

Abstract
Intracellular pathogens such as Shigella flexneri and Listeria monocytogenes achieve dissemination in the intestinal epithelium by displaying actin-based motility in the cytosol of infected cells. As they reach the cell periphery, motile bacteria form plasma membrane protrusions that resolve into vacuoles in adjacent cells, through a poorly understood mechanism. Here, we report on the role of the class II phosphatidylinositol 3-phosphate kinase PIK3C2A in S. flexneri dissemination. Time-lapse microscopy revealed that PIK3C2A was required for the resolution of protrusions into vacuoles through the formation of an intermediate membrane-bound compartment that we refer to as a vacuole-like protrusion (VLP). Genetic rescue of PIK3C2A depletion with RNA interference (RNAi)-resistant cDNA constructs demonstrated that VLP formation required the activity of PIK3C2A in primary infected cells. PIK3C2A expression was required for production of phosphatidylinositol 3-phosphate [PtdIns(3)P] at the plasma membrane surrounding protrusions. PtdIns(3)P production was not observed in the protrusions formed by L. monocytogenes, whose dissemination did not rely on PIK3C2A. PIK3C2A-mediated PtdIns(3)P production in S. flexneri protrusions was regulated by host cell tyrosine kinase signaling and relied on the integrity of the S. flexneri type 3 secretion system (T3SS). We suggest a model of S. flexneri dissemination in which the formation of VLPs is mediated by the PIK3C2A-dependent production of the signaling lipid PtdIns(3)P in the protrusion membrane, which relies on the T3SS-dependent activation of tyrosine kinase signaling in protrusions.
INTRODUCTION
Shigella flexneri and Listeria monocytogenes are food-borne pathogens that display the ability to invade nonphagocytic cells, such as epithelial cells, and spread from primary infected cells to adjacent cells (1–4). This dissemination process is supported by actin-based motility in primary infected cells (5, 6), which leads to the formation of membrane protrusions that project into adjacent cells, as motile bacteria reach the cell periphery (7, 8). The resolution of protrusions into vacuoles from which the pathogen escapes allows the bacteria to gain access to the cytosolic compartment of adjacent cells, thereby achieving cell-to-cell spread (7, 8). The mechanisms supporting L. monocytogenes and S. flexneri cytosolic motility are fairly well understood (9). Both pathogens achieve actin-based motility by recruiting to their surface a major nucleator of actin polymerization in eukaryotic cells, the ARP2/3 complex (10, 11). The expansion of the actin network formed at the bacterial surface generates forces that propel the bacterium throughout the cytosolic compartment (5, 6). In cells, the activity of the ARP2/3 complex is regulated by nucleation-promoting factors of the N-WASP/WAVE family (12, 13). S. flexneri engages the ARP2/3 complex through expression of IcsA (2, 14), a bacterial adaptor that recruits and activates N-WASP (15, 16). L. monocytogenes does not engage the ARP2/3 complex through N-WASP recruitment but through expression of ActA (17, 18), a bacterial factor that displays structural and regulatory mimicry with N-WASP (19–21).
In contrast to the mechanisms supporting cytosolic motility, the mechanisms supporting the formation and resolution of membrane protrusions are poorly understood. The sequence of events occurring during bacterial spread has been documented using time-lapse microscopy of epithelial cells infected with L. monocytogenes (22, 23). As motile bacteria encounter cell-cell junctions, they form protrusions that first elongate for a short period of time and then exist as nonelongating protrusions for an extended period of time, until resolution into vacuoles occurs. In addition to the ARP2/3-dependent assembly machinery and the AIP1/cofilin-dependent disassembly machinery (23), several studies have revealed the importance of various cellular components in bacterial dissemination. These include the cell-cell adhesion protein E-cadherin (24), the gap junction protein connexin 26 (25), the myosin light chain kinase and its target myosin II (26, 27), myosin 10 (28), the membrane-cytoskeleton linker ezrin (29), the dynamin binding protein Tuba (30), and actin nucleators of the formin family (31, 32). Recent studies have also revealed the importance of cellular signaling in bacterial dissemination (33–36). The massive accumulation of phosphotyrosine residues in protrusions suggested a role for tyrosine kinase signaling in S. flexneri dissemination (33). Accordingly, the tyrosine kinase inhibitor imatinib strongly impairs the accumulation of phosphotyrosine residues in protrusions, which results in severe defects in the resolution of protrusions into vacuoles (33). Interestingly, the integrity of the S. flexneri type 3 secretion system (T3SS) was required for tyrosine kinase signaling in protrusions and efficient resolution of protrusions into vacuoles (37). In addition to tyrosine kinase signaling, a role for phosphoinositol 3-kinase (PI3K)/AKT signaling in S. flexneri dissemination was proposed on the basis of the observation that the pan-PI3K inhibitors strongly inhibit the formation of S. flexneri infection foci (36). As inhibiting PI3K kinases did not impair protrusion formation, the authors proposed a role for PI3K signaling in vacuole formation through endocytosis of protrusions by adjacent cells (36).
Here, we investigated the mechanisms supporting the resolution of protrusions into vacuoles during S. flexneri dissemination. We uncovered that the resolution process relies on the formation of intermediate structures that we refer to as vacuole-like protrusions (VLPs). On the basis of our results, we propose a model of S. flexneri dissemination in which the formation of VLPs is mediated by the PIK3C2A-dependent production of the signaling lipid phosphatidylinositol 3-phosphate [PtdIns(3)P] in the protrusion membrane, which relies on the T3SS-dependent activation of tyrosine kinase signaling in protrusions.
MATERIALS AND METHODS
Cell lines and bacterial strains.
HT-29 cells (ATCC HTB-38) were cultured at 37°C with 5% CO2 in McCoy's 5A medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen). The wild-type Shigella flexneri strain used in this study is serotype 2a 2457T (38). The isogenic MxiG S. flexneri mutant strain was described previously (37). The Listeria monocytogenes strain used in this study is 10403S (39).
DNA constructs and cell transfection.
HT-29 cell lines stably expressing cyan fluorescent protein (CFP) membrane, yellow fluorescent protein (YFP) membrane, or green fluorescent protein (GFP) membrane markers were generated using the pLB vector from Addgene (Addgene plasmid 11619) as previously described (33). PIK3C2A was cloned into the XhoI and SmaI sites of the pDsRed-monomer C1 vector (Clontech). The PIK3C2Ares construct was constructed by swapping the AccI-NheI DNA fragment from the original GFP construct (40) into the pDsRed-PIK3C2A construct. Transient plasmid transfections were carried out using the X-treme gene HP reagent (Roche Applied Science) 48 h prior to the day of infection.
Immunofluorescence and antibodies.
Cells were fixed in phosphate-buffered saline (PBS)–4% formaldehyde (overnight at 4°C) and permeabilized in PBS-0.5% Triton for staining with antiphosphotyrosine (1:500; Cell Signaling) antibody and Alexa Fluor phalloidin (1:1,000; Invitrogen).
Bacterial infection.
S. flexneri was grown overnight in LB broth at 37°C with agitation. Twenty microliters of stationary-phase culture was used to inoculate 2 ml of LB, and the bacteria were grown to exponential phase for approximately 3 h at 37°C. Cells were infected with S. flexneri expressing GFP, CFP, or red fluorescent protein (RFP) under the control of an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter. Infection was initiated by centrifuging the plate at 1,000 rpm for 5 min, and internalization of the bacteria was allowed to proceed for 1 h at 37°C before gentamicin (50 μM final concentration) was added in order to kill the extracellular bacteria. Two hours before the infection was stopped, IPTG (4 mM final concentration) was added to the medium to induce GFP, CFP, or RFP expression. For quantification of protrusion formation, VLP formation, and vacuole resolution, infected cells were incubated at 37°C for 4 h and fixed in PBS-4% formaldehyde at 4°C overnight. For S. flexneri focus size analysis, infected cells were incubated at 37°C for 8 h. For PI3K and tyrosine kinase inhibition, LY294002 (10 μM) and imatinib (100 nM) were added 1.5 h postinfection. Dimethyl sulfoxide (DMSO) was used as a control for LY294002 treatment (1:1,000) and imatinib treatment (1:1,000).
Protrusion, VLP, and vacuole imaging.
For time-lapse microscopy, HT-29 cells were grown on 35-mm imaging dishes at 37°C in 5% CO2 (MatTek, Ashland, MA). Cells were infected with S. flexneri and imaged with a Nikon TE2000 spinning disc confocal microscope driven by the Volocity software package (Improvision). For analysis of protrusion-to-vacuole resolution and vacuole escape, Z-stacks were captured 2.5 h postinfection every 5 min for at least 160 min. For clarity, the Z plans corresponding to the basal plasma membrane were subsequently excluded from the merged Z-stacks. Protrusions were defined as plasma membrane extensions that formed as a result of bacteria reaching the cell cortex and projecting into adjacent cells. Vacuole-like protrusions (VLPs) were defined as an intermediate step between protrusions and vacuoles, characterized by a continuous membrane lining around the bacteria and a membranous tether <0.4 μm in width that derived from the former protrusion neck. Vacuoles were defined as membrane-bound compartments that derived from VLPs after disappearance of the membranous tether. As opposed to VLPs, vacuoles were therefore no longer connected to the primary infected cell. Free bacteria were defined as bacteria that were previously observed in vacuoles but were no longer surrounded by a continuous lining of the plasma membrane.
siRNA and quantitative real-time PCR.
Cells were transfected by reverse transfection with Dharmafect1 and individual small interfering RNAs (siRNAs) (D1, D2, D3, and D4; 50 nM final concentration) or a pool of the four silencing reagents (12.5 nM each, 50 nM total) or siRNA buffer alone (mock) and incubated for 72 h in a 384-well format or a 24-well plate format. Experiments for PIK3C2A knockdown (KD) were performed with a pool of silencing reagents, unless stated otherwise. For real-time PCR analysis, total RNA and first-strand cDNA synthesis was performed using the Cells-to-CT kit from Ambion. mRNA levels were determined by quantitative real-time PCR using the Universal Probe Library (Roche Biochemicals, Indianapolis, IN) and LightCycler 480 Probes Master (Roche Biochemicals, Indianapolis, IN). Thermal cycling was carried out using a Light Cycler 480 instrument (Roche Diagnostics) under the following conditions: 95°C for 5 min and 45 cycles at 95°C for 10 s and 60°C for 25 s.
Determination of infection focus size.
The size of infection foci formed in HT-29 cells infected with GFP-expressing S. flexneri was determined in a 384-well plate format. After fixation and 4′,6-diamidino-2-phenylindole (DAPI) staining, the plates were imaged using a TE2000 automated microscope (Nikon) equipped with a motorized stage (Prior), motorized filter wheels (Sutter Instrument, Inc.), and a 10× objective (Nikon) mounted on a piezo focus drive system (Physik Instrumente). Image acquisition was conducted using MetaMorph 7.1 software (Molecular Devices, Inc.), and image analysis was performed as previously described (33).
Quantification of YFP-PX probe enrichment in protrusions.
CFP membrane (Mb)-expressing HT-29 cells were transfected with the YFP-PX construct and infected with CFP-expressing S. flexneri for 4 h. Image analyses were performed on fixed samples with the Volocity software package (Improvision). The line profile analyses were computed from 2.0-μm-long lines drawn across each protrusion and across the plasma membrane next to the protrusion (see example depicted in Fig. 5A and B). Signal intensities for a given channel were recorded every pixel (at 0.16-μm intervals) along the line. The location of the plasma membrane was determined as the highest signal intensity along the line corresponding to the CFP channel. The corresponding signal intensities in the YFP channel were used to determine the enrichment of the YFP-PX probe in protrusions as the ratio of intensities in protrusions (see Fig. 5B and C, line 2) and at the plasma membrane next to the protrusion (see Fig. 5B and C, line 1) after subtraction of local background. In the example shown in Fig. 5A and B, signal intensity at the plasma membrane (line 1) was 6,000, signal intensity at the protrusion membrane (line 2) was 20,000, and local background was 2,500. The enrichment in protrusion was (20,000 − 2,500)/(6,000 − 2,500) = 5.
FIG 5.

PIK3C2A supports PtdIns(3)P production in membrane protrusions in HT-29 cells. (A) Representative image (left panel) of HT-29 cells transfected with the YFP-tagged PtdIns(3)P-binding PX domain construct (YFP-PX, green) and infected with RFP-expressing S. flexneri (red). Bar, 5 μm. The panels on the right show high-magnification images of the bacteria labeled 1 and 2 in the left image. Arrowheads indicate the recruitment of the YFP-PX probe to the membrane surrounding protrusions, and asterisks indicate the absence of the probe at the plasma membrane next to the protrusions. (B) Representative image of a protrusion formed in CFP membrane marker-expressing HT-29 cells (cyan) expressing the YFP-PX probe (yellow) and infected with RFP-expressing S. flexneri (red). Bar, 2 μm. (C) Graph showing line profile analysis of the signal corresponding to CFP membrane marker and the YFP-PX probe in the cytosol and at the plasma membrane (line 1) and in the protrusion membranes (line 2) as shown in panel B. (D) Graph showing statistical analysis of the relative enrichment of the YFP-PX probe in the protrusions formed in mock-treated (Mock) and PIK3C2A-depleted (PIK3C2A KD) HT-29 cells (****, P < 0.0001; unpaired t test).
RESULTS
PIK3C2A promotes S. flexneri dissemination.
We identified the class II phosphatidylinositol 3-phosphate kinase (PI3K) family member PIK3C2A in an RNA interference (RNAi) screen for host cell kinases required for S. flexneri dissemination in the human intestinal HT-29 cell line (A.-M. Dragoi and H. Agaisse, unpublished data). To further confirm the specific involvement of PIK3C2A in S. flexneri dissemination, we followed a validation strategy that relies on the use of independent siRNA duplexes in order to establish a strict correlation between the strength of the observed spreading defect and the silencing efficiency of the corresponding siRNA duplexes (41). The approach revealed that PIK3C2A-targeting duplexes D1 and D4 conferred the strongest spreading defects (Fig. 1A and B; 50 to 60% reduction in the size of infection foci), which correlated with the strongest silencing efficiency (Fig. 1C; 94% and 96% decrease in PIK3C2A mRNA expression for D1 and D4, respectively). We confirmed these genetic results by showing that, as expected, pan-inhibitors of PI3K activity, such as LY294002 and wortmannin, interfered with S. flexneri spread from cell to cell in a dose-dependent manner (see Fig. S1A and B in the supplemental material). Altogether, these results suggest a specific role for the class II phosphatidylinositol 3-phosphate kinase PIK3C2A in S. flexneri dissemination.
FIG 1.

PI3KC2A promotes S. flexneri dissemination in HT-29 cells through resolution of protrusion into vacuoles. (A) Representative images of HT-29 cells mock treated (Mock) or transfected with four individual siRNA duplexes (D1, D2, D3, and D4) targeting PIK3C2A and infected with GFP-expressing S. flexneri (green, Shigella; red, DNA). Bar, 100 μm. (B) Graph showing the relative size of infection foci in mock-treated and PIK3C2A-depleted cells for each individual siRNA duplex (D1, D2, D3, and D4). (C) Graph showing the relative silencing efficiency of individual siRNA duplexes as used in panel B. Values represent the mean ± standard deviation of three independent experiments (****, P < 0.0001; **, P < 0.0025; unpaired t test). (D) Graph showing the counts of protrusions, vacuoles, and free bacteria in adjacent cells in mock-treated (Mock) and PIK3C2A-depleted (PIK3C2A KD) cells. Values represent the mean of 3 independent experiments. Statistical analysis: PIK3C2A KD versus Mock protrusions, P < 0.0001; PIK3C2A KD versus Mock vacuoles, P < 0.0025; PIK3C2A KD versus Mock free bacteria, P < 0.0025; unpaired t test.
PIK3C2A promotes the resolution of membrane protrusions into vacuoles.
To further understand the role of PIK3C2A in S. flexneri dissemination, we first examined its potential involvement in cytosolic motility. Depletion of PIK3C2A did not affect the numbers of bacteria displaying actin tails (see Fig. S2A in the supplemental material; 64% in mock-treated cells versus 65% in PIK3C2-depleted cells). Moreover, the velocity of motile bacteria in the cytosol of PIK3C2A-depleted cells was similar to the velocity recorded in mock-treated cells (see Fig. S2B). We next investigated the potential involvement of PIK3C2A in protrusion resolution and vacuole formation. We determined whether spreading bacteria were located in membrane protrusions or in vacuoles or were free in the cytosol of adjacent cells, by visualizing the plasma membrane of primary infected cells with a plasma membrane-targeted version of CFP, as previously described by our group (see Fig. S3 in reference 33). The numbers of bacteria located in protrusions in PIK3C2A-depleted cells were greater than the numbers recorded in mock-treated cells (Fig. 1D, mock treated, 38%, versus PIK3C2A depleted, 78%). In addition, the depletion of PIK3C2A led to a decrease in the numbers of bacteria located in vacuoles (Fig. 1D, mock treated, 39%, versus PIK3C2A depleted, 13%) and a decrease in the numbers of bacteria no longer associated with membrane (free) in the cytosol of adjacent cells (Fig. 1D, mock treated, 23%, versus PIK3C2A depleted, 9%). These results suggest that PIK3C2A depletion affects the resolution of S. flexneri protrusions into vacuoles.
The resolution of S. flexneri protrusions occurs through formation of VLPs.
To further understand the process of protrusion resolution, we investigated the exact sequence of events leading to vacuole formation. Using time-lapse confocal microscopy (Fig. 2; also see Movie S1 in the supplemental material), we observed the formation of S. flexneri membrane protrusions that elongated into adjacent cells, forming a membrane-bound compartment that remained connected to the primary infected cell through a membranous neck (Fig. 2A, protrusion; see also Movie S1). As protrusions stopped elongating, their resolution into vacuoles occurred through the formation of an intermediate membrane-bound compartment that we refer to as a vacuole-like protrusion (Fig. 2A, VLP and vacuole; see also Movie S1). A distinctive feature of VLPs was the seemingly continuous lining of the membrane surrounding the bacteria, typical of vacuoles (Fig. 2A, VLP). Unlike vacuoles, however, VLPs were still tethered to the primary infected cells through the protrusion neck that apparently collapsed (Fig. 2A, protrusion and VLP, arrows). The subsequent disappearance of the membranous tether marked the formation of genuine vacuoles (Fig. 2A, vacuole; see also Movie S1). To quantify the protrusion-to-VLP transition, we monitored the protrusion neck width (PNW), 1 μm distal to the bacterial pole (Fig. 2A, protrusion and VLP, arrows). We computed that protrusions displayed a PNW greater than 0.4 μm, whereas VLPs, which display continuous lining of the membrane surrounding the bacteria, displayed a PNW less than 0.4 μm (Fig. 1B). In contrast to VLP formation, we also observed protrusions that failed to transition into the VLP stage, as determined by PNW measurements (PNW of >0.4 μm) (Fig. 2C and D; see also Movie S2 in the supplemental material). After a period of elongation, these protrusions usually retracted, bringing the pathogen back to the primary infected cells (Fig. 2C and D; see also Movie S2). Our tracking data for 30 protrusions in 10 infection foci and systematic quantification of PNW demonstrated that more than 75% (23/30) of the bacteria that formed a protrusion transitioned into the VLP stage (Fig. 3A). Importantly, almost all bacteria (22/23) that transitioned into the VLP stage ultimately formed a vacuole, from which the pathogen escaped (Fig. 3A). These results indicate that the commitment to the VLP stage is essential to the process of protrusion resolution into vacuoles.
FIG 2.

S. flexneri spreads from cell to cell in HT-29 cells through vacuole-like protrusion (VLP) formation. Time-lapse microscopy of HT-29 cells (green, GFP membrane marker) infected with RFP-expressing S. flexneri (red, Shigella). (A) Representative images showing a bacterium forming a membrane protrusion (Protrusion) that transitions into a vacuole-like protrusion (VLP), which further resolves into a vacuole (Vacuole). Bar, 2 μm. (B) Graph showing the quantification of the protrusion neck width (PNW; arrows in panel A) in images from Movie S1 in the supplemental material (75′ to 120′) and corresponding images (vertical arrows) as shown in panel A. Protrusion, light blue, PNW of >0.4 μm; VLPs, purple, PNW of <0.4 μm; vacuole yellow, no PN. (C) Representative images showing a bacterium forming a membrane protrusion (Protrusion) that fails to transition into a vacuole-like protrusion and retracts to the primary infected cell (Primary cell). Bar, 2 μm. (D) Graph showing the quantification of the protrusion neck width (PNW; arrows in panel A) in images from Movie S2 in the supplemental material (15′ to 65′) and corresponding images (vertical arrows) as shown in panel C. Protrusion, light blue, PNW of >0.4 μm; Primary cell, dark blue.
FIG 3.

PIK3C2A promotes vacuole-like protrusion (VLP) formation in HT-29 cells. Schematic representation of the dissemination process in mock-treated (A) or PIK3C2A-depleted (B) HT-29 cells. Each line represents the tracking of one bacterium every 5 min for 160 min, and the progression of the dissemination process was depicted using the color key displayed at the bottom of the panels: protrusion, light blue; VLP, purple; vacuole, yellow; free bacteria in the cytosol of adjacent cells, red; retraction to the primary infected cells, dark blue. Thirty bacteria were tracked in 10 independent infection foci.
PIK3C2A promotes VLP formation.
To further understand the role of PIK3C2A in the resolution of protrusions into vacuoles, we investigated the sequence of events occurring upon S. flexneri dissemination in PIK3C2A-depleted cells. In sharp contrast with the situation observed in mock-treated cells, where more than 75% of the bacteria successfully spread from cell to cell (Fig. 3A), our tracking data revealed that only 30% (9/30) of the bacteria that formed protrusions successfully spread from cell to cell in PIK3C2A-depleted cells (Fig. 3B). Strikingly, the vast majority of the bacteria formed protrusions that did not transition into the VLP stage and ultimately retracted, bringing the pathogen back to the primary infected cells (Fig. 3B). Thus, the activity of PIK3C2A is required for S. flexneri dissemination through VLP formation. In agreement with our tracking data, examination of fixed samples to determine the proportion of the formed protrusions and VLPs demonstrated a strong defect in VLP formation in PIK3C2A-depleted cells (Fig. 4A and B, mock versus PIK3C2A KD). Importantly, transient transfection of a PIK3C2A cDNA construct (PIK3C2Ares) resistant to RNAi treatment (40) rescued the defect in VLP formation observed in PIK3C2A-depleted cells (Fig. 4C, mock and PIK3C2Ares). In addition to providing additional evidence for the specific role of PIK3C2A, these results demonstrate that PIK3C2A is required in the primary infected cells for efficient VLP formation.
FIG 4.
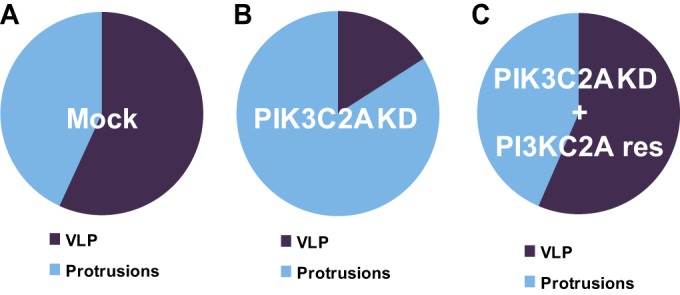
PIK3C2A is required in primary infected cells for VLP formation in HT-29 cells. Graphs showing the relative proportion of protrusions (Protrusions) and vacuole-like protrusions (VLP) in mock-treated (Mock) (A), PIK3C2A-depleted (PIK3C2A KD) (B), and PIK3C2A-depleted (C) HT-29 cells transfected with an RNAi-resistant construct (PIK3C2A res). Values represent the mean of 3 independent experiments. Statistical analysis: PIK3C2A KD versus Mock (VLP), P < 0.0001; PIK3C2A KD versus PIK3C2A res (VLP), P < 0.0005; unpaired t test.
PIK3C2A is required for PtdIns(3)P production in protrusions.
As class II phosphatidylinositol 3-kinases generate PtdIns(3)P in vitro and in vivo (42–44), we next investigated the potential site(s) of PtdIns(3)P production in cells infected with S. flexneri. To this end, we used the YFP-PX probe harboring the PX domain of the p40phox subunit of the superoxide-producing phagocyte NADPH-oxidase, which specifically binds PtdIns(3)P (45, 46). When transfected into HT-29 cells, the YFP-PX probe was weakly expressed in the cytosol and strongly colocalized with vesicular structures, reflecting expected production of PtdIns(3)P on a subset of endosomes (Fig. 5A, left panel). In addition to this endosomal localization, we observed a dramatic recruitment of the YFP-PX probe to the plasma membrane surrounding bacteria in protrusions (Fig. 5A, left panel, bacteria 1 and 2, and corresponding right panels, arrowheads), with little recruitment to the plasma membrane next to the protrusions (Fig. 5A, right panels, asterisks). The massive recruitment of the YFP-PX probe was dynamic and occurred within the first minutes of protrusion formation (see Fig. S3 in the supplemental material, arrowhead). We used computer-assisted image analyses to quantify the levels of the YFP-PX probe at the plasma membrane adjacent to protrusions (Fig. 5B and C, line 1) and at the plasma membrane surrounding protrusions (Fig. 5B and C, line 2). Statistical analyses showed a ∼5-fold enrichment in the YFP-PX signal recorded at the plasma membrane surrounding protrusions compared to the levels recorded at the plasma membrane adjacent to the protrusions (Fig. 5D, mock). This was in contrast with the situation observed in PIK3C2A-depleted cells where no significant enrichment was observed (Fig. 5D, PIK3C2A KD). These results indicate that PIK3C2A supports the production of PtdIns(3)P at the plasma membrane surrounding S. flexneri protrusions.
L. monocytogenes dissemination does not rely on PIK3C2A.
Similarly to S. flexneri, Listeria monocytogenes is an intracellular bacterial pathogen that invades intestinal epithelial cells and spreads from cell to cell through formation of membrane protrusions. To determine whether the signaling events observed in S. flexneri protrusions reflected a general property of pathogen-containing protrusions or were specific to S. flexneri-containing protrusions, we compared PtdIns(3)P production in cells infected with S. flexneri and in cells infected with L. monocytogenes (Fig. 6A). We did not observe any enrichment in the YFP-PX probe in the protrusion membrane formed by L. monocytogenes (Fig. 6A and B). We also determined that the sizes of the infection foci formed by L. monocytogenes were similar in mock-treated and PIK3C2A-depleted cells (Fig. 6C). These experiments indicate that the resolution of protrusions into vacuoles through PIK3C2A-dependent PtdIns(3)P production is a specific feature of S. flexneri dissemination.
FIG 6.

L. monocytogenes dissemination does not rely on PtdIns(3)P production in HT-29 cells. (A and B) Representative images of protrusions formed in CFP membrane marker-expressing HT-29 cells expressing the YFP-tagged PtdIns(3)P-binding PX domain construct and infected with RFP-expressing S. flexneri (A) or L. monocytogenes (B) for 4 h. Bar, 5 μm. (C) Graph showing statistical analyses of the relative enrichment of the YFP-PX probe in protrusions formed by S. flexneri or L. monocytogenes in HT-29 cells (****, P < 0.0001; unpaired t test). (D) Graph showing the quantification of L. monocytogenes infection focus size in mock-treated and PIK3C2A-depleted HT-29 cells 8 h postinfection.
PtdIns(3)P production relies on the S. flexneri T3SS.
We have previously shown that the type 3 secretion system (T3SS) is required for efficient resolution of S. flexneri protrusions into vacuoles (37), and we have now uncovered that the resolution process correlates with PtdIns(3)P production in protrusions. To test whether the T3SS may be responsible for PtdIns(3)P production in S. flexneri protrusions, we used an S. flexneri strain engineered to express the T3SS component MxiG under the control of an arabinose-inducible promoter (37). We induced the expression of MxiG with arabinose prior to infection in order to allow for T3SS-dependent invasion and then maintained or removed arabinose for the remaining time of the infection. We found that the YFP-PX probe was not enriched in S. flexneri protrusions when arabinose was removed from the medium (Fig. 7A), suggesting that the production of PtdIns(3)P in membrane protrusions relies on the integrity of the T3SS. As we previously showed that the T3SS is required for tyrosine kinase signaling in protrusions (33, 37), we next investigated the potential epistatic relationship between tyrosine kinase signaling and PtdIns(3)P production. We found that imatinib, an inhibitor of tyrosine kinase signaling that abolishes tyrosine phosphorylation in S. flexneri protrusions (33), also inhibited the recruitment of the YFP-PX probe (Fig. 7B). In contrast, treatment with the PI3K inhibitor LY294002 failed to inhibit tyrosine phosphorylation in S. flexneri protrusions (Fig. 7C; also see Fig. S4 in the supplemental material). These results suggest that S. flexneri dissemination relies on a signaling cascade involving T3SS-dependent activation of tyrosine kinase signaling and PIK3C2A-mediated production of PtdIns(3)P in protrusions (Fig. 7D).
FIG 7.

PtdIns(3)P production relies on the bacterial type 3 secretion system and host cell tyrosine kinase signaling in HT-29 cells. (A) Graph showing statistical analyses of the relative enrichment of the YFP-PX probe in HT-29 cells infected with wild-type S. flexneri (2457T WT) or ΔMxiG mutant (2457T ΔMxiG) (****, P < 0.0001; unpaired t test). (B) Graph showing statistical analyses of the relative enrichment of the YFP-PX probe in DMSO-treated (DMSO) and HT-29 cells treated with imatinib (100 nM) (Imatinib) and infected with wild-type S. flexneri (****, P < 0.0001; unpaired t test). (C) Graph showing the quantification of phosphotyrosine-positive and -negative protrusions in DMSO- and LY294002-treated HT-29 cells (10 μM) infected with wild-type S. flexneri (DMSO versus LY294002, P > 0.1; unpaired t test). (D) VLP formation in HT-29 cells is mediated by a signaling cascade that relies on the T3SS and activation of tyrosine kinase signaling in protrusions and the subsequent PIK3C2A-dependent production of PtdIns(3)P.
DISCUSSION
Intracellular pathogens such as Listeria monocytogenes and Shigella flexneri achieve dissemination by displaying actin-based motility in the cytosol of infected cells. As motile bacteria reach the plasma membrane of epithelial cells, they form protrusions that resolve into vacuoles in adjacent cells. Although the cellular processes supporting actin-based motility are now fairly well understood, the mechanisms supporting the actual spread from cell to cell remain poorly characterized. Here, we introduce the notion that S. flexneri spreads from cell to cell through formation of vacuole-like protrusions (VLPs). VLPs derive from protrusions that stopped elongating and whose neck underwent total collapse, leaving a vacuole-like structure that remained tethered to the primary infected cell through the former protrusion neck (Fig. 2A).
As the shape of membrane protrusions is determined by the underlying actin cytoskeleton, VLP formation most likely reflects a dramatic collapse of the actin cytoskeleton in S. flexneri protrusions. Interestingly, we uncovered a critical role for PIK3C2A and PtdIns(3)P production in VLP formation. Our cellular imaging and genetic experiments indicate that PIK3C2A is required in primary infected cells to generate PtdIns(3)P in the membrane surrounding bacteria in protrusions. We therefore propose that the accumulation of PtdIns(3)P at the plasma membrane regulates the dynamics of the underlying actin cytoskeleton in protrusions. The role of phosphoinositides, such as PtdIns(4,5)P2 and PtdIns(3,4,5)P3, in the regulation of cortical actin and plasma membrane extensions has been extensively documented. However, very few studies have reported on a role for PtdIns(3)P signaling at the plasma membrane in the regulation of the actin cytoskeleton dynamics in mammalian cells (42, 47). Future studies will therefore be required to determine the cellular processes supporting the PtdIns(3)P-dependent formation of the VLP and potential regulation of the actin cytoskeleton dynamics in S. flexneri protrusions. We also note that PtdIns(3)P plays a central role in cellular processes that involve membrane remodeling, such as fusion of endosomes (48). It is thus possible that, in addition to its potential regulatory role in the dynamics of the actin cytoskeleton, PtdIns(3)P may also contribute to vacuole formation through remodeling of the plasma membrane in protrusions and/or VLPs.
Our analyses using RNAi and related genetic rescues established that PtdIns(3)P production in S. flexneri protrusions is mediated by the activity of class II PIK3C2A. Several studies have documented a role for PIK3C2A-mediated production of PtdIns(3)P in insulin signaling at the cell cortex, demonstrating a connection between tyrosine kinase signaling and PIK3C2A activation (42, 47, 49). In agreement with the notion that tyrosine kinase signaling may regulate PIK3C2A activation in S. flexneri protrusions, we have shown previously that the tyrosine kinase inhibitor imatinib prevented protrusion resolution (33), and we now report that imatinib also inhibits PtdIns(3)P production. Moreover, we show that the PI3K inhibitor LY294002 does not inhibit tyrosine phosphorylation in S. flexneri protrusions, indicating that tyrosine kinase signaling acts upstream from PIK3C2A-mediated PtdIns(3)P production. Unlike that of class I PIK3CA, the activity of class II PIK3C2A is apparently not modulated through the recruitment of PI3K regulatory subunits, such as p85, to the plasma membrane upon activation of receptor tyrosine kinases. Thus, the exact mechanism leading to PIK3C2A activation in response to tyrosine kinase signaling remains to be determined. Together with our previous report showing that the bacterial T3SS is required for activation of tyrosine kinase signaling in protrusions (37), we propose a model of S. flexneri dissemination in which the T3SS-dependent activation of tyrosine kinase signaling leads to the PIK3C2A-mediated production of PtdIns(3)P, which may regulate cytoskeleton collapse and VLP formation (Fig. 7D).
In conclusion, our work reveals a critical role for PIK3C2A and PtdIns(3)P production in S. flexneri dissemination through VLP formation. In contrast, we showed that L. monocytogenes does not require PIK3C2A and PtdIns(3)P production for efficient dissemination. Thus, two bacterial pathogens that display similar strategies of actin cytoskeleton-mediated motility in the cytosol of infected cells have evolved strikingly different strategies of spread from cell to cell through resolution of protrusions into vacuoles. We note that, in addition to L. monocytogenes and S. flexneri, various bacterial pathogens, such as Burkholderia spp. and Rickettsia spp., display the ability to spread from cell to cell (50). Future studies may thus reveal a previously unappreciated diversity of strategies evolved by intracellular pathogens to achieve dissemination.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Agaisse laboratory for discussions and comments on the manuscript. We thank Arthur Talman for contributing Fig. S2B in the supplemental material. We thank York Posor and Volker Haucke for providing the RNAi-resistant version of PIK3C2A cDNA.
This work was supported by the National Institutes of Health grant R01AI073904 (H.A.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.03138-14.
REFERENCES
- 1.Havell EA. 1986. Synthesis and secretion of interferon by murine fibroblasts in response to intracellular Listeria monocytogenes. Infect Immun 54:787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makino S, Sasakawa C, Kamata K, Kurata T, Yoshikawa M. 1986. A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell 46:551–555. doi: 10.1016/0092-8674(86)90880-9. [DOI] [PubMed] [Google Scholar]
- 3.Pizarro-Cerda J, Cossart P. 2009. Listeria monocytogenes membrane trafficking and lifestyle: the exception or the rule? Annu Rev Cell Dev Biol 25:649–670. doi: 10.1146/annurev.cellbio.042308.113331. [DOI] [PubMed] [Google Scholar]
- 4.Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7:333–340. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- 5.Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. 1990. Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci U S A 87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. 1992. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature 357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- 7.Kadurugamuwa JL, Rohde M, Wehland J, Timmis KN. 1991. Intercellular spread of Shigella flexneri through a monolayer mediated by membranous protrusions and associated with reorganization of the cytoskeletal protein vinculin. Infect Immun 59:3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch MD, Way M. 2013. Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe 14:242–255. doi: 10.1016/j.chom.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. 1999. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- 11.Welch MD, Iwamatsu A, Mitchison TJ. 1997. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- 12.Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. 1999. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A 96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97:221–231. doi: 10.1016/S0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 14.Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A 86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF. 1999. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol 146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki T, Mimuro H, Suetsugu S, Miki H, Takenawa T, Sasakawa C. 2002. Neural Wiskott-Aldrich syndrome protein (N-WASP) is the specific ligand for Shigella VirG among the WASP family and determines the host cell type allowing actin-based spreading. Cell Microbiol 4:223–233. doi: 10.1046/j.1462-5822.2002.00185.x. [DOI] [PubMed] [Google Scholar]
- 17.Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, Leimeister-Wachter M, Wuenscher M, Chakraborty T. 1992. A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J 11:1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531. [DOI] [PubMed] [Google Scholar]
- 19.Chong R, Swiss R, Briones G, Stone KL, Gulcicek EE, Agaisse H. 2009. Regulatory mimicry in Listeria monocytogenes actin-based motility. Cell Host Microbe 6:268–278. doi: 10.1016/j.chom.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lasa I, Gouin E, Goethals M, Vancompernolle K, David V, Vandekerckhove J, Cossart P. 1997. Identification of two regions in the N-terminal domain of ActA involved in the actin comet tail formation by Listeria monocytogenes. EMBO J 16:1531–1540. doi: 10.1093/emboj/16.7.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skoble J, Portnoy DA, Welch MD. 2000. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J Cell Biol 150:527–538. doi: 10.1083/jcb.150.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbins JR, Barth AI, Marquis H, de Hostos EL, Nelson WJ, Theriot JA. 1999. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J Cell Biol 146:1333–1350. doi: 10.1083/jcb.146.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talman AM, Chong R, Chia J, Svitkina T, Agaisse H. 2014. Actin network disassembly powers dissemination of Listeria monocytogenes. J Cell Sci 127:240–249. doi: 10.1242/jcs.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sansonetti PJ, Mounier J, Prevost MC, Mege RM. 1994. Cadherin expression is required for the spread of Shigella flexneri between epithelial cells. Cell 76:829–839. doi: 10.1016/0092-8674(94)90358-1. [DOI] [PubMed] [Google Scholar]
- 25.Tran Van Nhieu G, Clair C, Bruzzone R, Mesnil M, Sansonetti P, Combettes L. 2003. Connexin-dependent inter-cellular communication increases invasion and dissemination of Shigella in epithelial cells. Nat Cell Biol 5:720–726. doi: 10.1038/ncb1021. [DOI] [PubMed] [Google Scholar]
- 26.Lum M, Morona R. 2014. Myosin IIA is essential for Shigella flexneri cell-to-cell spread. Pathog Dis 72:174–187. doi: 10.1111/2049-632X.12202. [DOI] [PubMed] [Google Scholar]
- 27.Rathman M, de Lanerolle P, Ohayon H, Gounon P, Sansonetti P. 2000. Myosin light chain kinase plays an essential role in S. flexneri dissemination. J Cell Sci 113:3375–3386. [DOI] [PubMed] [Google Scholar]
- 28.Bishai EA, Sidhu GS, Li W, Dhillon J, Bohil AB, Cheney RE, Hartwig JH, Southwick FS. 2013. Myosin-X facilitates Shigella-induced membrane protrusions and cell-to-cell spread. Cell Microbiol 15:353–367. doi: 10.1111/cmi.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pust S, Morrison H, Wehland J, Sechi AS, Herrlich P. 2005. Listeria monocytogenes exploits ERM protein functions to efficiently spread from cell to cell. EMBO J 24:1287–1300. doi: 10.1038/sj.emboj.7600595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajabian T, Gavicherla B, Heisig M, Muller-Altrock S, Goebel W, Gray-Owen SD, Ireton K. 2009. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol 11:1212–1218. doi: 10.1038/ncb1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fattouh R, Czuczman MA, Kwon H, Copeland JW, Pelletier L, Quinlan M, Muise AM, Higgins DE, Brumell JH. 3 October 2014. The Diaphanous-related formins promote protrusion formation and cell-to-cell spread of Listeria monocytogenes. J Infect Dis doi: 10.1093/infdis/jiu546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heindl JE, Saran I, Yi CR, Lesser CF, Goldberg MB. 2010. Requirement for formin-induced actin polymerization during spread of Shigella flexneri. Infect Immun 78:193–203. doi: 10.1128/IAI.00252-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dragoi AM, Agaisse H. 2014. The serine/threonine kinase STK11 promotes Shigella flexneri dissemination through establishment of cell-cell contacts competent for tyrosine kinase signaling. Infect Immun 82:4447–4457. doi: 10.1128/IAI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dragoi AM, Agaisse H. 2014. Tyrosine kinases, drugs, and Shigella flexneri dissemination. Gut Microbes 5:44–47. doi: 10.4161/gmic.26523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dragoi AM, Talman AM, Agaisse H. 2013. Bruton's tyrosine kinase regulates Shigella flexneri dissemination in HT-29 intestinal cells. Infect Immun 81:598–607. doi: 10.1128/IAI.00853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukumatsu M, Ogawa M, Arakawa S, Suzuki M, Nakayama K, Shimizu S, Kim M, Mimuro H, Sasakawa C. 2012. Shigella targets epithelial tricellular junctions and uses a noncanonical clathrin-dependent endocytic pathway to spread between cells. Cell Host Microbe 11:325–336. doi: 10.1016/j.chom.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Kuehl CJ, Dragoi AM, Agaisse H. 2014. The Shigella flexneri type 3 secretion system is required for tyrosine kinase-dependent protrusion resolution, and vacuole escape during bacterial dissemination. PLoS One 9:e112738. doi: 10.1371/journal.pone.0112738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labrec EH, Schneider H, Magnani TJ, Formal SB. 1964. Epithelial cell penetration as an essential step in the pathogenesis of bacillary dysentery. J Bacteriol 88:1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bishop DK, Hinrichs DJ. 1987. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol 139:2005–2009. [PubMed] [Google Scholar]
- 40.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, Muller R, Zarbakhsh S, Gulluni F, Hirsch E, Krauss M, Schultz C, Schmoranzer J, Noe F, Haucke V. 2013. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499:233–237. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 41.Chong R, Squires R, Swiss R, Agaisse H. 2011. RNAi screen reveals host cell kinases specifically involved in Listeria monocytogenes spread from cell to cell. PLoS One 6:e23399. doi: 10.1371/journal.pone.0023399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falasca M, Hughes WE, Dominguez V, Sala G, Fostira F, Fang MQ, Cazzolli R, Shepherd PR, James DE, Maffucci T. 2007. The role of phosphoinositide 3-kinase C2alpha in insulin signaling. J Biol Chem 282:28226–28236. doi: 10.1074/jbc.M704357200. [DOI] [PubMed] [Google Scholar]
- 43.MacDougall LK, Domin J, Waterfield MD. 1995. A family of phosphoinositide 3-kinases in Drosophila identifies a new mediator of signal transduction. Curr Biol 5:1404–1415. doi: 10.1016/S0960-9822(95)00278-8. [DOI] [PubMed] [Google Scholar]
- 44.Meunier FA, Osborne SL, Hammond GR, Cooke FT, Parker PJ, Domin J, Schiavo G. 2005. Phosphatidylinositol 3-kinase C2alpha is essential for ATP-dependent priming of neurosecretory granule exocytosis. Mol Biol Cell 16:4841–4851. doi: 10.1091/mbc.E05-02-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, Thuring JW, Cooper MA, Lim ZY, Holmes AB, Gaffney PR, Coadwell J, Chilvers ER, Hawkins PT, Stephens LR. 2001. PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox). Nat Cell Biol 3:679–682. doi: 10.1038/35083076. [DOI] [PubMed] [Google Scholar]
- 46.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB. 2001. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 47.Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M. 2003. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J 22:4178–4189. doi: 10.1093/emboj/cdg402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suetsugu S, Kurisu S, Takenawa T. 2014. Dynamic shaping of cellular membranes by phospholipids and membrane-deforming proteins. Physiol Rev 94:1219–1248. doi: 10.1152/physrev.00040.2013. [DOI] [PubMed] [Google Scholar]
- 49.Brown RA, Domin J, Arcaro A, Waterfield MD, Shepherd PR. 1999. Insulin activates the alpha isoform of class II phosphoinositide 3-kinase. J Biol Chem 274:14529–14532. doi: 10.1074/jbc.274.21.14529. [DOI] [PubMed] [Google Scholar]
- 50.Stevens JM, Galyov EE, Stevens MP. 2006. Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol 4:91–101. doi: 10.1038/nrmicro1320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.