

Abstract
This study combines for the first time two major approaches to understanding the function and structure of neural circuits: large-scale multielectrode recordings, and confocal imaging of labeled neurons. To achieve this end, we develop a novel approach to the central problem of anatomically identifying recorded cells, based on the electrical image: the spatiotemporal pattern of voltage deflections induced by spikes on a large-scale, high-density multielectrode array. Recordings were performed from identified ganglion cell types in the macaque retina. Anatomical images of cells in the same preparation were obtained using virally transfected fluorescent labeling or by immunolabeling after fixation. The electrical image was then used to locate recorded cell somas, axon initial segments, and axon trajectories, and these signatures were used to identify recorded cells. Comparison of anatomical and physiological measurements permitted visualization and physiological characterization of numerically dominant ganglion cell types with high efficiency in a single preparation.
Keywords: ganglion cells, immunohistochemistry, morphology, multielectrode array, retina, viral transfection
Introduction
To fully understand a neural circuit requires measurements of both its function and its structure. Among the many approaches currently in use, two approaches stand out for their completeness and precision. First, large-scale, high-density multielectrode extracellular recordings can probe the activity of many cells of different types with the efficiency, sensitivity, and spatiotemporal resolution necessary to understand the precise patterns of firing in a full network (Litke et al., 2004; Frechette et al., 2005; Marre et al., 2012). Second, 3D imaging and labeling can reveal the location, detailed morphology, and molecular identity of many individual cells of different types simultaneously (Wässle et al., 1981b; Bock et al., 2011; Briggman et al., 2011; Kleinfeld et al., 2011). Some methods, such as voltage or calcium imaging, can simultaneously provide some information about both structure and function. However, to achieve high resolution or signal-to-noise can require combining these methods with laborious methods, such as intracellular recordings or filling (Hofer et al., 2011). Thus, the more complete and precise measurements afforded by combining large-scale, high-resolution electrical recordings and optical imaging would likely advance understanding considerably, if they could be coordinated.
A fundamental challenge to this coordination is matching multielectrode recordings with anatomical measurements; that is, determining which electrical recordings correspond to which labeled cells in a given preparation. This will be referred to herein as the “matching problem.” Although it is usually possible to determine the approximate location of an extracellularly recorded cell by triangulation based on spike amplitude, the high density of neurons in many circuits, as well as the complex extracellular electric fields produced by spiking neurons, make a precise match in a real neural circuit difficult. The most direct solution to the matching problem is sequential intracellular (Dacey, 1993) or juxtacellular (Pinault, 1996) recordings and labeling in a single preparation, but this is inefficient for characterizing a large circuit.
A possible solution to the matching problem lies in the electrical image: the average pattern of voltage deflections introduced by a spike across the electrodes in a high-density array (Litke et al., 2004; Greschner et al., 2014). The electrical image can be measured by averaging over many spikes recorded from a single cell, revealing features of the soma, dendrites, and axon of the cell with striking resolution. This could permit substantially more accurate matching. Importantly, the electrical image can be obtained in parallel from all cells recorded simultaneously with a multielectrode array, a huge advantage for characterizing circuits composed of many neurons.
Here, we demonstrate that the electrical image is a powerful tool for solving the matching problem, forming an effective link between high-resolution electrophysiological and anatomical views of the neural circuitry of the retina. Large-scale, high-density multielectrode recordings in isolated primate retina were used to obtain the electrical image of hundreds of cells of identified types, simultaneously, while characterizing their light responses. Labeling with antibodies, and with rabies virus transfected GFP expression, was used to obtain morphological information about the cells in the recorded region of retina. Alignment and comparison of recordings with anatomical images revealed how the soma, spike initiation zone, and axon trajectory can be used to uniquely identify electrically recorded cells with their physical substrates. The confluence of methods made possible by electrical imaging thus permits a more comprehensive view of circuit structure and function.
Materials and Methods
Electrophysiological recording.
Retinas were obtained and recorded as described previously (Chichilnisky and Baylor, 1999; Field et al., 2007). Briefly, eyes were taken from terminally anesthetized macaque monkeys (Macaca mulatta, Macaca fascicularis, Macaca radiata), male and female, euthanized by other laboratories, in accordance with IACUC guidelines for the care and use of animals. Immediately after eye removal, the anterior portion of the eye and vitreous were removed in room light. Segments of peripheral (4.5–9 mm temporal equivalent; Chichilnisky and Kalmar, 2002) retina were dissected and placed flat, retinal ganglion cell (RGC) side down, on a planar array of extracellular microelectrodes. While recording, the retina was perfused with Ames' solution (31−36°C) bubbled with 95% O2 and 5% CO2, pH 7.4.
Two different electrode arrays equipped with custom designed integrated circuits were used. One consisted of 512 electrodes with 60 μm spacing, covering a rectangular region 1890 × 900 μm (Litke et al., 2004; Fig. 4). The other consisted of 519 electrodes with 30 μm spacing, covering a hexagonal region 450 μm on a side (Figs. 1, 2, 7; Gunning et al., 2007); 512 of these electrodes were used for recording and seven were disconnected. Electrodes were arranged in an isosceles triangular lattice with 60 μm (or 30 μm) interelectrode spacing within each row of electrodes, and 60 μm (or 30 μm) between rows. Extracellular voltages were digitized at 20 kHz per channel.
Figure 4.
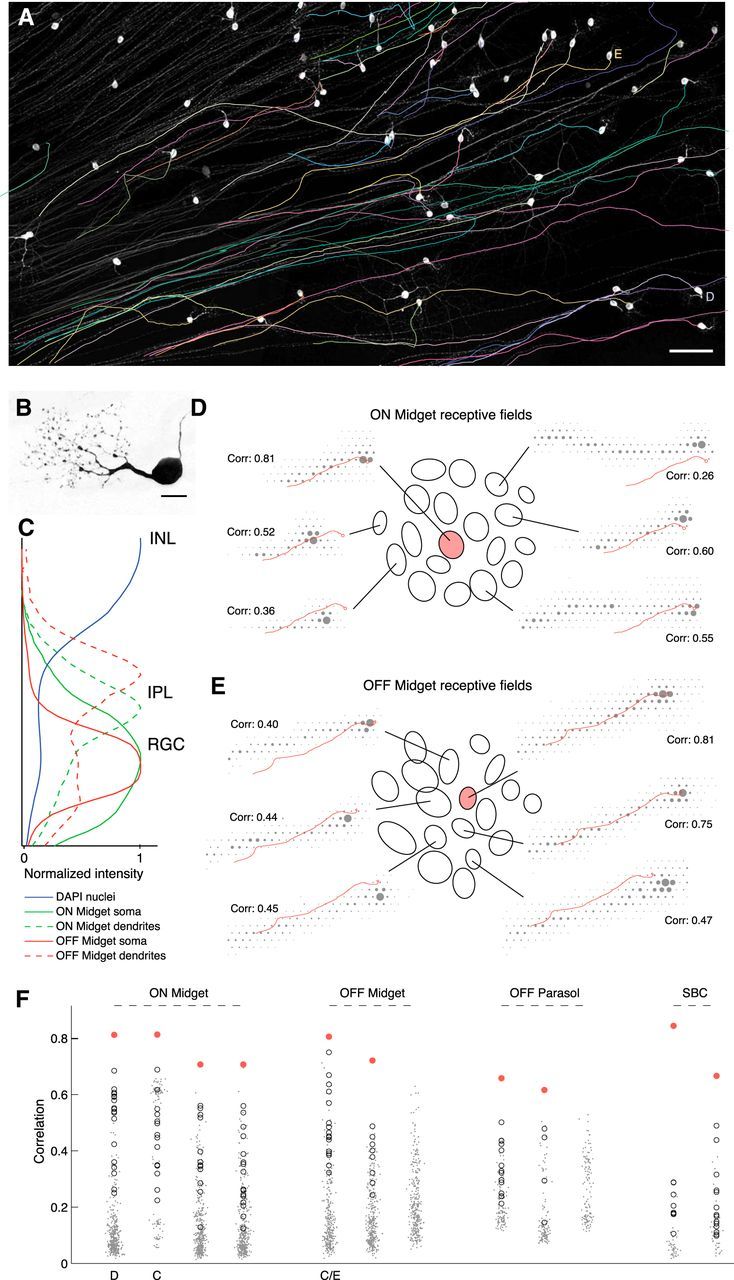
Matching axon trajectories. A, Traced axon trajectories (colored) are shown superimposed on grayscale image of GFP-labeled cells (see Materials and Methods). White somas and untraced axons are visible, traced axons obscured by traces. The cells used in D and E are labeled alongside. Scale bar, 100 μm. The imaged region corresponds closely to the borders of the rectangular recording array. B, Close-up image of a single labeled soma and dendrites with characteristic midget RGC morphology. Scale bar, 15 μm. This cell does not appear in other panels. C, Density of fluorescence over the normalized thickness of the inner nuclear layer (INL), inner plexiform layer (IPL), and RGC layer, within regions of interest local to two putative midget RGCs. DAPI signal is over the whole image and shows two peaks for nuclei dense regions at the centers of the RGC layer and INL. Density of somal GFP label peaks near the center of the RGC layer for both cells. Density of dendritic GFP label peaks in the proximal IPL for the putative ON midget, and in the distal IPL for the putative OFF midget, as expected for ON and OFF cell processes. D, Correspondence between a GFP-traced ON midget axon trajectory and selected recorded electrical images. Ellipses show Gaussian fits to the receptive fields of the putative matching RGC (shaded pink) and all surrounding RGCs of the same type (open) in the recording. For six of these RGCs, the electrical image is shown overlaid with the traced axon trajectory. Correlations indicated are between the recorded electrical image and the GFP-predicted electrical signal (see Materials and Methods). E, As in D, for an OFF midget axon tracing. F, Correlations for 12 axon tracings against full populations of recorded electrical images of the appropriate RGC type. In most cases, the best match (shaded pink) is clear relative to the rest of the population. The immediately surrounding RGCs, equivalent to the receptive field ellipses shown in C, D, are open circles. Remaining population are dots, with small random scatter on the x-axis to aid visualization. The cells used in C–E are labeled beneath. In two cases, there is no clear match among the recorded electrical images.
Figure 1.

Aligning confocal images of fixed tissue to live recordings. Left, Live retina over multielectrode array, peanut agglutinin labeling vasculature. Center, Same retina, fixed, peanut agglutinin still visible, overlapping HCN1 labeling channel (Fig. 5). Right, Fixed retina, same field as center, DAPI. Scale bars, 50 μm.
Figure 2.

Electrical image of retinal ganglion cell action potentials. A, Left, Raw voltage traces as a function of time recorded on six electrodes indicated in B. Times of easily identified spikes recorded from a single cell on Electrode 1 are identified as ticks at top. The average waveform of these five spikes and 2121 other similar spikes in the recording is shown at the right, expanded in time. Voltage traces for Electrodes 2–6 have the same time windows highlighted, corresponding to the time of the spike of the cell recorded on Electrode 1. Average waveforms on these electrodes, calculated on the same time windows, are shown at right. Distinct waveforms reveal characteristic average spike signatures for this cell on the different electrodes. Electrode 6 shows little or no voltage deflection associated with a spike in this cell. B, The maximum absolute amplitude of average voltage deflections from A are shown for each of the 519 electrodes in the hexagonal array, indicated by the diameter of the dot plotted at each electrode location. Large amplitudes near Electrode 1 are saturated. Apparent soma and dendrites are in cluster at top left, apparent axon extends down and right.
Figure 7.

Matching with multiple anatomical and physiological features. A, βIII-Tubulin immunolabeling, focused at level of RGC somas. Black nuclei surrounded by bright somal labeling mark cell locations. Bright striate labeling marks axon fascicles passing in and out of image plane. Some primary cellular processes are visible until they pass out of image plane. Putative parasol RGCs identified based on gross morphology (see text) marked with white diamonds. B, C, Highlighted areas from A, focused at level of axon initial segments. Immunolabeling for ankyrin-G marks multiple axon initial segments. DAPI marks nuclei, including many tubulin negative cells, putative amacrine cells, and blood vessels. White arrows show vector defined for a putative ON (B) and OFF (C) parasol cell, from the somal center to the center of the axon initial segment. Scale bar, 15 μm. D, E, Electrical images, putatively matching the cells in B, C. Color of each electrode disc indicates the timing of its peak negative signal relative to the time of the maximal amplitude somal spike. Black arrows show vector defined as pointing from the somal spike center of mass to the earlier peaking putative initial segment location. F, 2D histogram of linear assignment analysis (see text). The entire space of possible assignments was probed between anatomical and electrical image vectors for 14 putative parasol cells. Color shows density of possible assignments yielding cost metrics with the given amplitudes. Inset, The lower left extreme points, near optimal for both cost metrics. Global optimum circled in red. G, H, Same area as shown in A, focused on the primary dendritic elaborations of the ON (G, dashed yellow circles) and OFF (H, blue circles) parasol populations, as identified by the optimal point in F. Clean mosaic tiling confirms identification. ON parasol arbors lie closer to the RGC layer than OFF parasol arbors, as evident from additional somas still slightly in plane in G. Scale bar, 50 μm also applies to A.
Recordings were analyzed offline to isolate the spikes of different cells, as described previously (Litke et al., 2004; Field et al., 2007). Briefly, candidate spike events were detected using a threshold on each electrode, and the voltage waveforms on the electrode and nearby electrodes around the time of the spike were extracted. Clusters of similar spike waveforms were identified as candidate neurons if they exhibited a refractory period and an average rate >0.25 Hz. Duplicate recordings of the same cell were identified by temporal cross-correlation and removed.
Electrical images.
In a typical 30–60 min recording, each identified parasol, midget, or small bistratified cell fired 1000–100,000 identified action potentials. The electrical image for a given cell was computed from a 5 ms window starting 1 ms before the peak negative voltage sample for each spike, and was averaged over all recorded spikes. Frequently, the spatiotemporal electrical image was further collapsed across time by taking the maximal absolute voltage deflection at each electrode location, yielding a spatial representation as shown in Figures 2B, 4D,E, 7D,E.
In some cases, the electrical image was further processed to generate a current-source density representation. The differential sharpening effect of this calculation was particularly useful for manually identifying soma and initial segment locations in the electrical image (Fig. 7). Current-source density was calculated as the negative 2D Laplacian of the (arbitrarily scaled) electrical image (Plonsey and Barr, 2007). This assumes a uniform, passively resistive extracellular volume (Gold et al., 2006). In the present data, voltages were sampled at discrete points along a triangular lattice, with some points missing where electrodes were disconnected or damaged. Thus, an efficient method was used to calculate the Laplacian on arbitrarily scattered 2D data. For each electrode, the nearest neighboring electrodes were determined based on a Delaunay triangulation of all good electrodes for a given array. Gradients were calculated as the voltage difference between neighboring electrodes divided by their distance, and the divergence was calculated as the sum of gradients out of a given electrode, weighted by the length of the corresponding side in the dual Voronoi diagram. The Laplacian calculated this way is equivalent to assuming a planar interpolation of the initial data between each triplet of arbitrarily scattered electrode positions. Because the Laplacian is a linear function of the raw voltage, this procedure can be reduced to a sparse matrix, which was multiplied by each electrical image to generate its current-source density.
Spike propagation velocities for each cell were calculated from the average distance traveled by the putative axonal spike per sampling interval (Fig. 3). First, a temporal window corresponding to the triphasic axonal signal (Fig. 2A), with minimal contamination from the large preceding somal spike, was extracted from the full spatiotemporal electrical image. Depending on the speed of propagation and the distance to the edge of the electrode array, this window ranged from 0.45–2 ms (the corresponding total propagation distance range was 343–1817 μm, see below). The negative component of the electrical image within this window was then fit with a translating, radially symmetric 2D Gaussian, whose peak location was parameterized to move freely at each time step, and whose amplitude and variance were parameterized to take on a single best-fitting value for the entire window. The distance between consecutive fitted peak locations was taken as the spike propagation distance for each sampling interval, and the propagation velocity for each cell was reported as the average velocity over the window. No systematic changes in estimated velocity as a function of distance from the soma were observed.
Figure 3.

Axon conduction velocities estimated from electrical images. Histograms by cell type of estimated spike propagation velocity (see Materials and Methods) over the axonal portion of the electrical image for: 22 ON Parasol, 29 OFF Parasol, 124 ON Midget, 65 OFF Midget, and 15 small bistratified cells. Higher velocity in ON/OFF Parasol cells is consistent with larger axon caliber.
To quantify how well axon trajectories matched, cross-correlations were calculated between each recorded electrical image and a predicted electrical image based on the anatomically localized soma location and axon trajectory. The electrical image prediction consisted of only a maximal amplitude spatial profile (arbitrarily scaled) and did not evolve in time (Figs. 2B, 4D,E). Separate contributions were predicted for the somal and axonal signal at each electrode location (see below), and then on the assumption that somal and axonal signals would have little temporal overlap, the maximum of these two components was taken as the electrode maximum amplitude prediction.
The predicted somal and axonal contributions were computed as follows. Under a simple model of the action potential as a point sink of inward current propagating down the axon, and assuming a uniform, passively resistive extracellular volume, the somal and axonal contributions are given by: C(d2 + z2)−1/2, where d is the 2D distance (in μm) from the electrode to the soma or axon respectively, taken from the aligned anatomical tracing, z is a constant representing a static estimate of the depth of the soma or axon relative to the array (in μm), and C is a scaling constant to account for the relative amplitude of the somal and axonal currents. Cross-correlations were not highly sensitive to the estimated parameters C and z, and the following parameters yielded reasonable predictions across preparations and cell types (Fig. 4): for the axon: C = 1, z = 2.5; for the soma: C = 4, z = 5.
Virally mediated expression of GFP.
LGN-projecting retinal ganglion cells were retrogradely infected with glycoprotein-deleted rabies virus expressing GFP as described previously (Nassi and Callaway, 2007; Nhan and Callaway, 2012) except that viral injections were made into the LGN. Briefly, under isoflurane anesthesia a craniotomy was made above the LGN and electrical recordings were targeted to the expected stereotaxic coordinates of the LGN (Malpeli and Baker, 1975), while light was flashed into the eyes. Responses to light flashes as well as characteristic transitions between ipsilaterally and contralaterally driven responses in LGN layers indicated that the electrode was in the LGN and the corresponding stereotaxic coordinates were then used to target viral injections. The G-deleted rabies virus was injected by pressure at multiple locations in each LGN via a glass micropipette (tip diameter 20–50 μm, 0.5 μl per injection). Retinas were collected during terminal procedures 5–8 d later.
Immunohistochemistry.
Following recording, retinal pieces were fixed with 4% paraformaldehyde in PBS (10 mm) for 25–45 min at room temperature and then washed in PBS 3 × 10 min and left in PBS at 4°C for 6–48 h. Fixed tissue was incubated for several hours in blocking solution at room temperature and then incubated in blocking solution with primary antibody (Table 1) for 7 d at 4°C on a shaker. The blocking solution consisted of 5% normal donkey serum, 2% bovine serum albumin, and 0.5% Triton X-100 in PBS. In some cases, 0.05% sodium azide was added as a preservative.
Table 1.
Monoclonal antibodies used for immunohistochemistry
| Antigen | Dilution | Host Ig | Vendor | Catalog # |
|---|---|---|---|---|
| Rat HCN1 C-terminal aa 778–910 | 1:200 | Mouse IgG1 | Neuromab | 75-110 |
| Fusion ankyrin-G ∼1000 aa* | 1:200 | Mouse IgG2a | Neuromab | 75-146 |
| βIII-Tubulin C-terminal aa EAQGPK | 1:500 | Rabbit | Covance | MRB-435P |
| βIII-Tubulin C-terminal aa EAQGPK | 1:500 | Mouse IgG1 | Promega | G7121 |
*See datasheet.
Following incubation with primary antibody, tissue was washed in PBS 3 × 10 min and left in PBS at 4°C for 6–24 h. Tissue was then incubated with Cy3- or Cy5-conjugated secondary antibodies (Jackson ImmunoResearch) at 1:200 dilution for 2–3 d at 4°C on a shaker, washed in PBS 3 × 10 min, counterstained with DAPI 10 min, and mounted on slides in ProLong Gold antifade medium (Life Technologies).
Two monoclonal βIII-tubulin antibodies were tested, one raised in mouse (data not shown) and the other raised in rabbit (see Figs. 6, 7). In both cases, the antigen is six C-terminal amino acids (Table 1). The sequence of the Covance rabbit monoclonal was not published but was determined from the package labeling of the commercially available blocking peptide. Immunolabeling was indistinguishable between the two antibodies.
Figure 6.
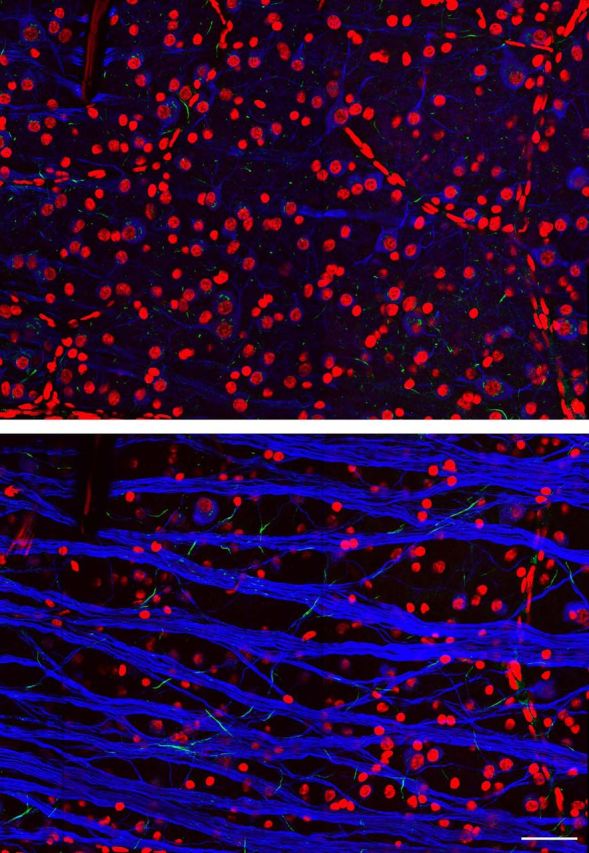
Immunolabeling RGC somas, axons, initial segments, and primary dendrites. RGB channels: DAPI, ankyrin-G, βIII-tubulin. Two z-slices show the top and bottom of the RGC layer. DAPI labeling indicates nuclei of putative RGCs, amacrine cells, and retinal blood vessels. βIII-tubulin labeling appears restricted to RGC somas, axons, and primary dendrites, with particularly bright labeling of axon fascicles. Ankyrin-G labels short segments of RGC axons near the soma, putative initial segments. In addition, ankyrin-G also labels numerous short βIII-tubulin-negative segments, suggesting axonal processes of non-RGCs. Scale bar, 50 μm.
Confocal microscopy, image processing, and alignment.
Imaging was performed on a modified Leica TCS SP2 confocal microscope (Leica) with a 63×, 1.40 numerical aperture oil-immersion objective. Montaged tilesets of image stacks were reassembled using the globally optimal stitching algorithms in ImageJ/FIJI (Preibisch et al., 2009). For Figure 4A, the 3D confocal stack was collapsed down to a 2D image via maximum intensity z-projection. For Figure 4B, the midget RGC image stack was collapsed via a SD z-projection.
For Figures 5, 6, and 7, image stacks were collapsed using a custom process. First, the stack was manually marked with scattered points in 3D, indicating the locations of image features to be highlighted. For example, several hundred points might be placed within the 3D volume to follow cellular features, such as primary dendritic arborizations (Fig. 7G,H). These scattered 3D points were then used to define the desired z-level for every x/y point over the 2D image area via linear interpolation. (For points without a valid interpolation; e.g., points along the outer edges of the x/y image area, the z-level of the nearest marked point was used.) These z-level values then defined the weighted blending of neighboring stack images used to calculate the final pixel value for each corresponding x/y image point. This approach allowed 2D renderings of image stacks to focus on arbitrary features within a given stack volume, so long as desired features did not exist at multiple z levels for a given x/y location (e.g., note the conflict between the cell nuclei and primary dendrites of some OFF parasol cells in Fig. 7H).
Figure 5.
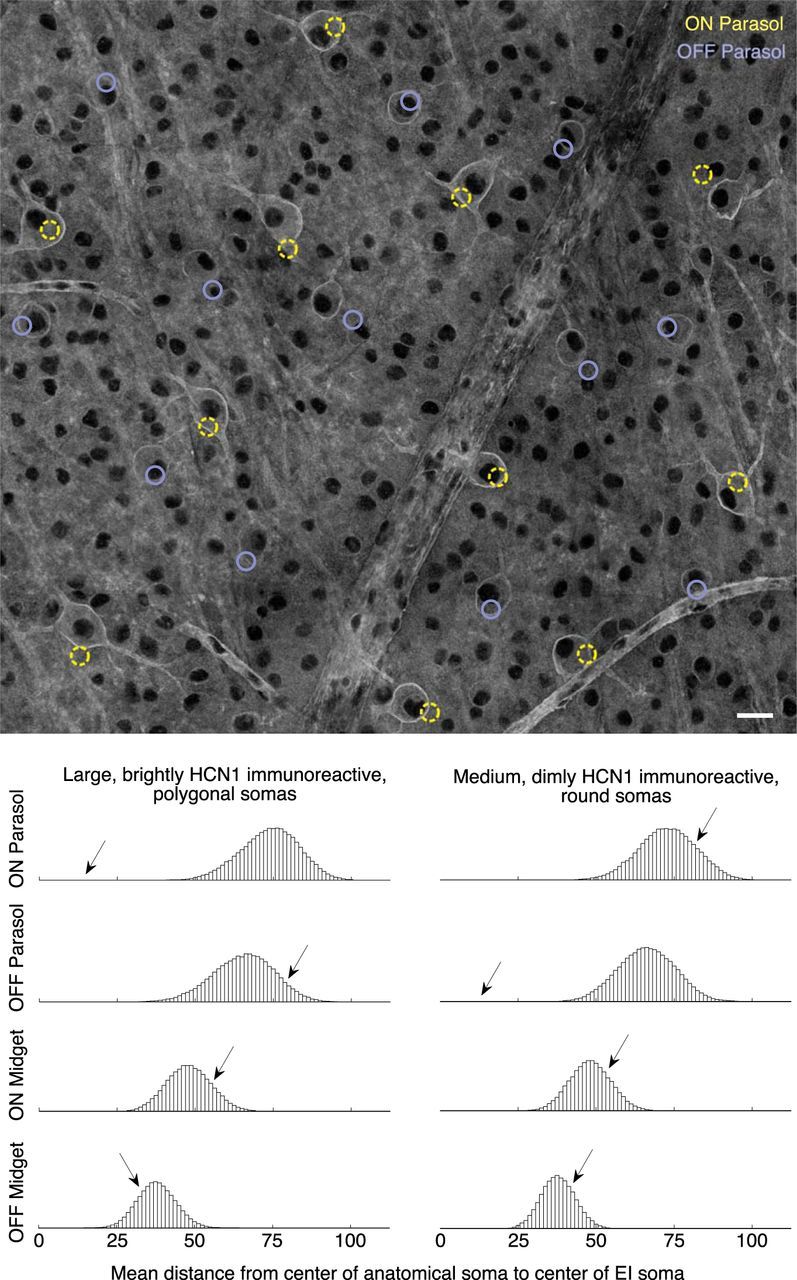
Matching molecularly identified subsets of cells. Top, Whole-mount HCN1 immunolabeled retina, at the level of the retinal ganglion cells. Black lacunae are cell nuclei, bright outlines are labeled cells. Rope-like structures are blood vessels (labeled by conjugated peanut lectin in the same channel). Colored circles represent the soma locations estimated from electrical images of ON and OFF parasol cells, with apparent correspondence to two distinct HCN1-labeled populations (see text). Scale bar, 25 μm. Bottom, Histograms showing results of permutation analysis (see Materials and Methods). Arrows indicate the location of the actual observed data; remaining data are from random permutations of electrical image-estimated locations. For most pairings, e.g., either HCN1-labeled population versus either population of midget electrical images, the actual data match no better than chance (arrows in the middle of permutation histograms). Two notable exceptions (arrows well outside permutation histograms) indicate probable matches.
Confocal images of fixed tissue were registered with electrical images from physiological recordings via alignment to epifluorescence images of the unfixed retina over the multielectrode array taken at the conclusion of the recording session (Fig. 1). The most common method for alignment was via the positions of retinal vasculature, visualized in the live retina by staining with fluorophore-conjugated peanut agglutinin (AlexaFluorophore 488 or 568, Life Technologies). Peanut agglutinin staining was often still visible in the fixed retina, or vasculature could alternatively be visualized in fixed retina via its DAPI signature of elongated nuclei in banded patterns (Fig. 1). In virally transfected tissue (Fig. 4), labeled cells were also visible in both live and fixed tissue, providing additional alignment points. Spatial distortions due to fixation were compensated by using a manually selected set of matching points in the images obtained before and after fixation, and mapping the complete images based on a polynomial fit of these matching points.
Permutation analyses.
Permutation tests were run to assess the probability that two apparently distinct populations of HCN1 immunolabeled RGCs (see Results) corresponded to known physiological classes (Fig. 5). Simulated mosaics of RGCs were generated based on soma locations identified in electrical images from selected RGC types. Actual recorded soma locations were randomized while maintaining their relative spacing by applying an arbitrary shift, rotation, and (in half the cases) a mirror inversion. Randomizations were constrained to keep the mosaic of soma locations covering the immunolabeled region of interest. Randomizations that were not sufficiently different from the original data (any randomized soma location <60 μm shifted from its original location) were discarded.
For each randomized mosaic, the mean distance from each randomized soma location to the nearest immunolabeled soma location was computed and the results of many repeated simulations were summarized as histograms (Fig. 5). The immunolabeled cells were divided into two populations based on their appearance (see Results), without reference to electrical recording data. A few cells that could not be confidently assigned to one population or the other were excluded. The p values for different matches were calculated in two ways. First, as the proportion of mosaics (all simulated plus one real) for which the mean distance was less than or equal to the mean distance for the real data. In the two cases of highly probable matches, there were no simulated data with lower mean distance, so the estimate of the probability of the observed mean distance based on chance arrangement was limited by the number of simulations performed (∼100,000). Thus, the reported value serves as an upper bound. As an alternative, p values were also calculated based on a one-sided test using an approximation of the distribution of simulations.
A permutation analysis was also performed to probe the space of possible assignments between 14 parasol electrical images (7 ON and 7 OFF) and 14 parasol cells identified based on their tubulin immunolabeled morphology (see Results; Fig. 7). Each possible permutation (14! = 87,178,291,200 total) of this linear assignment problem (Burkard et al., 2009) was generated iteratively in lexicographic order and for each permutation two distance-based cost metrics were calculated (see Results; Fig. 7F). This approach illustrated the full distribution of cost metrics for all possible assignments, but it is not feasible for larger match problems. For larger problems, polynomial time algorithms are known (Jonker and Volgenant, 1987) for efficiently finding the globally optimal assignment. Further searches of the space of possible assignments could then be initialized from this point.
Results
To characterize distinct retinal cell types and circuits, segments of peripheral macaque retina were recorded on large-scale, dense multielectrode arrays (512 channels, 30 or 60 μm electrode spacing; Litke et al., 2004), and visually stimulated with spatiotemporal noise. Spikes from different RGCs were sorted and then correlated with the visual stimulus to obtain the spike-triggered average stimulus for each cell, a characterization of the receptive field (Chichilnisky, 2001). Spatial, temporal, and chromatic properties of the receptive field were combined with other parameters, such as spike train autocorrelation, to segregate distinct RGC types (Chichilnisky and Kalmar, 2002; Field et al., 2007; Petrusca et al., 2007). Several RGC types identified this way each formed locally complete mosaics with their spatial receptive fields, tiling the retinal area over the array (Field et al., 2007; Fig. 4D,E) and confirming their correspondence to morphologically distinct cell types (Wässle et al., 1981a; Devries and Baylor, 1997). Light responses and density were used to identify the five numerically dominant RGC types in the primate retina: ON parasol, OFF parasol, ON midget, OFF midget, and small bistratified (Chichilnisky and Kalmar, 2002; Field et al., 2007).
To identify signatures of cellular morphology from the recordings, electrical images were calculated (Litke et al., 2004) by obtaining voltage waveforms recorded from every electrode on the array over a window of several milliseconds around each identified spike, and averaging these waveforms over all the spikes recorded from the cell. The electrical image thus calculated (Fig. 2) comprises several features indicative of gross cellular morphology. These features include a localized large biphasic spike waveform revealing the putative soma (Fig. 2A, trace 1), smaller opposite-sign spike waveforms in surrounding regions indicating the putative dendrites (Fig. 2A, trace 2), and a unidirectionally propagating triphasic spike indicating the putative axon (Fig. 2A, traces 3–5). The full spatiotemporal electrical image was summarized spatially by plotting discs centered at each electrode location with a diameter representing the peak amplitude of the signal (Fig. 2B).
The characterization of triphasic propagating signals (Fig. 2A, traces 3–5) as axonal was supported by analysis of propagation velocities (Fig. 3). The velocity of triphasic signal propagation clustered strongly by cell type, with parasol cells exhibiting significantly higher velocities than midget or small bistratified cells, as expected based on the larger axon caliber of parasol cells (see below) and from previous work (Dreher et al., 1976).
The putative cellular structures identified from electrical images provided a valuable tool for matching recorded cells with labeled cells. Three interrelated approaches to solving the matching problem based on electrical images were explored.
Matching axon trajectories of labeled and recorded cells
The extended trajectories of axons (∼100–1000 μm from the cell body) identified from electrical images provide one potential approach to solving the matching problem. The axons of most or all RGCs in any given preparation extended in a similar direction (Fig. 4A), reflecting their common trajectory toward the distant optic disc. However, the detailed trajectories for different cells were generally distinct (see below). These differences provided a distinguishing electrical signature for most cells.
To test the utility of the axon trajectory signature, we examined RGCs expressing GFP due to retrograde transfection with engineered rabies virus injected in the lateral geniculate nucleus (see Materials and Methods). GFP expression yielded clear images of a subset of cells over the electrode array (Fig. 4A), primarily ON and OFF midget and parasol cells, which together constitute ∼70% of RGCs in the primate retina (Dacey, 2004). To associate optically imaged and electrically recorded axons, the imaged axons were manually traced from confocal images obtained after the experiment, in fixed tissue (Fig. 4A, colored traces). The axon tracings from fixed tissue were then registered with the electrical images from physiological recordings via alignment to images of the multielectrode array, labeled cells, and retinal vasculature, taken at the end of the recording session (see Materials and Methods; Fig. 1). With this approach, electrical and anatomical images of axon trajectories could be compared.
Comparison of axon trajectories appeared to provide a solution to the matching problem in the conditions tested. The axons of each labeled RGC were compared with the electrical images obtained from all the recorded cells of the appropriate RGC type. Many candidate matches were excluded easily based on the obvious misalignment of the axon, soma, or both. Among the remaining recorded cells, one or more exhibited an electrical image that approximately aligned with the anatomical image (Fig. 4D,E). Closeness of alignment was quantified by computing the cross-correlation between each recorded electrical image and a predicted electrical image based on the traced anatomical soma and axon trajectory (see Materials and Methods). As expected, the electrical images of most cells exhibited an essentially random relationship to the predicted electrical image for the traced axon, and thus formed a broad distribution of cross-correlation values (Fig. 4F, small dots for each cell). Typically, however, several cells exhibited higher correlation coefficients, and one recorded cell stood out as the closest match to the traced axon (Fig. 4F, pink circles), suggesting it corresponded to the labeled cell. The correlation coefficient calculated this way generally corresponded to the visual impression of the most likely matching cell. More sophisticated simulated electrical images could improve this accuracy.
A potential problem with the above approach is that, because recording efficiency is generally lower than 100% of the cells in a region, the true matching cell might not have been recorded, in which case any identified match would be spurious. This concern could be eliminated if it could be demonstrated that: (1) the labeled cell was of a particular identified type, and (2) all the cells of that type were demonstrably recorded over the region. The mosaic organization of each RGC type was exploited to this end. Specifically, in the labeled cells found in one region of retina, ON midget cells were identified by their characteristic dendritic morphology (Watanabe and Rodieck, 1989; Dacey, 1993) and stratification (Famiglietti and Kolb, 1976; Nelson et al., 1978; Fig. 4B,C). In the recordings from the same region, a functionally homogeneous group of cells formed a nearly complete mosaic with their receptive fields (Fig. 4D, ellipses), indicating that they corresponded to a single morphological type (Wässle et al., 1981b). This cell type was identified as ON-midget, based on density and light response properties (Chichilnisky and Kalmar, 2002; Field et al., 2007). The traced axon of each labeled ON-midget cell matched the electrical image of one of the recorded ON-midget cells substantially more accurately than the rest (Fig. 4D), and the nearly complete mosaic of recorded ON-midget cells indicated that spurious matches were unlikely. Similar results were observed with OFF-midget cells (Figs. 4C,E), parasol cells, and small bistratified cells (Fig. 4F). Note two cases for which there was no clear match among the recorded cells.
In summary, comparison of the axon trajectory of labeled cells and the electrical images of recorded cells appeared to provide a solution to the matching problem, and the evidence for the accuracy of this solution was strengthened by complete recordings from identified cell types, limiting the concerns associated with unrecorded cells.
Matching somas of molecularly identified RGC types
When complete information about many axon paths is unavailable, another major feature of the electrical image that can potentially be exploited to solve the matching problem is the soma location. This information is more likely to produce a definitive match if the number of candidate matches can be restricted, for example by focusing on specific cell types. This approach was tested by comparing soma locations estimated from the electrical image of physiologically defined cell types to images of labeled cells of specific anatomically defined types.
Somal labeling of putative parasol cells was provided by a monoclonal antibody raised against the hyperpolarization-activated cyclic nucleotide gated channel protein HCN1 (Goloshchapov et al., 2009; Murray et al., 2012; Table 1). Confocal microscopy revealed two distinct immunoreactive RGC populations: one population with large polygonal somas and bright transmembrane label, and another population with slightly smaller, rounder somas and less bright label (Fig. 5A). The different soma shapes were qualitatively consistent with the expected differences in proximal dendrites of ON versus OFF parasol cells, which stratify in layers close to and far from the soma, respectively.
The soma locations of physiologically recorded ON and OFF parasol cells, blindly estimated by inspection of electrical images, approximately aligned with the bright and dim somas labeled by HCN1, respectively (Fig. 5A). The apparent alignment was quantified by calculating the distance from each immunolabeled soma to the nearest electrically localized soma of a given type. First, the immunolabeled somas were partitioned into two groups based on morphology alone: (1) brighter, larger, polygonal somas versus (2) dimmer, smaller, rounder somas. The mean distance from the bright somas to the nearest electrically localized ON parasol somas was 14 μm, whereas the mean distance to the nearest electrically localized OFF parasol somas was 73 μm. Conversely, the distance from dimmer somas to their nearest electrically localized ON or OFF parasol somas averaged 74 and 12 μm respectively. Thus, the alignment between ON parasol and bright somas, and between OFF parasol and dim somas, was much closer than the alternative combinations.
These results were inconsistent with chance alignment. To calculate the chance expectation, for each possible match, the electrically localized soma locations were transformed through random rotations, translations, and reflections (see Materials and Methods). After each such transformation, the mean distance between immunolabeled and electrically localized somas was recalculated. Histograms from repeated permutations illustrate the distribution of means expected for chance arrangements (Fig. 5B). Compared with this chance expectation, the match between bright somas and ON parasols was highly statistically significant (p < 0.00001; loose upper bound; see Materials and Methods), while the matches between bright somas and OFF parasol cells, ON midget cells, or OFF midget cells were consistent with chance arrangement (p ≈ 0.92, p ≈ 0.84, p ≈ 0.22). The match between dim somas and OFF parasol cells exhibited similar high statistical significance (p < 0.00001), whereas the match to ON parasol cells, ON midget cells, and OFF midget cells was consistent with chance (p ≈ 0.81, p ≈ 0.76, p ≈ 0.74). Another statistical test, based on a Gaussian approximation to the distributions in Figure 5, yielded probabilities of the two putative match mean distances occurring by chance of p < 2e-9, whereas the probabilities of the alternative configurations in Figure 5 were again in the range 0.2–0.9.
In summary, matching the soma locations of a subset of molecularly identified cells with soma locations inferred from electrical images of physiologically identified cell types provided a solution to the matching problem.
Matching soma, local structure, and axon initial segment
Although molecular identification of specific cell types can constrain the matching problem sufficiently to rely on soma locations, in some cases such molecular tags may not be available. In this case, matching could be augmented with information about the axon initial segment and other morphological features of the cells. This idea was tested by comparing additional cellular structure identified in electrical images with antibody staining that revealed axon initial segments and primary dendrites of multiple cell types.
Antibodies raised against β-III tubulin (Table 1) clearly labeled the long axons that distinguish RGCs from amacrine cells in the innermost layer of the retina, permitting a focus on RGCs (Fig. 6). They also clearly labeled additional RGC features, allowing a preliminary identification of 14 putative parasol RGCs (Fig. 7A, marks) based on their large somas, large nuclei, large caliber axon initial segments, and large caliber primary dendrites (Fig. 7A–C,G,H). Other RGC types were also preliminarily identified by distinct morphological features; for example, putative OFF midget cells could be distinguished based on brightly labeled primary dendrites which traced recursive, local paths into the distal inner plexiform layer (Watanabe and Rodieck, 1989; Dacey, 1993).
In addition, colabeling was performed with antibodies raised against the scaffolding protein ankyrin-G (Table 1), which localizes primarily to the axon initial segment, ∼30–80 μm from the soma (Fig. 7B,C). Tubulin and ankyrin colabeling permitted recovery of the relative locations of the soma and axon initial segment of most RGCs. Correspondingly, identification of the initial segment in physiological data was performed by manual inspection of the full spatiotemporal electrical image, including its current–source density representation (see Materials and Methods). The putative axon initial segment could be distinguished as a large negative voltage deflection that peaked slightly (0.3–0.2 ms) before the somal voltage deflection (Fig. 7D,E; see Discussion).
Plotting the soma location, initial segment location, and physiologically identified RGC subtype of every recorded cell over confocal images of the immunolabeled tissue allowed subjective manual matching of cells of several types. The logic of the matching procedure and verification is illustrated quantitatively below, focused on parasol cells.
First, a vector was defined for each parasol cell, connecting the soma location to the center of the ankyrin-labeled initial segment (Fig. 7B,C). Next, similar vectors were defined for each parasol cell electrical image (Fig. 7D,E), connecting the putative soma location to the apparent location of the initial segment based on the timing of voltage deflections. Over the field of view (Fig. 7A), this yielded 14 vectors for anatomical images of parasol cells, and 14 vectors for electrical images of parasol cells.
To explore possible matches, all assignments between the two sets of 14 cells were examined. For each assignment, two quantities reflecting spatial registration of the somas and initial segments were computed: (1) the mean-squared distance between electrical and anatomical soma locations (start of vectors) across cells, and (2) the mean-squared angular difference between the electrical and anatomical vector directions. The computed distribution of these two quantities for all 14! assignments occupied a broad range (Fig. 7F), with only a small number of assignments exhibiting low values for both quantities (inset). Neither somal distance nor angular distance yielded a clear best match alone, but a single assignment was identifiable as the best match by considering the two measures in combination (Fig. 7F, inset, point circled red).
This optimum assignment was corroborated by identifying ON and OFF parasol cells separately, both functionally (by polarity of light response) and morphologically (by manually tracing dendritic arbors). These tracings (Fig. 7G,H) revealed mosaic tiling of retinal area within each of the two subpopulations, with no overlap of the traced (major) dendrites within the ON or OFF parasol groups, but with significant overlap across groups, as expected (Dacey and Brace, 1992). Furthermore, the arbors of each cell stratified either relatively close to the RGC layer, as expected for ON cells (Fig. 7G) or relatively far into the inner plexiform layer, as expected for OFF cells (Fig. 7H; Famiglietti and Kolb, 1976; Nelson et al., 1978), and thus tended to exhibit either several comparable primary dendrites or a single long one. This morphological identification confirmed the match: the seven morphologically identified ON parasol cells corresponded to the seven physiologically defined ON parasol cells, and thus the same held for the OFF parasol cells (p < 0.0003).
Several extensions and caveats bear mention. Usually, the axon initial segment location identified in the electrical image simply corresponded to a point between the grossly identified soma location and initial axon trajectory (Figs. 2, 4, 7D,E). However, there were significant counter-examples (data not shown), in which the local excursions of the axon as it left the soma were apparently too fine relative to the electrode spacing to be revealed by the gross axon trajectory. In these cases, careful inspection of the temporal electrical image was critical. Also, aside from parasol cells, many midget cells were matched, as well as a few cells of other types: small bistratified cells, a sparser ON RGC type (data not shown, possibly the ON counterpart of the OFF upsilon type previously described; Petrusca et al., 2007), and polyaxonal amacrine cells (data not shown; Greschner et al., 2014). The numerically dominant ON and OFF midget cells were easily identified by their distinctive asymmetric morphology, and by their density in physiological recordings. Small bistratified cells could generally be matched in cases in which most of the surrounding midget cells had clear matches in the recording, and thus did not provide confounding potential matches. Additional, sparser cell types could also be tentatively matched as long as the surrounding denser cell types were already identified. The sparser cells exhibited dimmer tubulin immunoreactivity and distinctive morphologies, including an elongated soma smaller than parasol cells but larger than midget cells, and brighter primary dendrites that were thinner than parasol dendrites and farther reaching than midget dendrites. Finally, polyaxonal amacrine cells could be tentatively matched based on DAPI counterstaining of large nuclei, faint ankyrin-G immunoreactivity, and negligible tubulin labeling.
In summary, more complete information about the morphology of the soma, initial segment, and dendritic structure provided an approach to the matching problem that identified cells of several types to differing degrees.
Discussion
Three approaches were used to test whether the electrical image of spikes recorded on high-density multielectrode arrays could provide a link between large-scale functional and structural measurements in the retina. First, extended axon trajectories from different cells were strongly correlated in anatomical and electrical images. Second, soma locations from electrical and morphological images permitted alignment of molecularly identified cell types in a recording. Finally, the soma and proximal morphology of labeled cells were identified with the soma and initial segment location from electrical images, to varying degrees in several cell types. These findings suggest the promise of electrical imaging for associating functional and structural studies of neural circuits, and provide some specific approaches to doing so in the retina.
Axon matching
The unique trajectories taken by axons of different RGCs as they exit the soma and head toward the optic nerve provided one valuable matching approach. Usually, just one electrical image among those recorded exhibited close alignment to the full trajectory of a particular labeled axon, and others exhibited much less close alignment. This suggests that the physiological recording matching the labeled cell was correctly identified. In addition, when a subset of cells of a specific type were considered, via anatomical and electrophysiological cell type identification, the electrical image most closely aligned to a given anatomical image was clear. These findings by themselves do not imply that axon alignment correctly identifies every cell (for example, see failures in Fig. 4F), but provide evidence that the approach can work in many cases.
A drawback of this approach is that it requires painstaking identification of many axon trajectories in a region. In addition, in the retina, axons form bundles that can make it difficult to trace an axon over long distances, although this concern was alleviated by the fact that the initial portion of the axon tended to take a distinctive path toward the bundle. It remains to be seen whether this approach would be usable if all cells in a region were labeled; a very high density of labeled axons could make identification more difficult at standard confocal resolution.
Soma matching with identified cell types
Using the electrical image to identify soma locations permitted association of electrical and anatomical images of a subset of molecularly and physiologically identified RGCs. This technique is similar to triangulation based on spike amplitude recorded on different electrodes, but exploits the more complete information about kinetics and propagation of spikes available in the electrical image. Restriction of the matching problem to a subset of cells reduced the overall density of candidate matches, and thus rendered the problem more tractable. Although the restriction was accomplished here with immunolabeling, other cell-type-specific labeling methods, such as retrograde injections or restricted expression of identifying proteins based on cell type specific promoters, could also be used.
This approach also demonstrated the potential for using functionally identified cell types and electrical images to elucidate specificity of molecular expression. For example, the current results (Fig. 5) strongly suggested that the channel protein HCN1 is expressed selectively in parasol cells, consistent with earlier results (Goloshchapov et al., 2009; Murray et al., 2012), and further indicated that this expression is significantly stronger in ON parasol cells than in OFF parasol cells. These observations could be tested further by investigating membrane currents in patch-clamp recordings.
A limitation of this approach is having access to the appropriate molecular markers for the cell type of interest. This problem should be alleviated by the discovery of more markers, particularly in the mouse (Trimarchi et al., 2007; Siegert et al., 2009; Kim et al., 2010; Kay et al., 2011; Rivlin-Etzion et al., 2011; Dhande et al., 2013; Ivanova et al., 2013). It also requires ready functional classification of cell types, a capability that is also growing, particularly in rodent species, such as guinea pig (M. Grivich, M. Greschner, A. Litke, and E.J. Chichilnisky, unpublished data), rat (Ravi et al., 2013), and mouse (A. Sher and G. Field, unpublished data). Finally, the efficacy of the technique depends on how easily electrical images from different cells can be distinguished, which in turn depends on the density of the RGCs and the electrode array. These issues will likely require additional investigation, particularly for use in central retina or in species with denser RGC layers.
Soma, local structure, and initial segment matching
Labeling with antibodies for βIII-tubulin and ankyrin-G demonstrated that axon trajectory, initial segment location, and primary dendritic morphology was sufficient to segregate multiple cell types and identify matched features of the electrical image including the apparent initial segment. This combined evidence yielded a fairly clear set of matches for some cell types, and suggestive evidence for matches of others. Although the details of the approaches could differ in other neural circuits, the general concept of using several converging lines of evidence from detailed morphological characterization and analysis of electrical images may generalize. This kind of approach leverages the advantages of simultaneous recordings, which yield a generally unique spatial arrangement of cells of different types.
βIII-tubulin is a neuron-specific tubulin subtype (Sullivan, 1988) that has been used previously as a marker for early differentiating RGCs (Watanabe et al., 1991) or as a putative pan-RGC marker for counting surviving RGCs in optic nerve damage disease models (Cui et al., 2003). Figures 6, 7 illustrate that immunolabeling for βIII-tubulin can reveal detailed cellular morphology for many RGCs, while leaving most displaced amacrine cells and other cells in the RGC layer unlabeled. More subtly, βIII-tubulin appears to label parasol, midget, and small bistratified RGCs most strongly, whereas sparser RGC types are labeled more weakly (data not shown).
In the electrical image, the putative location of the initial segment was identified by the location of earliest peak voltage deflection. This earlier deflection could be a consequence of several factors, including: (1) earlier spike initiation at the initial segment, (2) more rapid voltage changes due to the passive properties of the initial segment, or (3) more rapid rebound of the initial spike voltage deflection due to active conductances in the initial segment. Regardless of its origin, the systematic early deflection was useful for matching.
A limitation of this approach is its complexity. Identification of cell types from their morphological features (e.g., dendritic structure) depended on extensive knowledge built on decades of retinal research. Physiological identification of multiple cell types relied on light response characterizations in the macaque retina, which may not be readily available in other circuits or species. This could be a limiting factor if only a small subset of cells are identifiable morphologically and physiologically. In addition, although electrical images strongly suggested the location of the axon initial segment, which corresponded roughly to the location of ankyrin-G staining, a more complete model of electrical image formation on the array would be necessary to exploit additional structure such as the shape and extent of the dendrites. Finally, double-labeling with βIII-tubulin and ankyrin-G limited the available spectral imaging channels for additional labels. One possible alternative may be a pan-sodium channel antibody (Boiko et al., 2003), which may allow identification of both the soma and axon initial segment with a single channel.
Future directions
A novel approach to simultaneous analysis of structure and function is 3D reconstruction of electron microscope images, coupled with multiphoton calcium imaging of neural activity (Bock et al., 2011; Briggman et al., 2011). Such an approach has strong advantages over the approach described here, including revealing morphology and connectivity at extremely high resolution and completeness, and the capacity to record complete populations of cells in 3D structures in vivo. On the other hand, reconstruction is extremely difficult and time-consuming, and calcium sensors fail to reveal the fine timing structure of electrical signals. Hybrid approaches, such as combining 3D electron microscopy with large-scale multielectrode recording, could be valuable in the future. Depending on how well the current approaches generalize beyond the retina, using flat or 3D electrode arrays, additional methods may be necessary to solve the matching problem in other neural circuits.
Other scalable and parallel approaches to solving the matching problem could supplant some of the techniques presented here. For example, simultaneous calcium imaging with extracellular recordings may permit calculation of the spike-triggered calcium signal, which would be expected to reveal the precise location of the somas of all cells recorded, rapidly and in living tissue. Such an approach would exploit parallel recordings, would involve minimal data analysis, and would permit additional targeted experimental manipulations in living tissue or postfixation immunolabeling for molecular identification of cell types.
Until such methods are developed, the electrical image provides a ready bridge between anatomical and physiological measurements that may enable advances in diverse areas. A basic application would be in providing ground truth for detailed biophysical understanding of extracellular potentials generated by neural activity (Gold et al., 2006). Other possible applications include structure–function studies, physiological characterization of anatomically defined cell types, and elucidation of molecular specificity. In particular, considerable effort is being devoted to developing new cell-specific labels (Trimarchi et al., 2007; Kim et al., 2010; Dhande et al., 2013; Ivanova et al., 2013), including several approaches that yield rich, combinatorial patterns of labeling (Marc and Jones, 2002; Micheva and Smith, 2007; Badea et al., 2009; Badea and Nathans, 2011). Matching these anatomical approaches with multielectrode recordings via the electrical image could reveal the physiological substrates of such patterns, and thus help clarify the relationship between molecular specificity and functional specializations of cell types.
Footnotes
This work was supported by NIH Grant EY017992 (E.J.C.), NSF Postdoctoral Research Fellowship 1003198 (P.H.L.), VolkswagenStiftung (M.G.), NIH Grant EY022577 and The Gatsby Charitable Foundation (E.M.C.), NIH National Research Service Award F31NS054519 and Chapman Foundation (J.L.G.), and the Helen Hay Whitney Foundation (G.D.F.). We thank Clare Hulse, Daniela Amado, Devon Sandel, Brittany Cruz, Matthew Grivich, Steve Barry, David Chambers, and Stephan Preibisch for technical assistance, and Lauren Jepson for helpful discussions.
The authors declare no competing financial interests.
References
- Badea TC, Nathans J. Morphologies of mouse retinal ganglion cells expressing transcription factors Brn3a, Brn3b, and Brn3c: analysis of wild type and mutant cells using genetically-directed sparse labeling. Vision Res. 2011;51:269–279. doi: 10.1016/j.visres.2010.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea TC, Cahill H, Ecker J, Hattar S, Nathans J. Distinct roles of transcription factors brn3a and brn3b in controlling the development, morphology, and function of retinal ganglion cells. Neuron. 2009;61:852–864. doi: 10.1016/j.neuron.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock DD, Lee WC, Kerlin AM, Andermann ML, Hood G, Wetzel AW, Yurgenson S, Soucy ER, Kim HS, Reid RC. Network anatomy and in vivo physiology of visual cortical neurons. Nature. 2011;471:177–182. doi: 10.1038/nature09802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci. 2003;23:2306–2313. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, Helmstaedter M, Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471:183–188. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- Burkard RE, Dell'Amico M, Martello S. Assignment problems. Philadelphia, PA: Society for Industrial and Applied Mathematics; 2009. [Google Scholar]
- Chichilnisky EJ. A simple white noise analysis of neuronal light responses. Network. 2001;12:199–213. doi: 10.1080/net.12.2.199.213. [DOI] [PubMed] [Google Scholar]
- Chichilnisky EJ, Baylor DA. Receptive-field microstructure of blue-yellow ganglion cells in primate retina. Nat Neurosci. 1999;2:889–893. doi: 10.1038/13189. [DOI] [PubMed] [Google Scholar]
- Chichilnisky EJ, Kalmar RS. Functional asymmetries in ON and OFF ganglion cells of primate retina. J Neurosci. 2002;22:2737–2747. doi: 10.1523/JNEUROSCI.22-07-02737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yip HK, Zhao RC, So KF, Harvey AR. Intraocular elevation of cyclic AMP potentiates ciliary neurotrophic factor-induced regeneration of adult rat retinal ganglion cell axons. Mol Cell Neurosci. 2003;22:49–61. doi: 10.1016/S1044-7431(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Dacey DM. The mosaic of midget ganglion cells in the human retina. J Neurosci. 1993;13:5334–5355. doi: 10.1523/JNEUROSCI.13-12-05334.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacey DM. Origins of perception: retinal ganglion cell diversity and the creation of parallel visual pathways. In: Gazzaniga MS, editor. The Cognitive Neurosciences. Cambridge, MA: MIT; 2004. pp. 281–301. [Google Scholar]
- Dacey DM, Brace S. A coupled network for parasol but not midget ganglion cells in the primate retina. Vis Neurosci. 1992;9:279–290. doi: 10.1017/S0952523800010695. [DOI] [PubMed] [Google Scholar]
- Devries SH, Baylor DA. Mosaic arrangement of ganglion cell receptive fields in rabbit retina. J Neurophysiol. 1997;78:2048–2060. doi: 10.1152/jn.1997.78.4.2048. [DOI] [PubMed] [Google Scholar]
- Dhande OS, Estevez ME, Quattrochi LE, El-Danaf RN, Nguyen PL, Berson DM, Huberman AD. Genetic dissection of retinal inputs to brainstem nuclei controlling image stabilization. J Neurosci. 2013;33:17797–17813. doi: 10.1523/JNEUROSCI.2778-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher B, Fukada Y, Rodieck RW. Identification, classification and anatomical segregation of cells with X-like and Y-like properties in the lateral geniculate nucleus of old-world primates. J Physiol. 1976;258:433–452. doi: 10.1113/jphysiol.1976.sp011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famiglietti EV, Jr, Kolb H. Structural basis for ON-and OFF-center responses in retinal ganglion cells. Science. 1976;194:193–195. doi: 10.1126/science.959847. [DOI] [PubMed] [Google Scholar]
- Field GD, Sher A, Gauthier JL, Greschner M, Shlens J, Litke AM, Chichilnisky EJ. Spatial properties and functional organization of small bistratified ganglion cells in primate retina. J Neurosci. 2007;27:13261–13272. doi: 10.1523/JNEUROSCI.3437-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frechette ES, Sher A, Grivich MI, Petrusca D, Litke AM, Chichilnisky EJ. Fidelity of the ensemble code for visual motion in primate retina. J Neurophysiol. 2005;94:119–135. doi: 10.1152/jn.01175.2004. [DOI] [PubMed] [Google Scholar]
- Gold C, Henze DA, Koch C, Buzsáki G. On the origin of the extracellular action potential waveform: A modeling study. J Neurophysiol. 2006;95:3113–3128. doi: 10.1152/jn.00979.2005. [DOI] [PubMed] [Google Scholar]
- Goloshchapov AV, van der List DA, Chalupa LM, Usrey WM. Kv1.2 and HCN1 potassium channels are selectively expressed in a subset of large retinal ganglion cells in the macaque monkey. Soc Neurosci Abstr. 2009;35:848.25. [Google Scholar]
- Greschner M, Field GD, Li PH, Schiff ML, Gauthier JL, Ahn D, Sher A, Litke AM, Chichilnisky EJ. A polyaxonal amacrine cell population in the primate retina. J Neurosci. 2014;34:3597–3606. doi: 10.1523/JNEUROSCI.3359-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning D, Chichilnisky E, Litke AM, O'Shea V, Smith KM, Mathieson K. Performance of ultra-high-density microelectrode arrays. Nucl Instrum Meth A. 2007;576:215–219. doi: 10.1016/j.nima.2007.01.155. [DOI] [Google Scholar]
- Hofer SB, Ko H, Pichler B, Vogelstein J, Ros H, Zeng H, Lein E, Lesica NA, Mrsic-Flogel TD. Differential connectivity and response dynamics of excitatory and inhibitory neurons in visual cortex. Nat Neurosci. 2011;14:1045–1052. doi: 10.1038/nn.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova E, Lee P, Pan ZH. Characterization of multiple bistratified retinal ganglion cells in a purkinje cell protein 2-Cre transgenic mouse line. J Comp Neurol. 2013;521:2165–2180. doi: 10.1002/cne.23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker R, Volgenant A. A shortest augmenting path algorithm for dense and sparse linear assignment problems. Computing. 1987;38:325–340. doi: 10.1007/BF02278710. [DOI] [Google Scholar]
- Kay JN, De la Huerta I, Kim IJ, Zhang Y, Yamagata M, Chu MW, Meister M, Sanes JR. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. J Neurosci. 2011;31:7753–7762. doi: 10.1523/JNEUROSCI.0907-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IJ, Zhang Y, Meister M, Sanes JR. Laminar restriction of retinal ganglion cell dendrites and axons: subtype-specific developmental patterns revealed with transgenic markers. J Neurosci. 2010;30:1452–1462. doi: 10.1523/JNEUROSCI.4779-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfeld D, Bharioke A, Blinder P, Bock DD, Briggman KL, Chklovskii DB, Denk W, Helmstaedter M, Kaufhold JP, Lee WC, Meyer HS, Micheva KD, Oberlaender M, Prohaska S, Reid RC, Smith SJ, Takemura S, Tsai PS, Sakmann B. Large-scale automated histology in the pursuit of connectomes. J Neurosci. 2011;31:16125–16138. doi: 10.1523/JNEUROSCI.4077-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litke AM, Bezayiff N, Chichilnisky EJ, Cunningham W, Dabrowski W, Grillo AA, Grivich M, Grybos P, Hottowy P, Kachiguine S. What does the eye tell the brain? Development of a system for the large-scale recording of retinal output activity. IEEE Trans Nuclear Sc. 2004;51:1434–1440. doi: 10.1109/TNS.2004.832706. [DOI] [Google Scholar]
- Malpeli JG, Baker FH. The representation of the visual field in the lateral geniculate nucleus of Macaca mulatta. J Comp Neurol. 1975;161:569–594. doi: 10.1002/cne.901610407. [DOI] [PubMed] [Google Scholar]
- Marc RE, Jones BW. Molecular phenotyping of retinal ganglion cells. J Neurosci. 2002;22:413–427. doi: 10.1523/JNEUROSCI.22-02-00413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marre O, Amodei D, Deshmukh N, Sadeghi K, Soo F, Holy TE, Berry MJ., 2nd Mapping a complete neural population in the retina. J Neurosci. 2012;32:14859–14873. doi: 10.1523/JNEUROSCI.0723-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KD, van der List DAM, UW Molecular specificity of monkey retinal ganglion cells during development. Soc Neurosci Abstr. 2012;38:880.27. [Google Scholar]
- Nassi JJ, Callaway EM. Specialized circuits from primary visual cortex to V2 and area MT. Neuron. 2007;55:799–808. doi: 10.1016/j.neuron.2007.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R, Famiglietti EV, Jr, Kolb H. Intracellular staining reveals different levels of stratification for on- and off-center ganglion cells in cat retina. J Neurophysiol. 1978;41:472–483. doi: 10.1152/jn.1978.41.2.472. [DOI] [PubMed] [Google Scholar]
- Nhan HL, Callaway EM. Morphology of superior colliculus- and middle temporal area-projecting neurons in primate primary visual cortex. J Comp Neurol. 2012;520:52–80. doi: 10.1002/cne.22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrusca D, Grivich MI, Sher A, Field GD, Gauthier JL, Greschner M, Shlens J, Chichilnisky EJ, Litke AM. Identification and characterization of a Y-like primate retinal ganglion cell type. J Neurosci. 2007;27:11019–11027. doi: 10.1523/JNEUROSCI.2836-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or neurobiotin. J Neurosci Methods. 1996;65:113–136. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- Plonsey R, Barr RC. Bioelectricity: a quantitative approach. New York: Springer; 2007. [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi S, Ahn DM, Litke A, Chichilnisky EJ, Field G. Toward a complete functional classification of ganglion cells in the rat retina. ARVO Abstr. 2013:3387–C0108. [Google Scholar]
- Rivlin-Etzion M, Zhou K, Wei W, Elstrott J, Nguyen PL, Barres BA, Huberman AD, Feller MB. Transgenic mice reveal unexpected diversity of on-off direction-selective retinal ganglion cell subtypes and brain structures involved in motion processing. J Neurosci. 2011;31:8760–8769. doi: 10.1523/JNEUROSCI.0564-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegert S, Scherf BG, Del Punta K, Didkovsky N, Heintz N, Roska B. Genetic address book for retinal cell types. Nat Neurosci. 2009;12:1197–1204. doi: 10.1038/nn.2370. [DOI] [PubMed] [Google Scholar]
- Sullivan KF. Structure and utilization of tubulin isotypes. Annu Rev Cell Biol. 1988;4:687–716. doi: 10.1146/annurev.cb.04.110188.003351. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Stadler MB, Roska B, Billings N, Sun B, Bartch B, Cepko CL. Molecular heterogeneity of developing retinal ganglion and amacrine cells revealed through single cell gene expression profiling. J Comp Neurol. 2007;502:1047–1065. doi: 10.1002/cne.21368. [DOI] [PubMed] [Google Scholar]
- Wässle H, Peichl L, Boycott BB. Dendritic territories of cat retinal ganglion cells. Nature. 1981a;292:344–345. doi: 10.1038/292344a0. [DOI] [PubMed] [Google Scholar]
- Wässle H, Peichl L, Boycott BB. Morphology and topography of on- and off-alpha cells in the cat retina. Proc R Soc Lond B Biol Sci. 1981b;212:157–175. doi: 10.1098/rspb.1981.0032. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Rodieck RW. Parasol and midget ganglion cells of the primate retina. J Comp Neurol. 1989;289:434–454. doi: 10.1002/cne.902890308. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Rutishauser U, Silver J. Formation of the retinal ganglion cell and optic fiber layers. J Neurobiol. 1991;22:85–96. doi: 10.1002/neu.480220109. [DOI] [PubMed] [Google Scholar]