

Abstract
Ischemic stroke results in the diverse phathophysiologies including blood brain barrier (BBB) disruption, brain edema, neuronal cell death, and synaptic loss in brain. Vitamin C has known as the potent anti-oxidant having multiple functions in various organs, as well as in brain. Dehydroascorbic acid (DHA) as the oxidized form of ascorbic acid (AA) acts as a cellular protector against oxidative stress and easily enters into the brain compared to AA. To determine the role of DHA on edema formation, neuronal cell death, and synaptic dysfunction following cerebral ischemia, we investigated the infarct size of ischemic brain tissue and measured the expression of aquaporin 1 (AQP-1) as the water channel protein. We also examined the expression of claudin 5 for confirming the BBB breakdown, and the expression of bcl 2 associated X protein (Bax), caspase-3, inducible nitric oxide synthase (iNOS) for checking the effect of DHA on the neurotoxicity. Finally, we examined postsynaptic density protein-95 (PSD-95) expression to confirm the effect of DHA on synaptic dysfunction following ischemic stroke. Based on our findings, we propose that DHA might alleviate the pathogenesis of ischemic brain injury by attenuating edema, neuronal loss, and by improving synaptic connection.
Keywords: Dehydroascorbic acid (DHA), Cerebral ischemia, Edema, Blood-brain barrier (BBB), Neurotoxicity, Synaptic dysfunction
INTRODUCTION
Ischemic stroke is the second leading cause of death worldwide accompanied by severe disability [1]. Cerebral ischemia and reperfusion injury leads to damage of brain tissues, inflammation as a result of the blood-brain barrier (BBB) disruption, oxidative damage [2], and apoptosis [3]. Brain tissue is highly vulnerable to oxidative damage because of its high use of oxygen [4] under cerebral ischemia. Cerebral ischemia leads to loss of tight junction proteins in brain endothelium, BBB disruption, and finally brain edema [5]. Brain edema leads to an imbalance in energy demand and influences on the postsynaptic effects of glutamate [6] and interruption of synaptic transmission in the penumbra after stroke [7,8]. Overall, excitotoxicity, inflammation and oxidative stress caused by ischemic stroke plays a crucial role in the pathophysiology of ischemic stroke [9,10]. To reduce the brain damage caused by cerebral ischemia, the solution for oxidative damage is the issue of the greatest importance. Vitamin C is the most important antioxidant for metabolic function of the brain [11,12,13] owing to its low redox potential which is capable of neutralizing diverse pro oxidants [14,15,16,17]. Mainly, vitamin C could be found in its form such as ascorbic acid (AA) and dehydroascorbic acid (DHA) (AA's oxidized form) [18,19]. According to earlier studies, lower levels of vitamin C are a risk factor of cerebral stroke [20,21] and actually, decreased vitamin C levels has been demonstrated in patients with ischemic stroke [22]. Recent study demonstrated that the treatment of AA prevented the disruption of BBB and sustained the BBB integrity in the cortex [23]. Neuroprotection by DHA has been demonstrated in several recent studies in both in vitro and in vivo. In in vitro study, DHA has been reported that it inhibits mitochondrial damage and cell death against oxidative injury [24]. Specifically, DHA among vitamin C could crosses the BBB through glucose transporter 1 (GLUT1) [25] and prevents cell death against oxidative damage by increasing glutathione (GSH) levels through glucose transporters [26,27]. In in vivo study, DHA have been reported to have protective effects as antioxidants in experimental neurological disease models such as stroke [19,28,29,30]. DHA administration attenuates oxidative stress markers and inflammation in hyperglycemic stroke models [31]. However, the study on the role of DHA administration through intra-peritoneal route in ischemic stroke animal model focused on edema formation, neurotoxicity, and synaptic dysfunction has not yet been determined. In present study, we investigated DHA's beneficial effect after ischemic brain injury in in vivo study. Our results show that DHA is involved in the prevention of brain edema formation, neurotoxicity, and synaptic dysfunction following ischemia injury. Thus, we suggest that DHA might mitigate stroke-induced pathological alterations following cerebral ischemic stroke.
MATERIALS AND METHODS
Animal model
Male Sprague-Dawley (SD) rat (Orient, GyeongGi-Do, Korea; 8 weeks old; 250-260 g) were subjected to transient focal cerebral ischemia by intraluminal middle cerebral artery blockade with a nylon suture, as previously described [32]. After 60 min of middle cerebral artery occlusion (MCAO), blood flow was restored by withdrawing the suture, and regional cerebral blood flow was monitored using a laser Doppler flow meter (Transonic Systems, Inc., Ithaca, NY, USA). All animal procedures and experiments were performed in accordance with the Guide to the Care and Use of Laboratory Animals and were approved by the Association for Assessment and Accreditation of Laboratory Animal Care.
Drug treatments
For each experiment, rats were given anesthesia (chloral hydrate 300 mg/kg,ip). DHA was purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA), and dissolved in normal saline (pH 7.5) and administered to rat through intra-peritoneal (i.p) route. Rats were injected with DHA (100 mg/kg/ml) treatment at a time for 10 min just after MCAO occlusion time. Control animals were given an equal volume of saline by the same procedure.
Evaluation of brain edema
For the evaluation of brain edema, mice were sacrificed at reperfusion 24 hr after MCAO injury. Brain slices (2 mm thick) between 22.00 mm and +4.00 mm from Bregma were incubated with 2% 2, 3, 5-triphenyltetraxolium chloride (TTC) (Sigma-Aldrich, St. Louis, MO, USA) at 37℃ for 10 min in the dark in a drying oven, and later photographed using a Nikon E950 digital camera attached to a dissecting microscope. Infarct volume was determined from digitized images using the Quantity One software package (Bio-Rad, CA, USA). Typically 3 such slices were used for analysis. The area of the cortical and striatal infarct was measured separately in all slices in the ischemic and non-ischemic hemisphere. The ipsilateral and contralateral hemispheres were used to calculate the percentage of brain edema [33].
Cresyl violet staining
At reperfusion 24 hr after MCAO injury, mice were sacrificed and brains were fixed in 3.7% formaldehyde and quickly frozen. Tissues were sectioned coronally at 20 µm thickness and sequentially dipped into xylene 5 min, 100% alcohol 5 min, 95% alcohol 5 min, and 70% alcohol 5 min. Samples were stained with cresyl violet (Sigma-Aldrich, St. Louis, MO, USA) for 3 min. After the staining, slides were reacted with 70% alcohol 5 min, 95% alcohol 5 min, 100% alcohol 5 min, and xylene 5 min. After these processes, sections were observed under a microscope equipped with a digital camera (Olympus, Tokyo, Japan).
Western blot analysis
At reperfusion 24 hr after MCAO injury, rat were sacrificed and brains were washed rapidly with ice-cold PBS, and collected. Tissues were lysed with ice-cold RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA). The lysates were centrifuged at 13,200 rpm for 1 hr at 4℃ to produce whole-cell extracts. Protein content was quantified using the BCA method (Pierce, Rockford, IL, USA). Protein (20 µg) was separated on a 10% SDS-polyacrylamide (PAGE) gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% bovine serum albumin, prepared in Tris-buffered saline/Tween (TBS-T; 20 nM Tris [pH 7.2], 150 mM NaCl, and 0.1% Tween 20), for 1hr at room temperature (RT), immunoblots were incubated overnight at 4℃ with primary antibodies that specifically detect Bcl 2 associated X protein (Bax) (1:1000, Abcam, Cambridge, MA, USA), Cleaved caspase-3 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), or β-actin (1:2000, Cell Signaling Technology, Danvers, MA, USA). Next, blots were incubated with HRP-linked anti-mouse and -rabbit IgG antibodies purchased from Abcam (Cambridge, UK) for 1 hr at RT. Enhanced chemiluminescence was performed by ECL (Pierce) [34].
Immunohistochemistry
Five-micrometer-thick frozen brain sections were cut onto clean glass slides (Thermo Scientific, Waltham, MA, USA), air-dried, and fixed in cold acetone for 10 min at -20℃. The slides were first washed in Tris-buffered saline (TBS) and then incubated with 0.3% H2O2 in methanol to quench endogenous peroxidase activity. Followed by a series of washes (three times with distilled water), the sections were blocked with 10% normal rabbit serum. Frozen brain sections (20 µm) were fixed in ice-cold acetone for 20 min. To block nonspecific labeling, sections were incubated in 5% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO, USA) diluted in PBS for 30 min before addition of primary and secondary antibodies. Primary antibodies for aquaporin-1 (AQP-1) (1:50, Abcam, Cambridge, MA, USA), inducible nitric oxide synthase (iNOS) (1:50, Millipore, Billerica, MA, USA), Bax (1:200, Abcam, Cambridge, MA, USA), Cleaved caspase-3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Claudin 5 (1:50, Invitrogen, Carlsbad, CA, USA), postsynaptic density protein-95 (PSD-95) (1:50, Abcam, Cambridge, MA, USA) were applied to the samples for 24 hr at 4℃, followed by a 90 min incubation with appropriate florescence secondary antibody (1:100, Invitrogen, Carlsbad, CA, USA) and three washes in PBS for 10 min each. After three washes in 0.1% PBS with Tween-20 (PBST), the sections were incubated with rhodamine-conjugated sheep anti-rabbit or FITC-conjugated sheep anti-mouse secondary antibody that was diluted to 1:200 with 5% BSA fraction V in 0.1% PBST for 2 hr in the dark at RT. After three washes in PBS, all sections were incubated with 1 µg/mL of 4',6-diamidino-2-phenylindole (Sigma-Aldrich, St. Louis, MO, USA) and 2 µg/mL of propidium iodide (Sigma-Aldrich, St. Louis, MO, USA) for a counter staining. Tissues were then visualized under a confocal microscope (Zeiss LSM 700, Carl Zeiss, Oberkochen, Germany). Positive cells percentage were measured by using Image J as following the previous study [35].
Statistical analysis
Statistical analyses were carried out using SPSS 18.0 software (IBM Corp., Armonk, NY, USA). All data are expressed as mean±S.E.M. Significant intergroup differences were determined by one-way analysis of variance followed by Bonferroni post hoc multiple comparison test. Statistical significance with the MCAO group (experimental control (EC) group) was determined by t-test. Each experiment conducted 3 replicates per condition. Differences were considered significant at *p < 0.05, **p < 0.01.
RESULTS
DHA reduced brain edema formation following cerebral ischemia
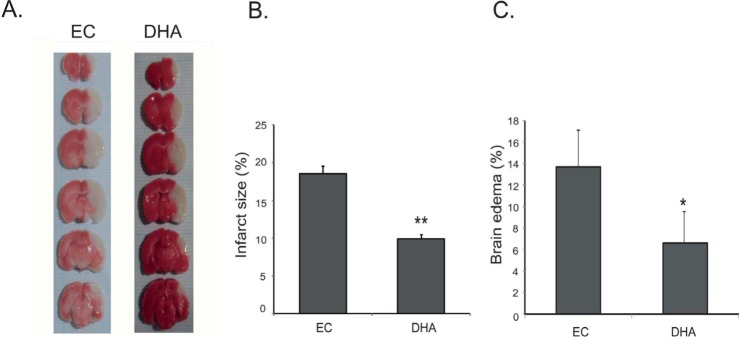
To investigate whether DHA affects vascular permeability in animal brain, we measured brain edema at reperfusion 24 hr after MCAO injury using TTC staining (Fig. 1A). White areas in brain are damaged brain areas due to ischemia (Fig. 1A). Fig. 1B shows the infarct size of brain in both groups (Fig. 1B). The graph shows the percentage of the ipsilateral hemisphere compared with the contralateral hemisphere both in the MCAO and DHA groups (Fig. 1C). The percentage of brain edema in the MCAO group was >12% whereas the percentage of brain edema after DHA treatment was <8% (Fig. 1C). Brain edema (%) was significantly reduced in the DHA group compared with the MCAO group. Our results indicate that the DHA treatment reduced brain edema formation after ischemic brain injury.
Fig. 1. Measurement of edema formation in MCAO mouse brain at reperfusion 24 hr after MCAO injury. (A) At reperfusion 24 hr after MCAO injury, TTC staining showed that white areas were damaged by ischemic injury. White areas reduced in the DHA treatment group compared to experimental control (EC) group. (B) Infarct size (%) was measured at reperfusion 24 hr after MCAO injury. The graph shows the infarct size was significantly attenuated in DHA treatment group. (C) The graph shows that brain edema (%) was significantly reduced in DHA treatment group compared with EC group. Differences were considered significant at *p<0.05. **p<0.0 (t-test via EC group). EC: reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.
DHA reduced the expression of AQP-1 as the marker of vascular permeability following cerebral ischemia
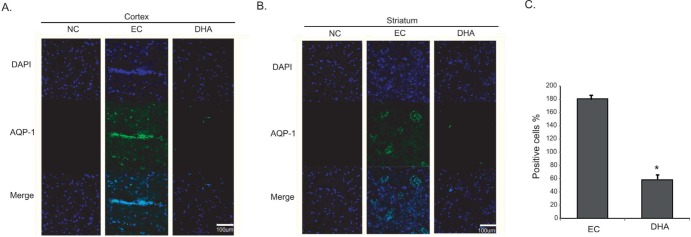
We performed immunohistochemistry using AQP-1 antibody at reperfusion 24 hr after MCAO injury to examine whether there were change of markers that affect vascular permeability both in cortex (Fig. 2A) and in striatum (Fig. 2B). We did not observe AQP-1 immunoreactivity in the cortex of the normal control (NC) group (Fig. 2A). However, AQP-1 positive cells were strongly expressed in the cortex in reperfusion 24hr after MCAO injury group (experimental control (EC) group) (Fig. 2A). In addition, DHA treated MCAO rat brain did not exhibit strongly the expression of AQP-1 compared to 24hr MCAO group (EC group). In striatum, AQP-1 expression showed the same pattern as the cortex (Fig. 2B). In addition, the water channel molecule AQP-1 was detected in rat brain cortex and striatum at 24 hr after MCAO injury (Fig. 2). Fig. 2C was significantly shown the decreased fluorescent intensity of AQP-1 over 3 times in the DHA treatment group compared to EC group (Fig. 2C). These data indicate that DHA affects the expression of AQP-1 in ischemic brain and may be involved in vascular permeability and edema after ischemia.
Fig. 2. Immunochemical image for measurement of AQP-1 expression by DHA treatment. (A) Immunochemical images showed that in DHA treatment group, AQP-1 positive cells (green) was decreased in rat cortex, compared with the EC group. (B) In DHA treated ischemic striatum, AQP-1's expression was decreased in striatum owing to DHA treatment. (C) The graph showed the percentage (%) of AQP-1 positive cells to compare the difference of AQP-1 fluorescence intensity between EC group and DHA group. Differences were considered significant at *p<0.05 (t-test via EC group). Scale bar=100 µm, AQP-1: aquaporin 1 (AQP-1); green, 4', 6-diamidino-2-phenylindole (DAPI): blue. NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.
DHA protects blood brain barrier (BBB) disruption following cerebral ischemia
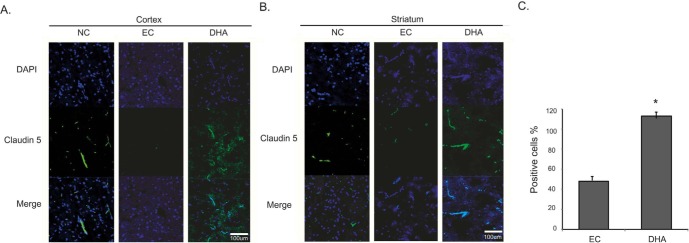
We conducted immunohistochemistry using Claudin 5 antibody at reperfusion 24 hr after MCAO injury to confirm whether or not there was change of marker as the component junction protein of blood brain barrier (BBB) both in cortex (Fig. 3A) and in striatum (Fig. 3B). In the NC group, Claudin 5 was considerably expressed both in cortex (Fig. 3A) and in striatum (Fig. 3B). However, Claudin 5 expression was evidently attenuated both in cortex (Fig. 3A) and in striatum (Fig. 3B) at reperfusion 24hr after MCAO injury group (EC group) (Fig. 3A). In the DHA treatment group, Claudin 5 expression was more increased both in cortex (Fig. 3A) and in striatum (Fig. 3B) compared to EC group. Fig. 3C was shown the significantly increased fluorescent intensity of Claudin 5 over 2 times in the DHA treatment group compared to EC group (Fig. 3C). Based on these results, we suggest that DHA may preserve the expression of Claudin 5 in ischemic brain and also protect the BBB disruption.
Fig. 3. Immunochemical image for confirmation of preserved Claudin 5 expression by DHA treatment. (A) Immunochemical images showed that Claudin 5-positive cells (green) were obviously reduced in rat cortex of experimental control (EC) group whereas in DHA treatment group, its expression was preserved in cortex. (B) Claudin 5-positive cells were retained in rat striatum owing to DHA treatment. (C) The graph showed the percentage (%) of Claudin 5 positive cells to compare the difference of Claudin 5 fluorescence intensity. Differences were considered significant at *p<0.05 (t-test via EC group). Scale bar=100 µm, Claudin 5: green, 4', 6-diamidino-2-phenylindole (DAPI): blue. NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.
Morphological alteration assessment using cresyl violet staining by DHA treatment
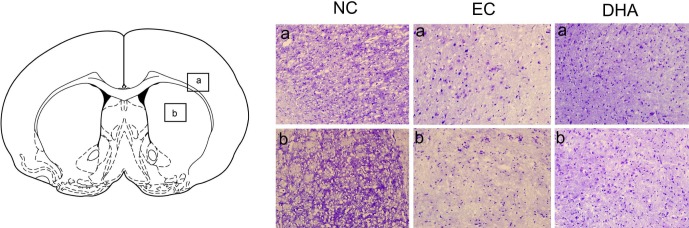
Cresyl violet staining was performed at reperfusion 24 hr after MCAO injury to assess the extent of ischemia-induced damage histologically in the striatum and cortex (Fig. 4). In the normal control group (without MCAO injury, without DHA treatment), intact cellular structure was observed in both the cortex and striatum. In the MCAO group (EC group), shrinked small cell bodies were detected, and also damaged tissue was observed in the ischemic cortex and striatum (Fig. 4). In DHA group treatment (DHA treatment and MCAO injury), damaged cells were reduced in number compared with EC group, and we observed healthy round cells in the ischemic cortex and striatum (Fig. 4).
Fig. 4. The histological assessment using cresyl violet staining after ischemic injury. Atlas of rat brain mainly presents the corpus callosum, cerebral cortex (a) and striatum (b). Cresyl violet staining indicated that severe cell loss was founded in the 24 hr MCAO group whereas more healthy and round cell bodies in striatum and cortex were observed in MCAO with DHA treatment group. a: cortex, b: striatum, NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.
DHA attenuates the cell damage against neurotoxicity following cerebral ischemia
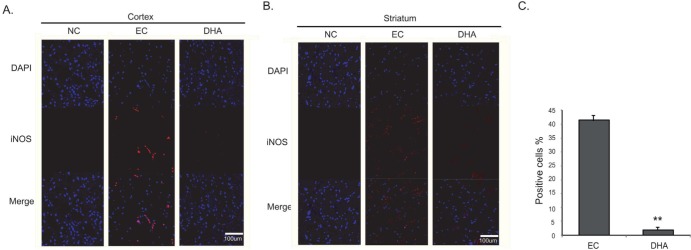
We performed immunohistochemical staining using cleaved caspase-3 antibody at reperfusion 24 hr after MCAO injury to examine whether DHA influences on the cell death both in cortex (Fig. 5A) and in striatum (Fig. 5B). Cleaved caspase-3 immunopositive cells were not observed in the rat cortex of the normal control (NC) group (Fig. 5A). However, cleaved caspase-3 positive cells were strongly expressed in the cortex in reperfusion 24 hr after MCAO injury group (EC group) (Fig. 5A). In addition, DHA treated rat cortex did not exhibit the expression of cleaved caspase-3 strongly compared to 24 hr MCAO group (EC group) (Fig. 5A). In striatum, cleaved caspase-3 expression showed the same pattern of the cortex (Fig. 5B). Fig. 5C was shown that the fluorescent intensity of cleaved caspase-3 was attenuated significantly over 4 times in the DHA treatment group compared to EC group (Fig. 5C). The western blot data (Fig.5D) also showed the reduction of cleaved caspase-3 expression in the DHA treatment group in spite of the MCAO injury. Following these data, DHA may inhibit the cell death under ischemic injury by attenuating the expression of cleaved caspase-3. Second, we conducted immunohistochemistry using Bax antibody at reperfusion 24 hr after MCAO injury to examine whether DHA influences on the alteration of marker that affects mitochondrial cell death both in cortex (Fig. 6A) and in striatum (Fig. 6B). We did not observe Bax immunoreactivity in the rat cortex of the normal control (NC) group (Fig. 6A). However, the expression of Bax was increased strongly in the cortex at reperfusion 24 hr after MCAO injury group (EC group) (Fig. 6A). In addition, the expression of Bax were not strongly exhibited in the cortex of DHA treated MCAO group compared to 24 hr MCAO group (EC group) (Fig. 6A). In striatum, Bax expression showed the same pattern as the cortex (Fig. 6B). Fig. 6C was shown that the significantly reduced fluorescent intensity of Bax over 2 times in the DHA treatment group compared to EC group (Fig. 6C). The western blot data (Fig. 6D) also showed the reduction of Bax expression in the DHA treatment group compared to the MCAO injury (Fig. 6D). Considering these data, DHA may influence on the cellular protection against the mitochondrial cell death under ischemic injury by attenuating the expression of Bax. Additionally, we conducted immunohistochemistry using iNOS antibody at reperfusion 24 hr after MCAO injury to confirm whether there was change of marker as the inflammatory mediator in both cortex (Fig. 7A) and striatum (Fig. 7B) or not. In the EC group, iNOS was considerably expressed in both in cortex (Fig. 7A) and striatum (Fig. 7B). However, iNOS expression was not found in both cortex (Fig. 7A) and striatum (Fig. 7B) at NC group. In the DHA treatment group, iNOS expression was decreased in both cortex (Fig. 7A) and striatum (Fig. 7B) compared to EC group. Fig. 7C was shown that the significantly decreased fluorescent intensity of iNOS over 9 times in the DHA treatment group compared to EC group (Fig. 7C). Based on these results, we suggest that DHA may inhibit the expression of iNOS in ischemic brain and also block the expression of inflammatory mediators.
Fig. 5. Immunochemical image for confirmation of reduced cleaved caspase-3 expression by DHA treatment. (A) Immunochemical images showed that cleaved caspase-3 positive cells (red) were densely expressed in the EC group. In DHA treatment group, cleaved caspase-3 expression was decreased in rat cortex, compared with the EC group. (B) In DHA treatment group, cleaved caspase-3 positive cells were decreased in rat striatum due to DHA treatment. (C) The graph showed the percentage (%) of cleaved caspase-3 positive cells to compare the difference of cleaved caspase-3 fluorescence intensity. Statistical significance with EC group was determined by t-test. (D) The graph of cleaved caspase-3 protein level showed the same pattern with immunochemical images. Differences were considered significant at *p<0.05, (ANOVA followed by Bonferroni post hoc multiple comparison test). Scale bar=100 µm, Cleaved caspase-3: red, 4', 6-diamidino-2-phenylindole (DAPI): blue. NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.

Fig. 6. Immunochemical image for confirmation of reduced Bax expression by DHA treatment. (A) Immunochemical images showed that Bax positive cells (red) were densely expressed in the EC group. In DHA treatment group, Bax expression was decreased in rat cortex, compared with the EC group. (B) In DHA treatment group, Bax positive cells were decreased in rat striatum due to DHA treatment. (C) The graph showed the percentage (%) of Bax positive cells to compare the difference of Bax fluorescence intensity. Statistical significance with EC group was determined by t-test. (D) The graph of Bax protein level showed the same pattern with immunochemical images. Differences were considered significant at *p<0.05, **p<0.01 (ANOVA followed by Bonferroni post hoc multiple comparison test). Scale bar=100 µm, Bax: bcl2 associated X protein (Bax); red, 4', 6-diamidino-2-phenylindole (DAPI): blue. NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.

Fig. 7. Immunochemical image for confirmation of attenuated iNOS expression by DHA treatment. (A) Immunochemical images showed that iNOS positive cells (red) were considerably expressed in the EC group. The rat cortex in DHA treatment group showed reduced iNOS expression compared with EC group. (B) In DHA treatment group, iNOS positive cells were decreased in rat striatum. (C) The graph showed the percentage (%) of iNOS positive cells to compare the difference of iNOS fluorescence intensity. Differences were considered significant at **p<0.01 (t-test via EC group). Scale bar=100 µm, iNOS: inducible nitric oxide synthase (iNOS); red, 4', 6-diamidino-2-phenylindole (DAPI): blue. NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.
DHA prevents the damage of post synaptic plasticity following cerebral ischemia
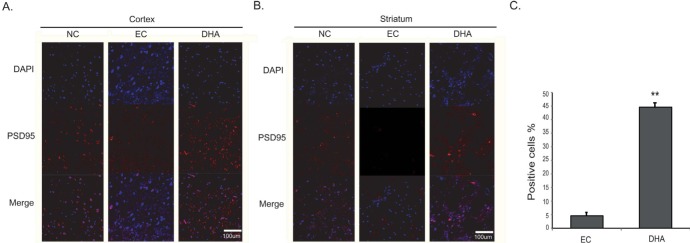
We performed immunohistochemistry using PSD-95 antibody at reperfusion 24 hr after MCAO injury to check whether DHA affect the postsynaptic plasticity's damage following cerebral ischemia (Fig. 8). We observed strong PSD-95 immunoreactivity in the cortex of the NC group (Fig. 8A). However, PSD-95 positive cells were substantially decreased in the cortex at reperfusion 24 hr after MCAO injury group (EC group) (Fig. 8A). In addition, it showed that strong expression of PSD-95 was observed in DHA treated rat cortex compared to 24 hr MCAO group (EC group). In striatum region, PSD-95 expression showed the same pattern as the cortex (Fig. 8B). Fig. 8C was significantly shown the increased fluorescent intensity of PSD-95 over 6 times in the DHA treatment group compared to EC group (Fig. 8C). Taken together, we speculate that DHA may improve the synaptic dysfunction in ischemic brain.
Fig. 8. Immunochemical image for confirmation of PSD-95 expression by DHA treatment. (A) Immunochemical images showed that PSD-95 positive cells (red) were few expressed in the EC group cortex. In DHA treatment group, PSD-95 expression was considerably increased in rat cortex compared to the EC group. (B) In DHA treatment group, PSD-95 positive cells were increased in rat striatum owing to DHA treatment. (C) The graph showed the percentage (%) of PSD-95 positive cells to compare the difference of PSD-95 fluorescence intensity. Differences were considered significant at **p<0.01 (t-test via EC group). Scale bar=100 µm, PSD-95: postsynaptic density protein 95 (PSD-95); red, 4', 6-diamidino-2-phenylindole (DAPI): blue. NC: normal control group, EC: experimental control; reperfusion 24 hr after MCAO injury, DHA: DHA treatment and reperfusion 24 hr after MCAO injury.
DISCUSSION
Ischemic stroke causes the blockage of cerebral blood vessels in the regions of brain, which can lead to human disability and death [36]. Subsequently, the blockage of blood vessels following stroke leads to diverse pathophysiologies including brain edema, neuronal loss, and cognitive dysfunction [37,38,39,40]. Cerebral cortex, hippocampus, and corpus striatum in the brain are the most vulnerable regions against oxidative stress and hypoxic injury induced by cerebral ischemia [41]. Many studies has reported that vitamin C among antioxidants is generally neuroprotective in response to brain ischemic injury [42,43,44,45]. Oral administration of AA to animal had demonstrated that it suppresses neuronal damage under cerebral ischemia-reperfusion [46]. Dehydroascorbic acid as AA's oxidized form [15,18,19] has been reported that it has a neuroprotective role [47] and is easily transported to the brain by mediating glucose transporter 1 (GLUT1) located in the endothelial cells of the BBB [48]. However, DHA did not fully be investigated in ischemic stroke animal model in spite of its advantages. We anticipated that DHA as an anti-oxidant may considerably affect on cerebral ischemia animal because it can rapidly pass through the brain than AA [25]. In present study, we investigated the neuroprotective effects of brain by DHA i.p administration in cerebral ischemia rat. First, we obtained the consequence that DHA treatment inhibits the brain edema formation in MCAO rat brain. Edema defined as an abnormal increase in brain water content is frequently observed in cerebral ischemia and also has a critical influence on morbidity and mortality [49]. Several studies reported that cerebral ischemia contributes to damage the integrity and permeability of the BBB [50,51]. Aquaporin (AQP) is the water channel protein that facilitates water transport through cell membranes [52,53]. Specifically AQP-1 is permeable only to water and is considered to participate in brain water homeostasis [54]. In addition, AQP 1 has been reported that it is involved in edema formation and cell death in the hippocampus following brain injury [55]. Following our results, we suggest that DHA may reduce osmotic water permeability following cerebral ischemia by inhibiting the expression of AQP-1. All BBB components have been reported to the association with the regulation of the BBB permeability including tight junctions of endothelial cells, glia cells [56,57,58]. The BBB is composed of the brain endothelial cells interconnected with transmembrane tight junction proteins such as claudin-5 [59] and regulates paracellular permeability [60,61]. In present study, our results indicated that claudin 5 as a tight junction protein in DHA treated MCAO rat brain was evidently preserved against ischemic injury. According to our results, DHA may protect the BBB integrity by preserving tight junction protein in response to ischemic injury. Cerebral ischemia induces the neurotoxic environment in brain and it could result in the severe neuronal cell damage, so we investigated the cell death marker such as Bax [62,63], caspase-3[64,65], and iNOS [66,67] in order to examine the protective effect of DHA against the neurotoxicity following ischemic stroke. In present study, DHA treatment reduced the expression of Bax and caspase-3 which is the marker of the mitochondrial cell death and iNOS in ischemic injured brain. Nitric oxide (NO) that causes neuronal cell death and exacerbates glutamate toxicity after cerebral ischemia [68] is synthesized by NO synthase such as iNOS [69]. Several studies demonstrated that inhibition of iNOS in cerebral ischemia improves neurological deficits and reduces infarct volume [70,71]. In consideration of Figure 1 result, our finding suggested that DHA attenuates the expression of iNOS and it may be linked to reduced infarct volume and improved cell death against hypoxic injury. Additionally, NO formed by iNOS has been reported the implicated in neurodegeneration [69]. Judging from our result regarding the reduced iNOS expression, we suggest the possibility of DHA regarding the improvement of cognitive function against ischemic stroke although we did not check the production of NO and behavior test considering that AA improves the cognitive decline in Alzheimer's disease [72]. As mentioned earlier, several studies demonstrated that DHA prevents cell death against ischemic injury [19,28,29,30]. However, previous studies have not yet been determined the effect of DHA on recovery of neuronal function in ischemia animal model. Therefore, we tried to examine the effect of DHA on neuronal function by measuring indirectly synaptic dysfunction in present study. In order to observe the effect of DHA on ischemia-induced synaptic connection alteration, we investigated the expression of PSD-95 protein in ischemic brain tissue. PSD-95 protein as a postsynaptic marker [73,74] is a member of the membrane-associated guanylate kinase family of synaptic molecules and is localized at excitatory synapses [75]. Postsynaptic densities (PSD) proteins are involved in regulation of synaptic function and in the transduction of synaptic signals to the postsynaptic cell [76,77,78]. Especially, PSD-95 has been implicated in the regulation of ion-channel function, synaptic activity, and intracellular signaling and finally cognitive impairment [79,80,81]. In addition, PSD-95 protein is implicated in promoting synapse stability and makes synaptic contacts more stable in neurons [75]. Recent studies suggested that the PSD-95 protein improves the neurophysiologic phenomenon after ischemic stroke involving MCAO [82,83]. Moreover, some researchers demonstrated that the decrease of PSD-95 protein immunoreactivity in the ischemic brain leads to a deficit of postsynaptic plasticity in the brain [84]. Several studies suggest that PSD-95's reduction is associated with cognitive impairment [85,86,87,88]. Based on our results, our findings indicate that DHA induced the increase of PSD-95 protein immunoreactivity in ischemic stroke brain and DHA may improve the ischemic-induced synaptic plasticity dysfunction. In addition, although we did not check the memory function related behavior test such as water maze, we carefully expect that DHA may improve the learning and memory dysfunction following cerebral ischemia by promoting the neuron's synapse stability. Taken together, our findings suggest three points that 1) DHA is involved in the inhibition of AQP-1 expression and the preservation of claudin 5, ultimately resulting in the reduction of edema formation induced by cerebral ischemia, 2) DHA is associated with the decrease of Bax, cleaved caspase-3 and iNOS expression, ultimately resulting in the protection of cell death against neurotoxicity following cerebral ischemia, 3) DHA is linked to the preservation of PSD-95 protein expression, ultimately resulting in the improvement of neuron's synaptic connection in cerebral ischemia. The present study has some limitations fully to prove the beneficial effect of DHA against ischemic injury. However, we suggest that this study is worthy in that it provide the need of the further study of DHA on ischemic stroke. Taken together, we propose that the DHA might be beneficial to alleviate clinical pathologies that occur after ischemic stroke.
ACKNOWLEDGEMENTS
This research was supported by the Brain Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Science, ICT & Future Planning (2012-0005827).
Footnotes
The authors declare no conflict of interest regarding the publication of this paper.
References
- 1.Luo Y, Yang YP, Liu J, Li WH, Yang J, Sui X, Yuan X, Nie ZY, Liu YQ, Chen D, Lin SH, Wang YA. Neuroprotective effects of madecassoside against focal cerebral ischemia reperfusion injury in rats. Brain Res. 2014;1565:37–47. doi: 10.1016/j.brainres.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008;28:1686–1696. doi: 10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaz-Ruiz A, Vacio-Adame P, Monroy-Noyola A, Méndez-Armenta M, Ortiz-Plata A, Montes S, Rios C. Metallothionein-II inhibits lipid peroxidation and improves functional recovery after transient brain ischemia and reperfusion in rats. Oxid Med Cell Longev. 2014;2014:436429. doi: 10.1155/2014/436429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahadik SP, Evans D, Lal H. Oxidative stress and role of antioxidant and omega-3 essential fatty acid supplementation in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:463–493. doi: 10.1016/s0278-5846(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 5.Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia - the ischemic penumbra. Stroke. 1981;12:723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- 8.Symon L. The relationship between CBF, evoked potentials and the clinical features in cerebral ischaemia. Acta Neurol Scand Suppl. 1980;78:175–190. [PubMed] [Google Scholar]
- 9.Castillo J, Dávalos A, Noya M. Progression of ischaemic stroke and excitotoxic aminoacids. Lancet. 1997;349:79–83. doi: 10.1016/S0140-6736(96)04453-4. [DOI] [PubMed] [Google Scholar]
- 10.Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53:135–159. [PubMed] [Google Scholar]
- 11.Carr A, Frei B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999;13:1007–1024. doi: 10.1096/fasebj.13.9.1007. [DOI] [PubMed] [Google Scholar]
- 12.Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 13.Savini I, Rossi A, Pierro C, Avigliano L, Catani MV. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids. 2008;34:347–355. doi: 10.1007/s00726-007-0555-7. [DOI] [PubMed] [Google Scholar]
- 14.May JM, Qu ZC, Whitesell RR. Generation of oxidant stress in cultured endothelial cells by methylene blue: protective effects of glucose and ascorbic acid. Biochem Pharmacol. 2003;66:777–784. doi: 10.1016/s0006-2952(03)00408-8. [DOI] [PubMed] [Google Scholar]
- 15.Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 16.Bowman GL. Ascorbic acid, cognitive function, and Alzheimer's disease: a current review and future direction. Biofactors. 2012;38:114–122. doi: 10.1002/biof.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lykkesfeldt J. Ascorbate and dehydroascorbic acid as biomarkers of oxidative stress: validity of clinical data depends on vacutainer system used. Nutr Res. 2012;32:66–69. doi: 10.1016/j.nutres.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. 1986;6:365–406. doi: 10.1146/annurev.nu.06.070186.002053. [DOI] [PubMed] [Google Scholar]
- 19.Wilson JX. The physiological role of dehydroascorbic acid. FEBS Lett. 2002;527:5–9. doi: 10.1016/s0014-5793(02)03167-8. [DOI] [PubMed] [Google Scholar]
- 20.Ness AR, Powles JW, Khaw KT. Vitamin C and cardiovascular disease: a systematic review. J Cardiovasc Risk. 1996;3:513–521. [PubMed] [Google Scholar]
- 21.Yokoyama T, Date C, Kokubo Y, Yoshiike N, Matsumura Y, Tanaka H. Serum vitamin C concentration was inversely associated with subsequent 20-year incidence of stroke in a Japanese rural community. The Shibata study. Stroke. 2000;31:2287–2294. doi: 10.1161/01.str.31.10.2287. [DOI] [PubMed] [Google Scholar]
- 22.Sánchez-Moreno C, Dashe JF, Scott T, Thaler D, Folstein MF, Martin A. Decreased levels of plasma vitamin C and increased concentrations of inflammatory and oxidative stress markers after stroke. Stroke. 2004;35:163–168. doi: 10.1161/01.STR.0000105391.62306.2E. [DOI] [PubMed] [Google Scholar]
- 23.Lin JL, Huang YH, Shen YC, Huang HC, Liu PH. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J Cereb Blood Flow Metab. 2010;30:1121–1136. doi: 10.1038/jcbfm.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puskas F, Gergely P, Jr, Banki K, Perl A. Stimulation of the pentose phosphate pathway and glutathione levels by dehydroascorbate, the oxidized form of vitamin C. FASEB J. 2000;14:1352–1361. doi: 10.1096/fj.14.10.1352. [DOI] [PubMed] [Google Scholar]
- 25.Agus DB, Gambhir SS, Pardridge WM, Spielholz C, Baselga J, Vera JC, Golde DW. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest. 1997;100:2842–2848. doi: 10.1172/JCI119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim EJ, Won R, Sohn JH, Chung MA, Nam TS, Lee HJ, Lee BH. Anti-oxidant effect of ascorbic and dehydroascorbic acids in hippocampal slice culture. Biochem Biophys Res Commun. 2008;366:8–14. doi: 10.1016/j.bbrc.2007.11.050. [DOI] [PubMed] [Google Scholar]
- 27.Kim EJ, Park YG, Baik EJ, Jung SJ, Won R, Nahm TS, Lee BH. Dehydroascorbic acid prevents oxidative cell death through a glutathione pathway in primary astrocytes. J Neurosci Res. 2005;79:670–679. doi: 10.1002/jnr.20384. [DOI] [PubMed] [Google Scholar]
- 28.Miura S, Ishida A, Nakajima W, Ohmura A, Kawamura M, Takada G. Intraventricular ascorbic acid administration decreases hypoxic-ischemic brain injury in newborn rats. Brain Res. 2006;1095:159–166. doi: 10.1016/j.brainres.2006.04.045. [DOI] [PubMed] [Google Scholar]
- 29.Santos LF, Freitas RL, Xavier SM, Saldanha GB, Freitas RM. Neuroprotective actions of vitamin C related to decreased lipid peroxidation and increased catalase activity in adult rats after pilocarpine-induced seizures. Pharmacol Biochem Behav. 2008;89:1–5. doi: 10.1016/j.pbb.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Deutsch JC. Dehydroascorbic acid. J Chromatogr A. 2000;881:299–307. doi: 10.1016/s0021-9673(00)00166-7. [DOI] [PubMed] [Google Scholar]
- 31.Bémeur C, Ste-Marie L, Desjardins P, Vachon L, Butterworth RF, Hazell AS, Montgomery J. Dehydroascorbic acid normalizes several markers of oxidative stress and inflammation in acute hyperglycemic focal cerebral ischemia in the rat. Neurochem Int. 2005;46:399–407. doi: 10.1016/j.neuint.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Selvamani A, Sohrabji F. Reproductive age modulates the impact of focal ischemia on the forebrain as well as the effects of estrogen treatment in female rats. Neurobiol Aging. 2010;31:1618–1628. doi: 10.1016/j.neurobiolaging.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohammadi MT, Shid-Moosavi SM, Dehghani GA. Contribution of nitric oxide synthase (NOS) in blood-brain barrier disruption during acute focal cerebral ischemia in normal rat. Pathophysiology. 2012;19:13–20. doi: 10.1016/j.pathophys.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Jung HJ, Jeon YH, Bokara KK, Koo BN, Lee WT, Park KA, Lee JE. Agmatine promotes the migration of murine brain endothelial cells via multiple signaling pathways. Life Sci. 2013;92:42–50. doi: 10.1016/j.lfs.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Papadopulos F, Spinelli M, Valente S, Foroni L, Orrico C, Alviano F, Pasquinelli G. Common tasks in microscopic and ultrastructural image analysis using ImageJ. Ultrastruct Pathol. 2007;31:401–407. doi: 10.1080/01913120701719189. [DOI] [PubMed] [Google Scholar]
- 36.Stapf C, Mohr JP. Ischemic stroke therapy. Annu Rev Med. 2002;53:453–475. doi: 10.1146/annurev.med.53.082901.104106. [DOI] [PubMed] [Google Scholar]
- 37.Damodaran T, Hassan Z, Navaratnam V, Muzaimi M, Ng G, Müller CP, Liao P, Dringenberg HC. Time course of motor and cognitive functions after chronic cerebral ischemia in rats. Behav Brain Res. 2014;275:252–258. doi: 10.1016/j.bbr.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 38.Wang YQ, Wu WW, Wang LK, Chen K, Li YH. Influence of hepatic ischemia-reperfusion on postoperative spatial cognitive function in mice. Genet Mol Res. 2014;13:5767–5777. doi: 10.4238/2014.July.29.4. [DOI] [PubMed] [Google Scholar]
- 39.Kamchatnov PR, Vorob'eva OV, Rachin AP. Treatment of emotional and cognitive disorders in patients with chronic cerebral ischemia. Zh Nevrol Psikhiatr Im S S Korsakova. 2014;114:52–56. [PubMed] [Google Scholar]
- 40.Lei Y, Guo Q, Li Y, Jiang H, Ni W, Gu Y. Characteristics of cognitive impairment in adults with cerebral ischemia. Zhonghua Yi Xue Za Zhi. 2014;94:984–989. [PubMed] [Google Scholar]
- 41.Stamford JA, Isaac D, Hicks CA, Ward MA, Osborne DJ, O'Neill MJ. Ascorbic acid is neuroprotective against global ischaemia in striatum but not hippocampus: histological and voltammetric data. Brain Res. 1999;835:229–240. doi: 10.1016/s0006-8993(99)01587-5. [DOI] [PubMed] [Google Scholar]
- 42.Block F, Schmitt W, Schwarz M. The antioxidant LY 231617 ameliorates functional and morphological sequelae induced by global ischemia in rats. Brain Res. 1995;694:308–311. doi: 10.1016/0006-8993(95)00821-7. [DOI] [PubMed] [Google Scholar]
- 43.Zamani M, Katebi M, Mehdizadeh M, Kafami L, Soleimani M. Combination therapy with A1 receptor agonist and vitamin C improved working memory in a mouse model of global ischemia-reperfusion. Basic Clin Neurosci. 2013;4:111–116. [PMC free article] [PubMed] [Google Scholar]
- 44.Chen GC, Lu DB, Pang Z, Liu QF. Vitamin C intake, circulating vitamin C and risk of stroke: a meta-analysis of prospective studies. J Am Heart Assoc. 2013;2:e000329. doi: 10.1161/JAHA.113.000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lagowska-Lenard M, Stelmasiak Z, Bartosik-Psujek H. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol Rep. 2010;62:751–756. doi: 10.1016/s1734-1140(10)70334-0. [DOI] [PubMed] [Google Scholar]
- 46.Iwata N, Okazaki M, Xuan M, Kamiuchi S, Matsuzaki H, Hibino Y. Orally administrated ascorbic acid suppresses neuronal damage and modifies expression of SVCT2 and GLUT1 in the brain of diabetic rats with cerebral ischemia-reperfusion. Nutrients. 2014;6:1554–1577. doi: 10.3390/nu6041554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.May JM. Vitamin C transport and its role in the central nervous system. Subcell Biochem. 2012;56:85–103. doi: 10.1007/978-94-007-2199-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang WW, Zhang L, Hou WK, Xu YX, Xu H, Lou FC, Zhang Y, Wang Q. Dynamic expression of glucose transporters 1 and 3 in the brain of diabetic rats with cerebral ischemia reperfusion. Chin Med J (Engl) 2009;122:1996–2001. [PubMed] [Google Scholar]
- 49.Klatzo I. Brain oedema following brain ischaemia and the influence of therapy. Br J Anaesth. 1985;57:18–22. doi: 10.1093/bja/57.1.18. [DOI] [PubMed] [Google Scholar]
- 50.Pluta R. Pathological opening of the blood-brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia-reperfusion brain injury. Chemotherapy. 2005;51:223–226. doi: 10.1159/000086924. [DOI] [PubMed] [Google Scholar]
- 51.Pluta R, Lossinsky AS, Wiśniewski HM, Mossakowski MJ. Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994;633:41–52. doi: 10.1016/0006-8993(94)91520-2. [DOI] [PubMed] [Google Scholar]
- 52.Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S. Aquaporin water channels--from atomic structure to clinical medicine. J Physiol. 2002;542:3–16. doi: 10.1113/jphysiol.2002.020818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zelenina M. Regulation of brain aquaporins. Neurochem Int. 2010;57:468–488. doi: 10.1016/j.neuint.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 54.Zador Z, Stiver S, Wang V, Manley GT. Role of aquaporin-4 in cerebral edema and stroke. Handb Exp Pharmacol. 2009:159–170. doi: 10.1007/978-3-540-79885-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qiu B, Li X, Sun X, Wang Y, Jing Z, Zhang X, Wang Y. Overexpression of aquaporin1 aggravates hippocampal damage in mouse traumatic brain injury models. Mol Med Rep. 2014;9:916–922. doi: 10.3892/mmr.2014.1899. [DOI] [PubMed] [Google Scholar]
- 56.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 57.Colgan OC, Ferguson G, Collins NT, Murphy RP, Meade G, Cahill PA, Cummins PM. Regulation of bovine brain microvascular endothelial tight junction assembly and barrier function by laminar shear stress. Am J Physiol Heart Circ Physiol. 2007;292:H3190–H3197. doi: 10.1152/ajpheart.01177.2006. [DOI] [PubMed] [Google Scholar]
- 58.Muellner A, Benz M, Kloss CU, Mautes A, Burggraf D, Hamann GF. Microvascular basal lamina antigen loss after traumatic brain injury in the rat. J Neurotrauma. 2003;20:745–754. doi: 10.1089/089771503767869971. [DOI] [PubMed] [Google Scholar]
- 59.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999;147:1351–1363. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96:511–516. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Q, Bi M, Bi W, Kang H, Yan L, Guo YL. WITHDRAWN: Edaravone protects brain tissue from apoptosis and oxidative stress after acute carbon monoxide poisoning. Am J Emerg Med. 2014 doi: 10.1016/j.ajem.2014.09.013. (in press) [DOI] [PubMed] [Google Scholar]
- 63.Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG, Faden AI. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34:10055–10071. doi: 10.1523/JNEUROSCI.1260-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yürüker V, Nazıroğlu M, Şenol N. Reduction in traumatic brain injury-induced oxidative stress, apoptosis, and calcium entry in rat hippocampus by melatonin: Possible involvement of TRPM2 channels. Metab Brain Dis. 2015;30:223–231. doi: 10.1007/s11011-014-9623-3. [DOI] [PubMed] [Google Scholar]
- 65.Kobeissy FH, Liu MC, Yang Z, Zhang Z, Zheng W, Glushakova O, Mondello S, Anagli J, Hayes RL, Wang KK. Degradation of βII-Spectrin Protein by Calpain-2 and Caspase-3 Under Neurotoxic and Traumatic Brain Injury Conditions. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8898-z. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Lee KF, Chen JH, Teng CC, Shen CH, Hsieh MC, Lu CC, Lee KC, Lee LY, Chen WP, Chen CC, Huang WS, Kuo HC. Protective effects of Hericium erinaceus mycelium and its isolated erinacine A against ischemia-injury-induced neuronal cell death via the inhibition of iNOS/p38 MAPK and nitrotyrosine. Int J Mol Sci. 2014;15:15073–15089. doi: 10.3390/ijms150915073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trollmann R, Richter M, Jung S, Walkinshaw G, Brackmann F. Pharmacologic stabilization of hypoxia-inducible transcription factors protects developing mouse brain from hypoxia-induced apoptotic cell death. Neuroscience. 2014;278:327–342. doi: 10.1016/j.neuroscience.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 68.Cui H, Lee JH, Kim JY, Koo BN, Lee JE. The neuroprotective effect of agmatine after focal cerebral ischemia in diabetic rats. J Neurosurg Anesthesiol. 2012;24:39–50. doi: 10.1097/ANA.0b013e318235af18. [DOI] [PubMed] [Google Scholar]
- 69.Toda N, Nakanishi-Toda M. Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin Eye Res. 2007;26:205–238. doi: 10.1016/j.preteyeres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 70.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koh PO. Ferulic acid modulates nitric oxide synthase expression in focal cerebral ischemia. Lab Anim Res. 2012;28:273–278. doi: 10.5625/lar.2012.28.4.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowman GL, Dodge H, Frei B, Calabrese C, Oken BS, Kaye JA, Quinn JF. Ascorbic acid and rates of cognitive decline in Alzheimer's disease. J Alzheimers Dis. 2009;16:93–98. doi: 10.3233/JAD-2009-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272:11943–11951. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- 74.Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, Han X, Weeber EJ, Bu G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feyder M, Karlsson RM, Mathur P, Lyman M, Bock R, Momenan R, Munasinghe J, Scattoni ML, Ihne J, Camp M, Graybeal C, Strathdee D, Begg A, Alvarez VA, Kirsch P, Rietschel M, Cichon S, Walter H, Meyer-Lindenberg A, Grant SG, Holmes A. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams' syndrome. Am J Psychiatry. 2010;167:1508–1517. doi: 10.1176/appi.ajp.2010.10040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 77.Kennedy MB. Signal transduction molecules at the glutamatergic postsynaptic membrane. Brain Res Brain Res Rev. 1998;26:243–257. doi: 10.1016/s0165-0173(97)00043-x. [DOI] [PubMed] [Google Scholar]
- 78.Ziff EB. Enlightening the postsynaptic density. Neuron. 1997;19:1163–1174. doi: 10.1016/s0896-6273(00)80409-2. [DOI] [PubMed] [Google Scholar]
- 79.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 80.Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 81.Yamada Y, Chochi Y, Takamiya K, Sobue K, Inui M. Modulation of the channel activity of the epsilon2/zeta1-subtype N-methyl D-aspartate receptor by PSD-95. J Biol Chem. 1999;274:6647–6652. doi: 10.1074/jbc.274.10.6647. [DOI] [PubMed] [Google Scholar]
- 82.Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483:213–217. doi: 10.1038/nature10841. [DOI] [PubMed] [Google Scholar]
- 83.Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, Milot G, Clark WM, Macdonald RL, Kelly ME, Boulton M, Fleetwood I, McDougall C, Gunnarsson T, Chow M, Lum C, Dodd R, Poublanc J, Krings T, Demchuk AM, Goyal M, Anderson R, Bishop J, Garman D, Tymianski M ENACT trial investigators. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:942–950. doi: 10.1016/S1474-4422(12)70225-9. [DOI] [PubMed] [Google Scholar]
- 84.Yan BC, Park JH, Ahn JH, Lee JC, Won MH, Kang IJ. Postsynaptic density protein (PSD)-95 expression is markedly decreased in the hippocampal CA1 region after experimental ischemia-reperfusion injury. J Neurol Sci. 2013;330:111–116. doi: 10.1016/j.jns.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 85.Zheng H, Dong Y, Xu Z, Crosby G, Culley DJ, Zhang Y, Xie Z. Sevoflurane anesthesia in pregnant mice induces neurotoxicity in fetal and offspring mice. Anesthesiology. 2013;118:516–526. doi: 10.1097/ALN.0b013e3182834d5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hongpaisan J, Sun MK, Alkon DL. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer's disease transgenic mice. J Neurosci. 2011;31:630–643. doi: 10.1523/JNEUROSCI.5209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 88.Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Jr, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]