

Abstract
Angelman syndrome (AS) is a neurogenetic disorder caused by deficiency of maternally expressed ubiquitin-protein ligase E3A (UBE3A), an E3 ligase that targets specific proteins for proteasomal degradation. Although motor function impairment occurs in all patients with AS, very little research has been done to understand and treat it. The present study focuses on Ube3A deficiency-induced alterations in signaling through the mechanistic target of rapamycin (mTOR) pathway in the cerebellum of the AS mouse model and on potential therapeutic applications of rapamycin. Levels of tuberous sclerosis complex 2 (TSC2), a negative regulator of mTOR, were increased in AS mice compared with wild-type mice; however, TSC2 inhibitory phosphorylation was also increased. Correspondingly, levels of phosphorylated/active mTOR were increased. Phosphorylation of the mTORC1 substrates S6 kinase 1 (S6K1) and S6 was elevated, whereas that of the mTORC2 substrates AKT and N-myc downstream regulated 1 was decreased, suggesting enhanced mTORC1 but inhibited mTORC2 signaling. Semi-chronic treatment of AS mice with rapamycin not only improved their motor performance but also normalized mTORC1 and mTORC2 signaling. Furthermore, inhibitory phosphorylation of rictor, a key regulatory/structural subunit of the mTORC2 complex, was increased in AS mice and decreased after rapamycin treatment. These results indicate that Ube3A deficiency leads to overactivation of the mTORC1–S6K1 pathway, which in turn inhibits rictor, resulting in decreased mTORC2 signaling in Purkinje neurons of AS mice. Finally, rapamycin treatment also improved dendritic spine morphology in AS mice, through inhibiting mTORC1 and possibly enhancing mTORC2-mediated regulation of synaptic cytoskeletal elements. Collectively, our results indicate that the imbalance between mTORC1 and mTORC2 activity may contribute to synaptic pathology and motor impairment in AS.
Keywords: Angelman syndrome, mTOR, phosphorylation, Purkinje neuron, rictor, TSC2
Introduction
Angelman syndrome (AS) is a genetic neurodevelopmental disorder characterized by severe developmental delay, language and cognition deficits, motor impairment manifested with gait imbalance and ataxia (Barry et al., 2005; Chamberlain et al., 2010), and, in some patients with AS, seizure activity (Peters et al., 2004; Barry et al., 2005; Dan, 2009). AS is caused by deficiency in expression of the maternally inherited ubiquitin-protein ligase E3A (UBE3A) gene (Lalande and Calciano, 2007). Mice with maternal Ube3A deficiency (AS mice) exhibit impairment in long-term potentiation, learning and memory (Jiang et al., 1998; van Woerden et al., 2007; Baudry et al., 2012; Kaphzan et al., 2012), and motor dysfunction (Jiang et al., 1998; Heck et al., 2008; Huang et al., 2013). Although several laboratories have studied hippocampal-related learning and memory impairments in AS mice (Weeber et al., 2003; Greer et al., 2010; Baudry et al., 2012; Cao et al., 2013), cerebellum-dependent motor dysfunction, which is a characteristic feature of AS, has not been addressed to the same extent.
The mechanistic target of rapamycin (mTOR) is an integrator of signaling from growth factors and energy status and plays critical roles in both brain development and synaptic plasticity. The two mTOR complexes, mTORC1, coupled to raptor, and mTORC2, coupled to rictor, respond to different signals and target different substrates; mTORC2 is mainly stimulated by growth factors, whereas mTORC1 responds to a number of signals, including growth factors, amino acids, oxygen, and energy status (Laplante and Sabatini, 2012). Abnormal mTOR signaling has been implicated previously in many neurodevelopmental disorders, including autism spectrum disorders (ASDs; for a recent review, see Ehninger and Silva, 2011).
Previous reports have linked abnormal levels of UBE3A expression to ASDs (Guffanti et al., 2011; Smith et al., 2011). Moreover, UBE3A has been shown to degrade tuberous sclerosis complex 2 (TSC2) (Zheng et al., 2008), a component of the TSC1/TSC2 complex that inhibits mTOR activation. Selective knock-out of TSC1 or TSC2 in Purkinje neurons alone induces autism-like behaviors in mice (Tsai et al., 2012; Reith et al., 2013), as well as Purkinje cell degeneration (Reith et al., 2011; Tsai et al., 2012). Therefore, we investigated whether maternal Ube3A deficiency affected levels of TSC2 and altered mTOR signaling in the cerebellum of AS mice through modifications of specific protein kinase(s), because TSC2 activity has also been shown to be regulated by several kinases, including AKT, ERK1/2, and p90 ribosomal S6 kinase 1 (RSK1; for review, see Huang and Manning, 2008). Here we report that Ube3A deficiency resulted in increased levels of TSC2 and its inhibitory phosphorylation. The inhibitory TSC2 phosphorylation was mediated by a rapamycin-sensitive kinase. TSC2 inhibition was associated with increased mTORC1 activity as evidenced by increased S6 kinase 1 (S6K1) and S6 phosphorylation but decreased mTORC2 activity with reduced AKT and N-myc downstream regulated 1 (NDRG1) phosphorylation. Furthermore, mTORC2 inhibition was linked to S6K1-mediated inhibitory phosphorylation of its regulator, rictor. These results support the notion that, in AS mice, overactivation of mTORC1 as a consequence of TSC2 inhibition suppresses mTORC2 via S6K1-mediated rictor phosphorylation. Finally, rapamycin treatment improved dendritic spine morphology of Purkinje neurons and motor function in AS mice.
Materials and Methods
Animals
Ube3Atm1Alb/J mice (AS mice) were purchased from The Jackson Laboratory. Wild-type (WT) and AS mice were obtained in-house through breeding of heterozygous females with WT males. Genotype was determined as described previously (Baudry et al., 2012). Animal handling and experimental use followed protocols approved by the local Institutional Animal Care and Use Committee. In all experiments, male AS mice aged 2–4 months were used. Control mice were age-matched, male, WT littermates. Mice were raised on a 12 h light/dark cycle, with food and water available ad libitum, and were housed in groups of two to three per cage.
Rapamycin treatment
Rapamycin (LC Laboratories) was dissolved in 100% DMSO and stored in a stock solution of 5 mg/ml at −20°C. A working solution was prepared immediately before injection with a final concentration of 0.5 mg/ml rapamycin, 10% DMSO, 5% Tween-80, and 5% polyethylene glycol 400. Two- to 3-month-old male AS and WT mice were injected intraperitoneally with either rapamycin (5 mg/kg) or vehicle once a day for a total of 7 d before behavioral tests.
Behavioral tests
Each mouse underwent the behavioral tests in the same order, starting with the gait analysis, followed by the rotarod test.
Gait analysis.
For footprint gait analysis, mice paws were dipped in nontoxic paint, and mice were placed at an open end of a wooden tunnel (40 × 5 cm) lined with white paper. Mice were trained for 2 d to walk through the tunnel and then tested for two trials. Two to four steps from the middle portion of each run were analyzed for hind-stride length and hind-base width (distance between the right and left hindlimb strides, sway distance). At least five steps were measured for each mouse. Mean values were used for statistical analysis.
Rotarod test.
The experimental procedure was adapted from procedures described previously (Chen et al., 2009; Mulherkar and Jana, 2010) using a rotarod apparatus (Med Associates). The total testing period lasted 8 d: 1 training day, followed by 7 trial days. Rapamycin or vehicle was given 30 min before the rotarod test. For day 1 training, the rotor was set at a constant speed of 4 rpm, and animals were placed on the rod for 30 s. If the animal fell off the rod before the end of the 30 s, they were placed back on the rod. This was repeated until the animal could maintain itself on the rod for the full 30 s duration. Trial days 1 through 7 consisted of four trials of 5 min each. The rod was set to ramp up from 4 to 40 rpm over the course of 5 min. Four mice per trial were placed on the stationary rod, which was turned on once all mice were in place. The trial ended when the animal fell off the rod or at the end of 5 min, and the latency to fall was recorded in seconds. Trials were repeated four times per day with at least a 15 min rest period for each animal between trials. Data were expressed as the average time, in seconds (means ± SEMs), of the latency to fall.
Immunohistochemistry
Twenty-four hours after the last rapamycin treatment, mice were killed under deep anesthesia using pentobarbital (administered intraperitoneally) and were perfused through the left cardiac ventricle with PBS, followed by 4% paraformaldehyde. Sagittal cerebellar sections (25 μm) were prepared with a cryomicrotome. Free-floating sections were processed for immunohistochemical staining as described previously (Qin et al., 2009), with few modifications. In brief, tissue sections were blocked for 1 h at room temperature (RT) in 5% goat serum with 0.3% Triton X-100 in PBS. After blocking, sections were incubated in primary antibody diluted in a solution of 1% BSA with 0.3% Triton X-100 in PBS at 4°C overnight. Antibodies for the following were used: UBE3A (1:400; catalog #E8655; Sigma), calbindin (1:400; catalog #13176; Cell Signaling Technology), calbindin (1:500; catalog #300; Swant), phosphorylated (p) mTOR–Ser2481 (1:100; catalog #2974; Cell Signaling Technology), p-S6K1–Thr389 (1:100; catalog #9205; Cell Signaling Technology), and p-AKT–Ser473 (1:100; catalog #4060; Cell Signaling Technology). The following day, sections were briefly washed at RT three times in PBS, followed by incubation with secondary antibodies (Alexa Fluor-488 anti-mouse, 1:500; Alexa Fluor-594 anti-rabbit, 1:500; and Alexa Fluor-555 anti-rabbit, 1:500) diluted in a solution of 1% BSA with 0.3% Triton X-100 in PBS at RT for 2 h with gentle rocking. Sections were then washed four times at RT in PBS and mounted using Vectashield mounting media with DAPI to stain cell nuclei. Brain sections were imaged using a Nikon confocal microscope with a 60× objective.
Tissue collection, P2/S2 fractionation, and Western blot analysis
Twenty-four hours after the last rapamycin treatment, mice were deeply anesthetized using gaseous isoflurane and then decapitated. The whole brain was removed, and various brain regions including the cerebellum were dissected. Brain tissues were immediately frozen on dry ice and stored at −80°C until processed further.
For P2/S2 fractionation, frozen cerebellum tissue was homogenized in ice-cold HEPES-buffered sucrose solution (0.32 m sucrose and 4 mm HEPES, pH 7.4) with protease inhibitors. Homogenates were centrifuged at 900 × g for 10 min to remove large debris (P1). The supernatant (S1) was then centrifuged at 11,000 × g for 20 min to obtain the crude synaptosomal (P2) and cytosolic (S2) fractions. The P2 pellets were sonicated in RIPA buffer (10 mm Tris, pH 8, 140 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS). For whole homogenates, cerebellum tissue was homogenized in RIPA buffer. Protein concentrations were determined with a BCA protein assay kit (Pierce).
Western blots were performed according to published protocols (Wang et al., 2014). Briefly, 10–25 μg of total protein was separated on 8% SDS-PAGE gels and then transferred to PVDF membrane (Millipore). After blocking with 3% BSA for 1 h, membranes were incubated with specific antibodies overnight at 4°C. Antibodies for the following were used: UBE3A (1:2000; catalog #E8655; Sigma), raptor (1:1000; catalog #2280; Cell Signaling Technology), p-mTOR–Ser2448 (1:1000; catalog #2971; Cell Signaling Technology), p-mTOR–Ser2481 (1:1000; catalog #2974; Cell Signaling Technology), mTOR (1:1000; catalog #2972; Cell Signaling Technology), p-S6K1–Thr389 (1:1000; catalog #9205; Cell Signaling Technology), S6K1 (1:1000; catalog #9202; Cell Signaling Technology), p-S6–Ser235/236 (1:1000; catalog #2211; Cell Signaling Technology), p-S6–Ser240/244 (1:1000; catalog #2215; Cell Signaling Technology), S6 (1:1000; catalog #2217; Cell Signaling Technology), p-AKT–Ser473 (1:1000; catalog #4060; Cell Signaling Technology), p-AKT–Thr308 (1:500; catalog #4056; Cell Signaling Technology), AKT (1:1000; catalog #2920; Cell Signaling Technology), p-NDRG1–Thr346 (1:1000; catalog #3217; Cell Signaling Technology), p-TSC2–Thr1462 (1:1000; catalog #3611; Cell Signaling Technology), TSC2 (1:1000; catalog #4308; Cell Signaling Technology), p-rictor–Thr1135 (1:1000; catalog #3806; Cell Signaling Technology), rictor (1:1000; catalog #A300-459A; Bethyl), p-insulin receptor substrate 1 (IRS1)–Ser636/639 (1:1000; catalog #2388; Cell Signaling Technology), IRS1 (1:1000; catalog #sc-559; Santa Cruz Biotechnology), p-ERK (1:1000; catalog #9106; Cell Signaling Technology), ERK (1:1000; catalog #9102; Cell Signaling Technology), and β-actin (used as loading control; 1:10,000; catalog #A5541; Sigma). After incubation in the primary antibody, membranes were then incubated with IRDye secondary antibodies for 2 h at RT. Antibody binding was detected with the Odyssey imaging system. Images were analyzed for differences in levels using LI-COR Image Studio Software.
Dendritic spine analysis
Twenty-four hours after the last rapamycin treatment, mice were deeply anesthetized using gaseous isoflurane and then decapitated. The cerebellum was quickly removed, and Golgi impregnation was performed according to the protocol outlined in the FD Rapid GolgiStain Kit (FD Neurotechnologies). One hundred micrometer sagittal sections of the cerebellum were cut and mounted. Images of dendritic branches of cerebellar Purkinje cells were acquired using a Zeiss light microscope with a 60× objective. The number of spines located on randomly selected dendritic branches was counted manually by an investigator blind to genotype and treatment. Spine density was calculated by dividing the number of spines on a segment by the length of the segment and was expressed as the number of spines per micrometer of dendrite. Five to seven distal dendritic branches between 10 and 20 μm in length were analyzed and averaged for a slice mean.
Statistical analysis
Error bars indicate SEM. To compute p values, unpaired Student's t test and two-way ANOVA with Newman–Keuls post hoc test were used, as indicated in the figure legends.
Results
Abnormal mTOR pathway signaling in cerebellar Purkinje neurons of AS mice
We first determined the expression pattern of Ube3A in the cerebellum. Immunofluorescence staining showed that, in WT mice, Ube3A was mostly expressed in cell bodies and dendrites of Purkinje neurons, which were immunopositive for calbindin (Fig. 1A). In contrast, only very weak Ube3A immunoreactivity was detected in Purkinje neurons of AS mice. Ube3A deficiency in AS mice was confirmed by Western blot (Fig. 1B). Western blot analysis of whole homogenates from the cerebellum also showed that TSC2 levels were significantly higher in AS mice compared with WT mice. However, levels of p-mTOR at Ser2448 and Ser2481, the active isoforms of mTOR (Hoeffer and Klann, 2010), were also increased in AS mice (Fig. 1B,C), although levels of raptor (Fig. 1B,C), a component of mTORC1, were not different between WT and AS mice. Furthermore, levels of p-S6K1 (p-S6K1–Thr389) and S6 (p-S6–Ser235/236 and p-S6–Ser240/244), two downstream target proteins of mTORC1, were also significantly increased, suggesting that the activity of mTORC1 was enhanced in AS mice compared with WT mice. Conversely, levels of p-NDRG1 (p-NDRG1–Thr346), a downstream target protein of mTORC2, were significantly decreased, suggesting that the activity of mTORC2 was reduced in AS mice compared with WT mice. Immunostaining results confirmed that levels of both p-mTOR and p-S6K1 were increased in Purkinje neurons of AS mice (Fig. 1D,E). Furthermore, both p-mTOR and p-S6K1 showed puncta-like staining patterns along the dendrites of Purkinje neurons (Fig. 1D,E).
Figure 1.

Characterization of the mTOR pathway in the cerebellum of WT and AS mice. A, Expression of Ube3A in cerebellar Purkinje neurons immunostained with anti-calbindin antibody. Scale bar, 30 μm. B, Western blot analysis of the mTOR pathway proteins and their phosphorylation status. Cerebellar tissue homogenates prepared from 2- to 3-month-old WT and AS mice were processed for Western blot as described in Materials and Methods. C, Quantitative data of Western blots shown in B (means ± SEMs from 3–5 mice; *p < 0.05, Student's t test). D, E, Cerebellar sections from AS and WT mice were immunostained with antibodies recognizing mTOR phosphorylated at Ser2481 (red, D) or S6K1 phosphorylated at Thr389 (red, E) and the Purkinje cell marker calbindin (green, D, E). Higher levels of p-mTOR and p-S6K1 were observed in Purkinje neurons of AS mice compared with WT mice. Scale bar: top, 50 μm; bottom, 10 μm.
Increased mTORC1 activity contributes to motor dysfunction in AS mice
To test whether increased mTOR activation in Purkinje neurons could contribute to motor function impairment in AS mice, we used the rotarod paradigm and footprint gait analysis, which have been shown to be altered in AS mice (Jiang et al., 1998; Heck et al., 2008; Huang et al., 2013), to determine whether mTOR inhibition with rapamycin affected motor function in AS mice. AS mice and their WT littermates were treated daily with either rapamycin (5 mg/kg) or vehicle for 1 week, and the treatment was continued for another week during the rotarod test. Compared with vehicle-treated WT mice, vehicle-treated AS mice showed a shorter latency to fall each trial day (Fig. 2A). Rapamycin-treated AS mice performed significantly better than vehicle-treated AS mice starting from day 2 of the 7 d trial (Fig. 2A), although they did not perform as well as the WT mice. Rapamycin treatment did not affect WT mice performance. Gait was analyzed by footprint measurement at the end of the first week of rapamycin or vehicle treatment. Sway distance was significantly increased (Fig. 2B), whereas stride length was reduced (Fig. 2C), resulting in a wider and shorter gait in AS mice compared with WT mice. Rapamycin treatment decreased sway distance and increased stride length in AS mice, revealing a significantly improved motor coordination (Fig. 2B,C; no difference from WT mice), but it had no effect on WT mice. These results suggest that increased cerebellar mTOR activity contributes, at least partially, to motor dysfunction in AS mice.
Figure 2.
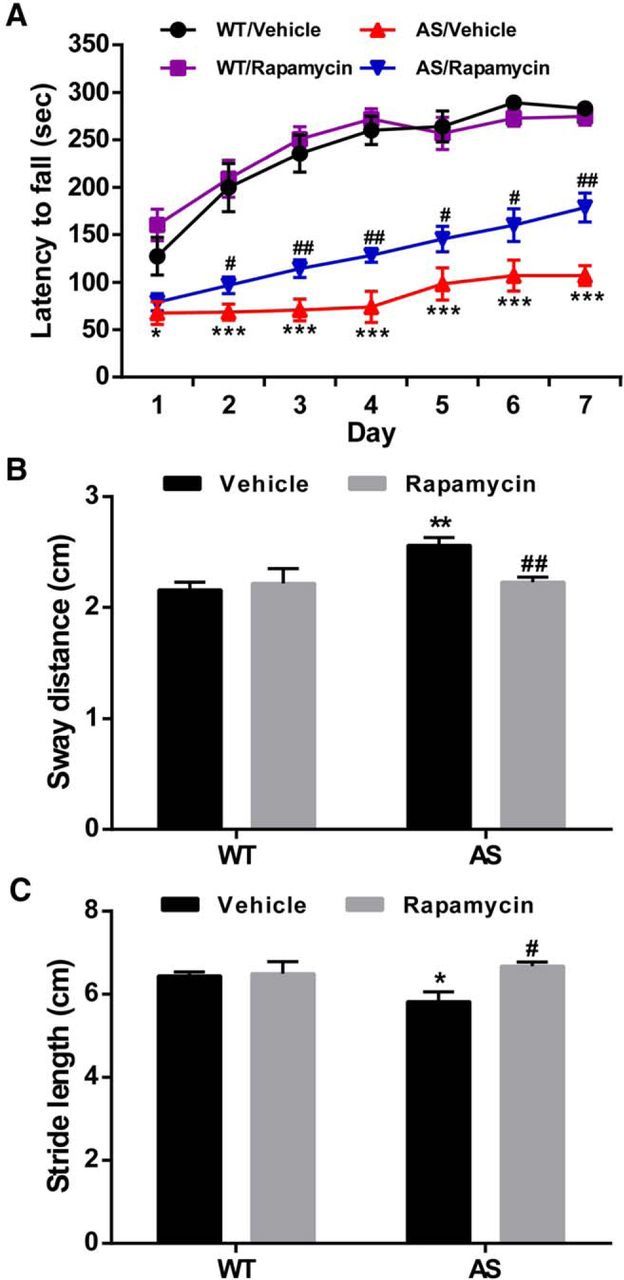
Effect of semi-chronic rapamycin treatment on motor function in WT and AS mice. Adult male WT and AS mice were treated daily with rapamycin (5 mg/kg, i.p.) and were tested for motor function on a rotarod apparatus and footprint analysis, as described in Materials and Methods. A, Rapamycin treatment increased the performance of AS mice on the rotarod test (means ± SEMs; *p < 0.05, ***p < 0.001 compared with vehicle-treated WT mice, n = 10; #p < 0.05, ##p < 0.01 compared with vehicle-treated AS mice, n = 10–12, Student's t test). B, Sway distance was increased in AS mice and reduced by rapamycin treatment (**p < 0.01 compared with vehicle-treated WT mice, n = 8–11; ##p < 0.01 compared with vehicle-treated AS mice, n = 6–11). Two-way ANOVA with Newman–Keuls post hoc test. C, Stride length was decreased in AS mice and increased by rapamycin treatment (*p < 0.05 compared with vehicle-treated WT mice, n = 8–11; #p < 0.05 compared with vehicle-treated AS mice, n = 6–11). Two-way ANOVA with Newman–Keuls post hoc test. Rapamycin treatment did not affect WT mouse performance.
Increased levels of TSC2 and its inhibitory phosphorylation in AS mouse cerebellum
Because mTOR activity was increased despite high TSC2 levels in AS mice, we determined TSC2 phosphorylation in AS mice, because recent reports indicated that phosphorylation of TSC2 at Thr1462 inhibits its activity (for recent reviews, see Huang and Manning, 2008; Laplante and Sabatini, 2012). In this set of experiments, crude membrane fraction (P2) was used. Like in whole homogenates, TSC2 levels in the P2 fraction were higher in AS mice compared with WT mice. However, phosphorylation of TSC2 at Thr1462 (p-TSC2) and the ratio of p-TSC2/total TSC2 were also increased in the P2 fraction of AS mice (Fig. 3). Rapamycin treatment significantly reduced p-TSC2 levels in AS and WT mice but did not alter total TSC2 levels (Fig. 3). These results suggest that increased TSC2 inhibitory phosphorylation may reduce its inhibition on mTOR and that phosphorylation of TSC2 at Thr1462 is mediated by a rapamycin-sensitive kinase. ERK1/2 has been shown to phosphorylate and inhibit TSC2; however, Western blot results showed that ERK1/2 activation/phosphorylation was increased in the cytosolic fraction in AS mice, but the increase was not reduced by rapamycin treatment (Table 1).
Figure 3.

Effect of rapamycin treatment on TSC2 and its phosphorylation in the cerebellum of WT and AS mice. WT and AS mice were treated with vehicle (VEH) or rapamycin (RAPA) as described in Materials and Methods. Animals were killed at the end of the 7 d treatment, the cerebella were dissected, and P2 and S2 fractions were prepared. A, Representative images of Western blots labeled with p-TSC2 (1462) and total TSC2 (β-actin as a loading control). B, Quantitative data of Western blots shown in A (means ± SEMs from 3–5 mice; *p < 0.05, **p < 0.01, ***p < 0.001 compared with vehicle-treated WT mice; ###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test).
Table 1.
Effects of rapamycin treatment on mTOR signaling pathway in the cerebellum of WT and AS mice
| WT/vehicle | WT/rapamycin | AS/vehicle | AS/rapamycin | |
|---|---|---|---|---|
| mTOR/β-actin | ||||
| P2 | 100.0 ± 2.6 | 117.6 ± 8.4 | 122.2 ± 11.44 | 105.0 ± 11.7 |
| S2 | 100.0 ± 3.7 | 66.0 ± 3.0** | 85.0 ± 4.6 | 59.2 ± 3.9## |
| S6K1/β-actin | ||||
| P2 | 100.0 ± 11.4 | 104.4 ± 4.7 | 91.0 ± 11.3 | 93.0 ± 6.6 |
| S2 | 100.0 ± 6.3 | 119.6 ± 4.8 | 105.6 ± 3.7 | 116.3 ± 6.2 |
| AKT/β-actin | ||||
| P2 | 100.0 ± 0.8 | 115.6 ± 6.4 | 107.2 ± 2.2 | 107.3 ± 3.4 |
| S2 | 100.0 ± 3.6 | 93.2 ± 4.0 | 97.9 ± 11.7 | 82.5 ± 3.9 |
| rictor/β-actin | ||||
| P2 | 100.0 ± 2.7 | 62.4 ± 6.2** | 97.6 ± 1.8 | 72.2 ± 8.1# |
| S2 | 100.0 ± 7.2 | 95.3 ± 6.7 | 108.4 ± 1.7 | 75.8 ± 5.4## |
| p-AKT 308/AKT | ||||
| P2 | 100.0 ± 2.6 | 118.0 ± 11.0 | 78.7 ± 7.0* | 150.5 ± 1.6### |
| S2 | 100.0 ± 4.6 | 104.5 ± 7.0 | 53.8 ± 8.9* | 84.4 ± 4.9# |
| p-IRS1/IRS1 | ||||
| P2 | 100.0 ± 14.3 | 138.3 ± 7.7 | 288.0 ± 50.7* | 246.3 ± 62.1 |
| S2 | 100.0 ± 21.1 | 44.5 ± 6. | 84.7 ± 27.2 | 77.8 ± 19.4 |
| p-ERK/ERK | ||||
| P2 | 100.0 ± 2.8 | 149.8 ± 14.6* | 110.1 ± 11.0 | 131.9 ± 11.9 |
| S2 | 100.0 ± 10.6 | 155.5 ± 11.5* | 165.9 ± 10.5** | 228.3 ± 31.5 |
All quantitative data of Western blot analyses are presented as means ± SEMs from 3–5 mice;
*p < 0.05,
**p < 0.01 compared with vehicle-treated WT mice;
#p < 0.05,
##p < 0.01,
###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test.
Increased mTORC1 and reduced mTORC2 activity in cerebellum of AS mice are normalized by rapamycin treatment
To determine whether rapamycin treatment-mediated improvements in motor function were related to its effects on the mTOR pathway, we analyzed cerebellar crude membrane (P2) and cytosolic (S2) fractions from rapamycin-treated mice and compared them with those of vehicle-treated mice. As with whole homogenates, levels of mTOR phosphorylated at Ser2448 and Ser2481 were increased in both P2 and S2 fractions from vehicle-treated AS mice (Fig. 4). Rapamycin treatment significantly reduced mTOR phosphorylation at Ser2448 and Ser2481 in both P2 and S2 in fractions of AS mice (Fig. 4). Rapamycin treatment also reduced levels of mTOR phosphorylated at Ser2481 but not at Ser2448 in both P2 and S2 of WT mice, which is in agreement with the notion that Ser2481 is an autophosphorylation site.
Figure 4.

Effects of rapamycin treatment on mTOR phosphorylation in the cerebellum of WT and AS mice. A, Representative images of Western blots labeled with p-mTOR (2448 and 2481) and mTOR in P2 fractions. B, Quantitative analysis of Western blots shown in A (means ± SEMs from 3–5 mice; *p < 0.05, **p < 0.01 compared with vehicle-treated WT mice; ###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test). C, Representative images of Western blots labeled with p-mTOR (2448 and 2481) and mTOR in S2 fractions. D, Quantitative analysis of Western blots shown in C (means ± SEMs from 3–5 mice; *p < 0.05, **p < 0.01 compared with vehicle-treated WT mice; ##p < 0.01, ###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test). VEH, Vehicle; RAPA, rapamycin.
We next evaluated mTORC1 and mTORC2 activity by analyzing the phosphorylation status of some of their respective downstream substrates. Levels of p-S6K1–Thr389 and p-S6–Ser235/236/p-S6–Ser240/244 were significantly increased in both P2 and S2 fractions of AS mice compared with WT mice (Fig. 5), and the increases in AS mice were significantly reduced in both P2 and S2 fractions by rapamycin treatment (Fig. 5). These results confirmed that mTORC1 is overactive in AS mice compared with WT mice. Rapamycin treatment also significantly reduced S6K1 and S6 phosphorylation in WT mice (Fig. 5), suggesting that active mTORC1 signaling is present in the cerebellum of WT mice under physiological conditions.
Figure 5.

Effects of rapamycin treatment on S6K1 and S6 phosphorylation in the cerebellum of WT and AS mice. A, C, Representative images of Western blot processed for p-S6K1 (389), S6K1, p-S6 (235/236), p-S6 (240/244), and S6 in P2 (A) and S2 (C) fractions. B, D, Quantitative analysis of Western blots shown in A and C (means ± SEMs from 3–5 mice; *p < 0.05, **p < 0.01, ***p < 0.001 compared with vehicle-treated WT mice; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test). VEH, Vehicle; RAPA, rapamycin.
We then determined levels of AKT phosphorylated at Ser473, a specific site for mTORC2. p-AKT–Ser473 levels were significantly reduced in both P2 and S2 fractions in AS mice compared with WT mice, which is consistent with our p-NDRG1 results. Interestingly, the reduction in AKT phosphorylation was reversed by rapamycin treatment (Fig. 6). Rapamycin treatment also increased levels of p-AKT–Ser473 in WT mice but only in the membrane fraction. Immunofluorescence examination confirmed the Western blot results (Fig. 6D). Furthermore, p-AKT–Ser473 showed a puncta-like staining pattern along the dendrites of Purkinje neurons, and the decrease in immunoreactive puncta in AS mice was reversed by rapamycin treatment (insets in Fig. 6D). Notably, AKT phosphorylation at Thr308 was also significantly reduced in both P2 and S2 fractions in AS mice compared with WT mice, and the reduction was reversed by rapamycin treatment (Table 1). These results suggest that, unlike mTORC1, mTORC2 activity was reduced in AS mice and that the reduction was related to mTORC1 activation, because rapamycin treatment not only reduced mTORC1 activity but also normalized mTORC2 activity. Furthermore, mTORC1-dependent suppression of mTORC2 also occurred in WT mice, suggesting that this cross-regulation between the two mTOR complexes may play important roles under physiological conditions. It is noteworthy that total protein levels of mTOR and mTORC1 and mTORC2 substrates were not significantly altered in AS mice (Table 1), further confirming changes in mTORC1 and mTORC2 activities but not protein levels.
Figure 6.

Effects of rapamycin treatment on AKT phosphorylation in the cerebellum of WT and AS mice. A, B, Representative images of Western blot processed for p-AKT and AKT in P2 (A) and S2 (B) fractions. C, Quantitative analysis of Western blots shown in A (means ± SEMs from 3–5 mice; *p < 0.05, **p < 0.01 compared with vehicle-treated WT mice; ##p < 0.01, ###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test). D, Representative images of p-AKT expression in the Purkinje cell layer in the cerebellum of WT or AS mice treated with vehicle (VEH) or rapamycin (RAPA). Insets are enlarged images of p-AKT-immunoreactive puncta along the dendrites. Scale bars: left, 30 μm; inset, 10 μm.
mTORC1 inhibits mTORC2 through S6K1-mediated phosphorylation of rictor
We next determined the molecular mechanism underlying mTORC1-mediated mTORC2 inhibition. Previous studies have shown that S6K1 phosphorylates and inhibits rictor, a key regulatory/structural subunit of the mTORC2 complex. S6K1 phosphorylates rictor at Thr1135, thereby inhibiting mTORC2-dependent phosphorylation of AKT at Ser473 (Julien et al., 2010). Therefore, we determined levels of rictor phosphorylated at Thr1135 (p-rictor). Levels of p-rictor were significantly increased in both P2 and S2 fractions of AS mice compared with WT mice, and the increase was reversed by rapamycin treatment (Fig. 7). Rapamycin treatment also decreased levels of p-rictor in both P2 and S2 fractions of WT mice (Fig. 7), an effect similar to that observed for p-S6K1, especially in P2 fraction. Conversely, phosphorylation at Ser636/639 of IRS1, another feedback regulation target of active S6K1, was only significantly increased in P2 fraction of AS mice compared with WT mice, and the increase was not affected by rapamycin treatment (Table 1). Therefore, these results suggest that S6K1-mediated rictor inhibitory phosphorylation is likely responsible for reduced mTORC2 activity in AS mice. The inhibitory phosphorylation also occurs in WT mice, suggesting a potential physiological function of this regulation.
Figure 7.

Effect of rapamycin treatment on rictor phosphorylation in the cerebellum of WT and AS mice. A, B, Representative images of Western blot labeled for p-rictor (1135) and rictor in P2 (A) and S2 (B) fractions. C, Quantitative analysis of Western blots shown in A and B (means ± SEMs from 3–5 mice; **p < 0.01, ***p < 0.001 compared with vehicle-treated WT mice; ###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test). VEH, Vehicle; RAPA, rapamycin.
Rapamycin treatment improves Purkinje neuron dendritic spine morphology in AS mice
Previous studies have suggested that morphological changes in dendritic spines of Purkinje cells are associated with motor learning impairment (Lee et al., 2007). Therefore, we performed Golgi staining in cerebellum of WT and AS mice treated with rapamycin or vehicle to determine the role of alterations in mTOR signaling in abnormal dendritic spine morphology and density. As reported previously (Dindot et al., 2008), dendritic spines of Purkinje neurons appeared “mushroom like,” with large heads and thin, short necks studded along dendrites in WT mice, whereas dendritic spines displayed an inconsistent morphology in AS mice. The density of spines was lower in AS mice compared with WT mice (Fig. 8), and rapamycin treatment normalized the number and appearance of Purkinje neuron dendritic spines in AS mice but it did not affect these parameters in WT mice. Quantification revealed that AS mice showed a significant reduction in spine density compared with WT mice (1.56 ± 0.05 vs 2.10 ± 0.07/μm, means ± SEMs; ***p < 0.001). Rapamycin treatment significantly increased spine density in AS mice (1.87 ± 0.09 vs 1.56 ± 0.05; #p < 0.05 compared with vehicle-treated AS mice and not significant for vehicle-treated WT mice), whereas no significant changes were observed in rapamycin-treated WT mice relative to vehicle-treated WT mice.
Figure 8.
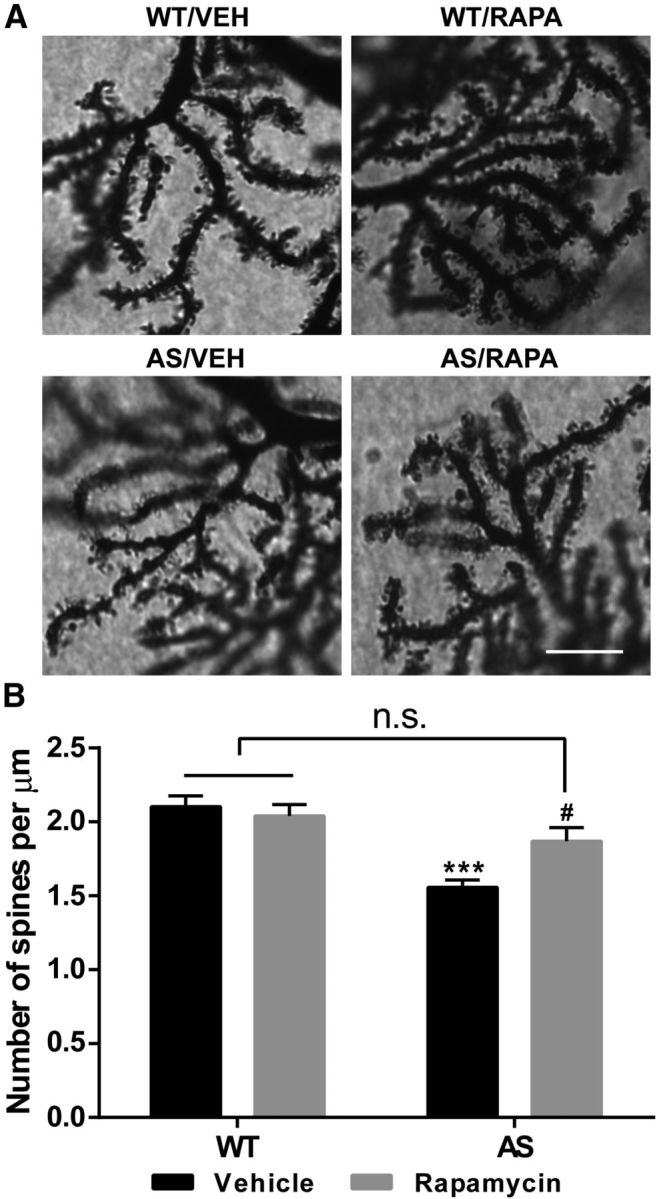
Effects of rapamycin treatment on Purkinje neuron dendrites and spines in AS and WT mice. A, Representative light micrograph images from Golgi-impregnated Purkinje cell. Scale bar, 10 μm. B, Quantitative analysis of dendritic spine density shown in A (means ± SEMs from 10 slices; ***p < 0.001 compared with vehicle-treated WT mice; #p < 0.05 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test). VEH, Vehicle; RAPA, rapamycin; n.s., not significant.
Recently, it has been reported that the striatum may also regulate rotarod performance (Rothwell et al., 2014). We thus characterized levels and phosphorylation of proteins in the mTORC1 and mTORC2 signaling pathways in the striatum. Western blot results showed that, unlike in the cerebellum, Ube3A deficiency was not associated with imbalanced activation of mTORC1 and mTORC2, although rapamycin treatment significantly reduced mTORC1 signaling in both WT and AS mice (Table 2).
Table 2.
Effects of rapamycin treatment on mTOR signaling pathway in the striatum of WT and AS mice
| WT/vehicle | WT/rapamycin | AS/vehicle | AS/rapamycin | |
|---|---|---|---|---|
| p-TSC2/TSC2 | 100.0 ± 13.4 | 67.8 ± 7.0 | 88.1 ± 7.8 | 87.6 ± 11.0 |
| p-mTOR 2448/mTOR | 100.0 ± 5.0 | 68.6 ± 2.2* | 64.0 ± 6.3* | 67.0 ± 8.8 |
| p-mTOR 2481/mTOR | 100.0 ± 6.8 | 84.3 ± 5.6 | 90.5 ± 3.6 | 57.1 ± 3.0## |
| p-S6K1/S6K1 | 100.0 ± 32.1 | 60.6 ± 11.5 | 168.0 ± 58.0 | 114.4 ± 44.2 |
| p-S6 235/236/S6 | 100.0 ± 11.8 | 23.6 ± 2.8*** | 92.5 ± 11.1 | 22.7 ± 4.6### |
| p-S6 240/244/S6 | 100.0 ± 18.0 | 29.9 ± 4.5** | 118.0 ± 7.9 | 32.1 ± 3.8### |
| p-AKT 473/AKT | 100.0 ± 7.4 | 94.9 ± 9.3 | 112.2 ± 10.8 | 97.7 ± 1.6 |
WT and AS mice were treated with vehicle or rapamycin as described in Materials and Methods. Animals were killed at the end of the 7 d treatment, the striatum was dissected, and the P2 fraction was prepared. All quantitative data of Western blot analyses are presented as means ± SEMs from 3–5 mice;
*p < 0.05,
**p < 0.01,
***p < 0.001 compared with vehicle-treated WT mice;
##p < 0.01,
###p < 0.001 compared with vehicle-treated AS mice, two-way ANOVA with Newman–Keuls post hoc test.
Discussion
It has been known that UBE3A deficiency results in AS since 1997 (Kishino et al., 1997; Matsuura et al., 1997), and yet the precise mechanisms linking UBE3A deficiency to AS pathological features remain to be elucidated fully. Likewise, although motor dysfunction characterized with gait imbalance and ataxia occurs in almost all AS patients and in AS mice (Jiang et al., 1998; Heck et al., 2008; Huang et al., 2013), the underlying mechanism remains unknown. In the present study, we found that abnormal overactivation of mTORC1 signaling in parallel with decreased mTORC2 activity in the cerebellum is critical for motor function impairments in AS mice, including rotarod performance and gait abnormalities. mTORC1 phosphorylation at Ser2448 and Ser2481 was enhanced, and phosphorylation of its downstream target proteins, S6K1 and S6, was also increased, with S6K1 highly increased in Purkinje neurons. Both effects, i.e., mTOR autophosphorylation and S6K1 phosphorylation, were reduced significantly by rapamycin treatment, suggesting that they are both mTORC1 mediated. Conversely, phosphorylation of the mTORC2 substrates AKT and NDRG1 was decreased, and the inhibitory phosphorylation of the mTORC2 regulator rictor was increased in AS mice; again, both effects were reversed by rapamycin treatment. These results indicate that, although mTORC1-mediated signaling is increased in AS mice, that of mTORC2 is reduced. Because both effects were significantly reversed by semi-chronic rapamycin treatment, these results also suggest that mTORC1 imposes a chronic inhibition on mTORC2, possibly through S6K1-mediated rictor phosphorylation. Collectively, our results indicate that balanced mTORC1 and mTORC2 activation in the cerebellum is important for motor function and that imbalanced mTORC1/mTORC2 signaling plays a critical role in motor function impairment in AS.
Activity of mTORC1 is increased in AS cerebellum
Accumulating evidence has shown that mTORC1 and mTORC2 complexes respond to different upstream signals and trigger different downstream cascades. Thus, mTORC1 is activated by growth factor signaling through the phosphatidylinositol 3-kinase (PI3K)–AKT pathway and is inhibited by energy/nutrient deficiency via the liver kinase B1–AMP-activated protein kinase cascade. In contrast, mTORC2 responds mainly to growth factor stimulation and is insensitive to energy/nutrient status. Phosphorylation of mTOR at Ser2481 is considered to be an autophosphorylation site (Copp et al., 2009; Soliman et al., 2010) and has been shown to increase mTOR activity (Caron et al., 2010; Soliman et al., 2010; although this remains controversial), whereas phosphorylation at Ser2448 was shown to be PI3K/S6K1 dependent and sensitive to rapamycin treatment (Chiang and Abraham, 2005; Copp et al., 2009). Therefore, S6K1 could be the kinase phosphorylating mTOR at Ser2448, which is supported by the fact that levels of mTOR–Ser2448 were reduced by rapamycin treatment.
How mTORC1 is activated in AS mice is not completely clear. The TSC1/2 complex is a major negative regulator of mTOR and TSC2 levels have been shown to be regulated by UBE3A. We found that TSC2 levels were increased in AS mice, a result in agreement with the literature (Zheng et al., 2008). However, increased levels of TSC2 would inhibit mTOR instead of stimulating it. TSC2 activity is also regulated by inhibitory phosphorylation at several sites, including Thr1462. Our results showed that levels of p-TSC2-Thr1462 were increased in AS mice, indicating that the inhibitory activity of the TSC1/2 complex may be reduced, resulting in mTORC1 overactivation. Because rapamycin treatment significantly reduced TSC2 inhibitory phosphorylation and mTORC1 activity without affecting total TSC2, increased TSC2 inhibitory phosphorylation in AS mice could account for mTORC1 overactivation. AKT was first identified as the kinase phosphorylating TSC2 at Thr1462 (Inoki et al., 2002; Manning et al., 2002; Potter et al., 2002). However, AKT is not likely the kinase phosphorylating TSC2 because its activity is reduced in AS mice, whereas TSC2 phosphorylation is increased in AS mice. In addition to AKT, ERK1/2 can inhibit TSC2 by phosphorylation at Thr1462, either directly or via the activation of RSK1 (Huang and Manning, 2008). Thus, it is conceivable that increased ERK1/2 activation (Table 1) contributes to increased TSC2 phosphorylation in AS mice. However, p-TSC2 levels were reduced after rapamycin treatment, whereas active ERK1/2 levels were not affected (Table 1). Therefore, a kinase sensitive to rapamycin treatment is likely to contribute to TSC2 phosphorylation at Thr1462. However, the identity of this kinase is currently unknown. Nevertheless, our results indicate that increased inhibitory phosphorylation of TSC2 contributes to overactivation of the mTORC1 pathway in AS mice. Once mTORC1 is activated, autophosphorylation at Ser2481 and phosphorylation at Ser2448 by S6K1 could keep mTORC1 in an active state.
Increased mTORC1/S6K1 activation inhibits mTORC2 signaling
In contrast to the increased phosphorylation of the mTORC1 downstream substrate S6K1, phosphorylation of the mTORC2 substrates AKT and NDRG1 was decreased. These results indicate that mTORC2 activity was decreased despite the release of inhibition normally provided by the TSC1/2 complex. This situation is reminiscent of what has been reported in cells lacking the TSC1/2 complex, in which inappropriate activation of mTORC1/S6K1 blocks growth factor-stimulated phosphorylation of AKT at Ser473. It has been proposed that S6K1 overactivation through feedback inhibition suppresses cell response to growth factor stimulation, possibly through IRS degradation (Manning, 2004). Additional studies showed that mTORC2 kinase activity was significantly reduced in a variety of cells lacking the TSC1/2 complex (Huang et al., 2008). The same group showed that the effect of TSC1/2 on mTORC2 was independent of its effect on both Ras homolog enriched in brain and mTORC1, indicating that the TSC1/2 complex may directly stimulate mTORC2 activity (Huang et al., 2008). Conversely, persistent mTORC1/S6K1 activity can inhibit mTORC2 through direct phosphorylation of members of the mTORC2 complex. For instance, S6K1 phosphorylates rictor at Thr1135 (Julien et al., 2010) and mammalian stress-activated MAP kinase-interacting protein 1 at Thr86 and Thr398 (Liu et al., 2014), thereby inhibiting mTORC2-dependent AKT phosphorylation. Therefore, the lower mTORC2 activity in AS mice could be a consequence of TSC2 inhibitory phosphorylation or increased feedback inhibition by S6K1 on the mTORC2 regulator rictor. Because rapamycin treatment corrected both lower AKT phosphorylation and higher TSC2 inhibitory phosphorylation, mTORC1–S6K1 overactivation is likely the key event in AS mice (Fig. 9).
Figure 9.

Altered mTORC1 and mTORC2 signaling in AS mice. Schematic representation showing the essential elements in mTORC1 and mTORC2 signaling pathways and their alterations in AS mice. TSC2 is a negative regulator of mTORC1 and is generally degraded in an Ube3A-dependent manner. Ube3A deficiency results in increased TSC2 levels and its inhibitory phosphorylation, which produces an overactivation of mTORC1. mTORC1–S6K1 overactivation suppresses mTORC2 by phosphorylating rictor and potentially (dash line) inhibits TSC1/2 activity by phosphorylating TSC2. Red line, Increased activity in AS mice; gray line, decreased activity in AS mice. PIP2, Phosphatidylinositol-4,5-biphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate; PTEN, phosphatase and tensin homolog; PDK1, pyruvate dehydrogenase lipoamide kinase isozyme 1; 4EBP1, 4E-binding protein 1.
Decreased mTORC2–AKT activity impairs spine development in Purkinje neurons and contributes to motor dysfunction
Although the roles of mTORC1 in cell growth and proliferation have been well characterized (Wullschleger et al., 2006; Laplante and Sabatini, 2012), mTORC2 roles are not as clear. Emerging experimental data in yeast and mammalian cell cultures indicate that mTORC2 may play critical roles in regulating the actin network and cell motility (Jacinto et al., 2004; Zhou and Huang, 2012). Early studies in yeast showed that mTORC2 regulates actin cytoskeleton through activation of Rho family GTPases (Schmidt et al., 1997). Studies with mammalian cells showed that the same pathway is also responsible for mTORC2-mediated regulation of actin filaments (Jacinto et al., 2004). Additionally, it has been shown that mTORC2 regulates the actin cytoskeleton through PKCα activation. However, additional research seems to indicate that the effects of AKT on cell motility are cell and tissue specific (García-Martinez and Alessi, 2008). AKT has been shown to play important roles in neuronal survival and synaptic plasticity. Deletion of the mTORC2 component rictor results in severe microcephaly, with decreased neuronal size and altered neurite organization, and motor function impairment (Thomanetz et al., 2013). Furthermore, selective knock-out of rictor in Purkinje neurons leads to abnormal morphology of these neurons and motor dysfunction (Thomanetz et al., 2013). Thus, it is conceivable that decreased mTORC2 activity together with increased mTORC1 activity are at least partially responsible for the reported immature dendritic spine phenotype in AS mice (Dindot et al., 2008) and the decreased spine density of Purkinje neurons in our study. This notion is supported by our results showing that rapamycin treatment not only significantly reduced alterations in the two mTOR signaling pathways but also improved dendritic spine morphology. Normalized mTOR signaling and improved Purkinje neuron morphology could contribute to rapamycin-induced improved motor performance and gaits. Purkinje neurons receive excitatory inputs from the climbing fiber and parallel fibers, as well as regulatory inputs from various inhibitory interneurons; they send inhibitory signals to cerebellar nuclei, which control the final output of the cerebellum (White and Sillitoe, 2013) and motor function. Emerging evidence indicates that cerebellar motor learning involves coordination of synaptic and intrinsic plasticity along the Purkinje neuron circuit (Gao et al., 2012; D'Angelo, 2014). How alterations in mTORC1 and mTORC2 signaling within Purkinje neurons translate to changes in motor performance and possibly non-motor function learning remains a topic for future research. Moreover, Gustin et al. (2010) showed recently that, in addition to the hippocampus and cerebellum, Ube3A deficiency was observed in almost all brain regions, including the striatum, thalamus, midbrain, and cerebral cortex. Whether these other regions also contribute, directly or indirectly, to motor dysfunction in AS remains an open question. Furthermore, because AS is a complex disorder, multiple factors are likely to be involved (Weeber et al., 2003; Greer et al., 2010; Margolis et al., 2010; Baudry et al., 2012; Kaphzan et al., 2012, 2013; Cao et al., 2013), which may explain the partial rescue of rapamycin treatment in the rotarod performance.
Conclusion
Ube3A deficiency in the cerebellum of AS mice was associated with increased levels and inhibitory phosphorylation of TSC2, an mTOR negative regulator, resulting in reduced mTORC1 inhibition. Consequently, activities of mTORC1 and its downstream target kinase S6K1 were increased, which reduced the activities of mTORC2 and AKT through rictor phosphorylation/inhibition. Imbalanced mTORC1 and mTORC2 activation plays important roles in the motor dysfunction and abnormal dendritic spine morphology of Purkinje neurons in AS mice, because semi-chronic rapamycin treatment not only corrected the activity of both complexes but also significantly improved motor function and dendritic spine morphology. These results not only shed light on the pathogenesis of cerebellum-dependent motor impairment but also could lead to new treatment for patients with AS directed at reestablishing a normal balanced activation of mTORC1 and mTORC2.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant P01NS045260-01 (principle investigator: Dr. C. M. Gall). X.B. was also supported by funds from the Daljit and the Elaine Sarkaria Chair. We thank Dr. Qingyu Qin, Dr. Guanghong Liao, and Kevin Chen for their contribution during the initial experiments.
The authors declare no competing financial interests.
References
- Barry RJ, Leitner RP, Clarke AR, Einfeld SL. Behavioral aspects of Angelman syndrome: a case control study. Am J Med Genet A. 2005;132A:8–12. doi: 10.1002/ajmg.a.30154. [DOI] [PubMed] [Google Scholar]
- Baudry M, Kramar E, Xu X, Zadran H, Moreno S, Lynch G, Gall C, Bi X. Ampakines promote spine actin polymerization, long-term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol Dis. 2012;47:210–215. doi: 10.1016/j.nbd.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K, Spaller MR, Goebel DJ, Marshall J. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013;11:e1001478. doi: 10.1371/journal.pbio.1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Ghosh S, Matsuoka Y, Ashton-Beaucage D, Therrien M, Lemieux S, Perreault C, Roux PP, Kitano H. A comprehensive map of the mTOR signaling network. Mol Syst Biol. 2010;6:453. doi: 10.1038/msb.2010.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Peto CA, Shelton GD, Mizisin A, Sawchenko PE, Schubert D. Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J Neurosci. 2009;29:118–130. doi: 10.1523/JNEUROSCI.3985-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821–1827. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan B. Angelman syndrome: current understanding and research prospects. Epilepsia. 2009;50:2331–2339. doi: 10.1111/j.1528-1167.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- D'Angelo E. The organization of plasticity in the cerebellar cortex: from synapses to control. Prog Brain Res. 2014;210:31–58. doi: 10.1016/B978-0-444-63356-9.00002-9. [DOI] [PubMed] [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Silva AJ. Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol Med. 2011;17:78–87. doi: 10.1016/j.molmed.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, van Beugen BJ, De Zeeuw CI. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci. 2012;13:619–635. doi: 10.1038/nrn3312. [DOI] [PubMed] [Google Scholar]
- García-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, Ploegh HL, Chowdhury S, Worley PF, Steen J, Greenberg ME. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guffanti G, Strik Lievers L, Bonati MT, Marchi M, Geronazzo L, Nardocci N, Estienne M, Larizza L, Macciardi F, Russo S. Role of UBE3A and ATP10A genes in autism susceptibility region 15q11–q13 in an Italian population: a positive replication for UBE3A. Psychiatry Res. 2011;185:33–38. doi: 10.1016/j.psychres.2010.04.057. [DOI] [PubMed] [Google Scholar]
- Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39:283–291. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck DH, Zhao Y, Roy S, LeDoux MS, Reiter LT. Analysis of cerebellar function in Ube3a-deficient mice reveals novel genotype-specific behaviors. Hum Mol Genet. 2008;17:2181–2189. doi: 10.1093/hmg/ddn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Burns AJ, Nonneman RJ, Baker LK, Riddick NV, Nikolova VD, Riday TT, Yashiro K, Philpot BD, Moy SS. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–90. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/S0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Hernandez P, Jung JI, Cowansage KK, Deinhardt K, Chao MV, Abel T, Klann E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol Psychiat. 2012;72:182–190. doi: 10.1016/j.biopsych.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, Klann E. Genetic reduction of the alpha1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. 2013;4:405–412. doi: 10.1016/j.celrep.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64:947–960. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Jung JG, Arii T, Imoto K, Rhyu IJ. Morphological changes in dendritic spines of Purkinje cells associated with motor learning. Neurobiol Learn Mem. 2007;88:445–450. doi: 10.1016/j.nlm.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Liu P, Guo J, Gan W, Wei W. Dual phosphorylation of Sin1 at T86 and T398 negatively regulates mTORC2 complex integrity and activity. Protein Cell. 2014;5:171–177. doi: 10.1007/s13238-014-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/S1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Margolis SS, Salogiannis J, Lipton DM, Mandel-Brehm C, Wills ZP, Mardinly AR, Hu L, Greer PL, Bikoff JB, Ho HY, Soskis MJ, Sahin M, Greenberg ME. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143:442–455. doi: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- Mulherkar SA, Jana NR. Loss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndrome. Neurobiol Dis. 2010;40:586–592. doi: 10.1016/j.nbd.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66:530–536. doi: 10.1111/j.1399-0004.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Qin Q, Baudry M, Liao G, Noniyev A, Galeano J, Bi X. A novel function for p53: regulation of growth cone motility through interaction with Rho kinase. J Neurosci. 2009;29:5183–5192. doi: 10.1523/JNEUROSCI.0420-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, Way S, McKenna J, 3rd, Haines K, Gambello MJ. Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol Dis. 2011;43:113–122. doi: 10.1016/j.nbd.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, Gambello MJ. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2013;51:93–103. doi: 10.1016/j.nbd.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Bickle M, Beck T, Hall MN. The yeast phosphatidylinositol kinase homolog TOR2 activates RHO1 and RHO2 via the exchange factor ROM2. Cell. 1997;88:531–542. doi: 10.1016/S0092-8674(00)81893-0. [DOI] [PubMed] [Google Scholar]
- Smith SE, Zhou YD, Zhang G, Jin Z, Stoppel DC, Anderson MP. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med. 2011;3:103ra197. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem. 2010;285:7866–7879. doi: 10.1074/jbc.M109.096222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomanetz V, Angliker N, Cloëtta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Rüegg MA. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201:293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhu G, Briz V, Hsu YT, Bi X, Baudry M. A molecular brake controls the magnitude of long-term potentiation. Nat Commun. 2014;5:3051. doi: 10.1038/ncomms4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL, Sweatt JD. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Sillitoe RV. Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol. 2013;2:149–164. doi: 10.1002/wdev.65. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zheng L, Ding H, Lu Z, Li Y, Pan Y, Ning T, Ke Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells. 2008;13:285–294. doi: 10.1111/j.1365-2443.2008.01162.x. [DOI] [PubMed] [Google Scholar]
- Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer. 2012;31:8–18. doi: 10.5732/cjc.011.10281. [DOI] [PMC free article] [PubMed] [Google Scholar]