populations or one way ANOVA tests for multiple comparisons with Bonferroni's or Tukey's post tests.
Supplementary Material
ACKNOWLEDGMENTS
We want to thank the large number of intellectually generous colleagues that provided suggestions for this manuscript, especially Drs, M. Neutra and M.-H. Jouvin. We thank M. Pardo, S. Schopoff, M. Stout, B. Sallis and C. Schultz for technical assistance, Dr. O. Burton for assistance with the staining of mucosal mast cells, and Dr. E. Dehlink for providing human tissue samples. This work was supported by grants from the National Institutes of Health: K01DK093597 (to B.P), DK53056 (to R.S.B), R37GM053950 (to J.-P. K.), R01HL091957 (to R.M.B.) and AI075037 (to E.F.). K.B was supported by the Canadian Institutes of Health Research. W.S. L. was supported by grants from the Ter Meulen Fund, the Royal Netherlands Academy of Arts and Sciences and the Banning de Jong Fund. This work was also supported by the Harvard Digestive Diseases Center Grant P30DK034854.
Footnotes
Author contributions
B.P., K.B., M.P.V., K.S., M.P., W.S.L., D.T., S.O.V., and E.F. performed experiments and analyzed data. J.-P.K., D.M., R.M.B., and R.S.B. provided mice, reagents and intellectual input during the study. B.P. and E.F. designed the study and wrote the manuscript. All authors discussed the results and edited the paper.
Competing Financial Interests: The authors declare no competing financial interests.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/mi
REFERENCES
- 1.Pawankar R, Canonica GW, Holgate ST, Lockey RF. Allergic diseases and asthma: a major global health concern. Current opinion in allergy and clinical immunology. 2012;12:39–41. doi: 10.1097/ACI.0b013e32834ec13b. [DOI] [PubMed] [Google Scholar]
- 2.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nature reviews. Immunology. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 3.Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nature medicine. 2012;18:693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinet JP. The essential role of mast cells in orchestrating inflammation. Immunological reviews. 2007;217:5–7. doi: 10.1111/j.1600-065X.2007.00528.x. [DOI] [PubMed] [Google Scholar]
- 5.Cruse G, et al. A truncated splice-variant of the FcepsilonRIbeta receptor subunit is critical for microtubule formation and degranulation in mast cells. Immunity. 2013;38:906–917. doi: 10.1016/j.immuni.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maurer D, et al. Peripheral blood dendritic cells express Fc epsilon RI as a complex composed of Fc epsilon RI alpha- and Fc epsilon RI gamma-chains and can use this receptor for IgE-mediated allergen presentation. Journal of immunology. 1996;157:607–616. [PubMed] [Google Scholar]
- 7.Cheung DS, et al. Cutting edge: CD49d+ neutrophils induce FcepsilonRI expression on lung dendritic cells in a mouse model of postviral asthma. Journal of immunology. 2010;185:4983–4987. doi: 10.4049/jimmunol.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurer D, et al. Expression of functional high affinity immunoglobulin E receptors (Fc epsilon RI) on monocytes of atopic individuals. The Journal of experimental medicine. 1994;179:745–750. doi: 10.1084/jem.179.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aaij R, et al. Search for the lepton-flavor-violating decays B(s)0-->e(+/−)mu(−/+) and B0-->e(+/−)mu(−/+). Physical review letters. 2013;111:141801. doi: 10.1103/PhysRevLett.111.141801. [DOI] [PubMed] [Google Scholar]
- 10.Porcherie A, et al. Critical role of the neutrophil-associated high-affinity receptor for IgE in the pathogenesis of experimental cerebral malaria. The Journal of experimental medicine. 2011;208:2225–2236. doi: 10.1084/jem.20110845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehlink E, Baker AH, Yen E, Nurko S, Fiebiger E. Relationships between levels of serum IgE, cell-bound IgE, and IgE-receptors on peripheral blood cells in a pediatric population. PloS one. 2010;5:e12204. doi: 10.1371/journal.pone.0012204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasudev M, et al. Expression of high-affinity IgE receptor on human peripheral blood dendritic cells in children. PloS one. 2012;7:e32556. doi: 10.1371/journal.pone.0032556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holloway JA, Holgate ST, Semper AE. Expression of the high-affinity IgE receptor on peripheral blood dendritic cells: differential binding of IgE in atopic asthma. The Journal of allergy and clinical immunology. 2001;107:1009–1018. doi: 10.1067/mai.2001.115039. [DOI] [PubMed] [Google Scholar]
- 14.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nature immunology. 2010;11:647–655. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 15.Gill MA. The role of dendritic cells in asthma. The Journal of allergy and clinical immunology. 2012;129:889–901. doi: 10.1016/j.jaci.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 16.Ruiter B, Shreffler WG. The role of dendritic cells in food allergy. The Journal of allergy and clinical immunology. 2012;129:921–928. doi: 10.1016/j.jaci.2012.01.080. [DOI] [PubMed] [Google Scholar]
- 17.van Rijt LS, Lambrecht BN. Dendritic cells in asthma: a function beyond sensitization. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2005;35:1125–1134. doi: 10.1111/j.1365-2222.2005.02321.x. [DOI] [PubMed] [Google Scholar]
- 18.van Rijt LS, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. The Journal of experimental medicine. 2005;201:981–991. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambrecht BN. Dendritic cells and the regulation of the allergic immune response. Allergy. 2005;60:271–282. doi: 10.1111/j.1398-9995.2005.00708.x. [DOI] [PubMed] [Google Scholar]
- 20.Nakano H, et al. Pulmonary CD103(+) dendritic cells prime Th2 responses to inhaled allergens. Mucosal immunology. 2012;5:53–65. doi: 10.1038/mi.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colonna M, Pulendran B, Iwasaki A. Dendritic cells at the host-pathogen interface. Nature immunology. 2006;7:117–120. doi: 10.1038/ni0206-117. [DOI] [PubMed] [Google Scholar]
- 22.Hammad H, et al. Inflammatory dendritic cells--not basophils--are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. The Journal of experimental medicine. 2010;207:2097–2111. doi: 10.1084/jem.20101563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao Y, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity. 2013;39:722–732. doi: 10.1016/j.immuni.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nature reviews. Immunology. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novak N, et al. FcepsilonRI engagement of Langerhans cell-like dendritic cells and inflammatory dendritic epidermal cell-like dendritic cells induces chemotactic signals and different T-cell phenotypes in vitro. The Journal of allergy and clinical immunology. 2004;113:949–957. doi: 10.1016/j.jaci.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Kraft S, Wessendorf JH, Hanau D, Bieber T. Regulation of the high affinity receptor for IgE on human epidermal Langerhans cells. Journal of immunology. 1998;161:1000–1006. [PubMed] [Google Scholar]
- 27.Novak N, et al. Evidence for a differential expression of the FcepsilonRIgamma chain in dendritic cells of atopic and nonatopic donors. The Journal of clinical investigation. 2003;111:1047–1056. doi: 10.1172/JCI15932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prussin C, et al. Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. The Journal of allergy and clinical immunology. 2003;112:1147–1154. doi: 10.1016/j.jaci.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Foster B, Metcalfe DD, Prussin C. Human dendritic cell 1 and dendritic cell 2 subsets express FcepsilonRI: correlation with serum IgE and allergic asthma. The Journal of allergy and clinical immunology. 2003;112:1132–1138. doi: 10.1016/j.jaci.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Greer AM, et al. Serum IgE clearance is facilitated by human FcepsilonRI internalization. The Journal of clinical investigation. 2014;124:1187–1198. doi: 10.1172/JCI68964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sallmann E, et al. High-affinity IgE receptors on dendritic cells exacerbate Th2-dependent inflammation. Journal of immunology. 2011;187:164–171. doi: 10.4049/jimmunol.1003392. [DOI] [PubMed] [Google Scholar]
- 32.Amoah AS, et al. Peanut-specific IgE antibodies in asymptomatic Ghanaian children possibly caused by carbohydrate determinant cross-reactivity. The Journal of allergy and clinical immunology. 2013 doi: 10.1016/j.jaci.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robbins SH, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome biology. 2008;9:R17. doi: 10.1186/gb-2008-9-1-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crozat K, et al. Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunological reviews. 2010;234:177–198. doi: 10.1111/j.0105-2896.2009.00868.x. [DOI] [PubMed] [Google Scholar]
- 35.Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunological reviews. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dombrowicz D, et al. Anaphylaxis mediated through a humanized high affinity IgE receptor. Journal of immunology. 1996;157:1645–1651. [PubMed] [Google Scholar]
- 37.Nakajima T, et al. Gene expression screening of human mast cells and eosinophils using high-density oligonucleotide probe arrays: abundant expression of major basic protein in mast cells. Blood. 2001;98:1127–1134. doi: 10.1182/blood.v98.4.1127. [DOI] [PubMed] [Google Scholar]
- 38.Geiss GK, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature biotechnology. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 39.Ahrens R, et al. Intestinal mast cell levels control severity of oral antigen-induced anaphylaxis in mice. The American journal of pathology. 2012;180:1535–1546. doi: 10.1016/j.ajpath.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grayson MH, et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. The Journal of experimental medicine. 2007;204:2759–2769. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khodoun MV, Strait R, Armstrong L, Yanase N, Finkelman FD. Identification of markers that distinguish IgE- from IgG-mediated anaphylaxis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12413–12418. doi: 10.1073/pnas.1105695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. The Journal of experimental medicine. 2000;192:455–462. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu M, et al. Mast cells can promote the development of multiple features of chronic asthma in mice. The Journal of clinical investigation. 2006;116:1633–1641. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willart MA, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. The Journal of experimental medicine. 2012;209:1505–1517. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dombrowicz D, et al. Absence of Fc epsilonRI alpha chain results in upregulation of Fc gammaRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between Fc epsilonRI and Fc gammaRIII for limiting amounts of FcR beta and gamma chains. The Journal of clinical investigation. 1997;99:915–925. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novak N, Kraft S, Bieber T. Unraveling the mission of FcepsilonRI on antigen-presenting cells. The Journal of allergy and clinical immunology. 2003;111:38–44. doi: 10.1067/mai.2003.2. [DOI] [PubMed] [Google Scholar]
- 47.Maurer D, et al. The high affinity IgE receptor (Fc epsilon RI) mediates IgE-dependent allergen presentation. Journal of immunology. 1995;154:6285–6290. [PubMed] [Google Scholar]
- 48.Tang H, et al. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nature immunology. 2010;11:608–617. doi: 10.1038/ni.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisenbarth SC, et al. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. The Journal of experimental medicine. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aloulou M, et al. IgG1 and IVIg induce inhibitory ITAM signaling through FcgammaRIII controlling inflammatory responses. Blood. 2012;119:3084–3096. doi: 10.1182/blood-2011-08-376046. [DOI] [PubMed] [Google Scholar]
- 51.Chensue SW, et al. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formation: relationship to local inflammation, Th cell expression, and IL-12 production. Journal of immunology. 1996;157:4602–4608. [PubMed] [Google Scholar]
- 52.Collington SJ, et al. The role of the CCL2/CCR2 axis in mouse mast cell migration in vitro and in vivo. Journal of immunology. 2010;184:6114–6123. doi: 10.4049/jimmunol.0904177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taub D, et al. Bone marrow-derived murine mast cells migrate, but do not degranulate, in response to chemokines. Journal of immunology. 1995;154:2393–2402. [PubMed] [Google Scholar]
- 54.Taub DD, et al. Monocyte chemotactic protein-1 (MCP-1), -2, and -3 are chemotactic for human T lymphocytes. The Journal of clinical investigation. 1995;95:1370–1376. doi: 10.1172/JCI117788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Legrand N, et al. Humanized mice for modeling human infectious disease: challenges, progress, and outlook. Cell host & microbe. 2009;6:5–9. doi: 10.1016/j.chom.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greer AM, et al. Serum IgE clearance is facilitated by human FcepsilonRI internalization. The Journal of clinical investigation. 2014 doi: 10.1172/JCI68964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campbell EM, et al. Monocyte chemoattractant protein-1 mediates cockroach allergen-induced bronchial hyperreactivity in normal but not CCR2−/− mice: the role of mast cells. Journal of immunology. 1999;163:2160–2167. [PubMed] [Google Scholar]
- 58.Conti P, et al. Impact of Rantes and MCP-1 chemokines on in vivo basophilic cell recruitment in rat skin injection model and their role in modifying the protein and mRNA levels for histidine decarboxylase. Blood. 1997;89:4120–4127. [PubMed] [Google Scholar]
- 59.Hallgren J, Gurish MF. Mast cell progenitor trafficking and maturation. Advances in experimental medicine and biology. 2011;716:14–28. doi: 10.1007/978-1-4419-9533-9_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rose CE, Jr., Sung SS, Fu SM. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation. 2003;10:273–288. doi: 10.1038/sj.mn.7800193. [DOI] [PubMed] [Google Scholar]
- 61.Blank U, Launay P, Benhamou M, Monteiro RC. Inhibitory ITAMs as novel regulators of immunity. Immunological reviews. 2009;232:59–71. doi: 10.1111/j.1600-065X.2009.00832.x. [DOI] [PubMed] [Google Scholar]
- 62.Blasius AL, Cella M, Maldonado J, Takai T, Colonna M. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood. 2006;107:2474–2476. doi: 10.1182/blood-2005-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fuchs A, Cella M, Kondo T, Colonna M. Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood. 2005;106:2076–2082. doi: 10.1182/blood-2004-12-4802. [DOI] [PubMed] [Google Scholar]
- 64.Cao W, et al. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. The Journal of experimental medicine. 2006;203:1399–1405. doi: 10.1084/jem.20052454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rock J, et al. CD303 (BDCA-2) signals in plasmacytoid dendritic cells via a BCR-like signalosome involving Syk, Slp65 and PLCgamma2. European journal of immunology. 2007;37:3564–3575. doi: 10.1002/eji.200737711. [DOI] [PubMed] [Google Scholar]
- 66.Pfirsch-Maisonnas S, et al. Inhibitory ITAM signaling traps activating receptors with the phosphatase SHP-1 to form polarized “inhibisome” clusters. Science signaling. 2011;4:ra24. doi: 10.1126/scisignal.2001309. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity. 2010;32:518–530. doi: 10.1016/j.immuni.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han C, et al. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nature immunology. 2010;11:734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 69.Lin YC, Huang DY, Chu CL, Lin YL, Lin WW. The tyrosine kinase Syk differentially regulates Toll-like receptor signaling downstream of the adaptor molecules TRAF6 and TRAF3. Science signaling. 2013;6:ra71. doi: 10.1126/scisignal.2003973. [DOI] [PubMed] [Google Scholar]
- 70.Hamerman JA, Lanier LL. Inhibition of immune responses by ITAM-bearing receptors. Science's STKE : signal transduction knowledge environment. 2006;2006:re1. doi: 10.1126/stke.3202006re1. [DOI] [PubMed] [Google Scholar]
- 71.Pasquier B, et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 72.Baravalle G, Greer AM, Laflam TN, Shin JS. Antigen-Conjugated Human IgE Induces Antigen-Specific T Cell Tolerance in a Humanized Mouse Model. Journal of immunology. 2014 doi: 10.4049/jimmunol.1301751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maizels RM, Hewitson JP, Smith KA. Susceptibility and immunity to helminth parasites. Current opinion in immunology. 2012;24:459–466. doi: 10.1016/j.coi.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nature reviews. Immunology. 2013;13:607–614. doi: 10.1038/nri3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 2000;96:3029–3039. [PubMed] [Google Scholar]
- 76.Naik SH, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nature immunology. 2007;8:1217–1226. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 77.Brandt EB, et al. Mast cells are required for experimental oral allergen-induced diarrhea. The Journal of clinical investigation. 2003;112:1666–1677. doi: 10.1172/JCI19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai PS, et al. Chronic endotoxin exposure produces airflow obstruction and lung dendritic cell expansion. American journal of respiratory cell and molecular biology. 2012;47:209–217. doi: 10.1165/rcmb.2011-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bannert C, et al. Fc-Epsilon-RI, the High Affinity IgE-Receptor, Is Robustly Expressed in the Upper Gastrointestinal Tract and Modulated by Mucosal Inflammation. PloS one. 2012;7:e42066. doi: 10.1371/journal.pone.0042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.