

Abstract
Toll-like receptors (TLRs) are evolutionarily conserved host proteins that are essential for effective host defense against pathogens. However, recent studies suggest that some TLRs can negatively regulate immune responses. We observed here that TLR2 and TLR9 played opposite roles in regulating innate immunity against oral infection of Salmonella enterica serovar Typhimurium in mice. While TLR9−/− mice exhibited shortened survival, an increased cytokine storm, and more severe Salmonella hepatitis than wild-type (WT) mice, TLR2−/− mice exhibited the opposite phenomenon. Further studies demonstrated that TLR2 deficiency and TLR9 deficiency in macrophages both disrupted NK cell cytotoxicity against S. Typhimurium-infected macrophages by downregulating NK cell degranulation and gamma interferon (IFN-γ) production through decreased macrophage expression of the RAE-1 NKG2D ligand. But more importantly, we found that S. Typhimurium-infected TLR2−/− macrophages upregulated inducible nitric oxide synthase (iNOS) expression, resulting in a lower bacterial load than that in WT macrophages in vitro and livers in vivo as well as low proinflammatory cytokine levels. In contrast, TLR9−/− macrophages showed decreased reactive oxygen species (ROS) expression concomitant with a high bacterial load in the macrophages and in livers of TLR9−/− mice. TLR9−/− macrophages were also more susceptible than WT macrophages to S. Typhimurium-induced necroptosis in vitro, likely contributing to bacterial spread and transmission in vivo. Collectively, these findings indicate that TLR2 negatively regulates anti-S. Typhimurium immunity, whereas TLR9 is vital to host defense and survival against S. Typhimurium invasion. TLR2 antagonists or TLR9 agonists may thus serve as potential anti-S. Typhimurium therapeutic agents.
INTRODUCTION
During the early stages of infection, the innate immune system serves as the first line of defense against microbial replication and spread before an adaptive response is mounted (1). As one mechanism to elicit a rapid and appropriate innate immune response against pathogen infection, pattern recognition receptors (PRRs) evolved in the host to recognize various microbial molecular patterns. In response, pathogens evolved to use many different tactics, including subversion of some of these host antimicrobial immune mechanisms, in order to ensure their multiplication, survival, and persistence in the host (2). This ongoing interplay between host innate immune responses and pathogen virulence factors largely determines the outcome of most infections.
As the most widely studied PRRs, Toll-like receptors (TLRs) play a crucial role in pathogen recognition and the induction of immune responses. Currently, 11 and 13 TLRs have been identified in humans and mice, respectively. They are widely expressed on many cell types, such as macrophages, neutrophils, dendritic cells (DCs), and mucosal epithelial cells. TLR signaling results in the induction of reactive oxygen species (ROS) and the activation of the transcription factor NF-κB, which in turn induces proinflammatory cytokine production as well as the upregulation of costimulatory molecules, inducible nitric oxide synthase (iNOS), antimicrobial peptides, and other defense molecules (3). Although all TLRs share many common functions, further research has shown that individual TLR signaling pathways diverge and have unique functions. For instance, during intestinal bacterial infection, TLR2 activation may increase the severity of intestinal barrier disruption through a specific effect on the permeability of epithelial tight junctions in the intestine (4). In sharp contrast, TLR9 expressed on antigen-presenting cells (APCs) is essential for APC expansion in the mesenteric lymph nodes of mice during infection (5). Indeed, the contribution of each individual TLR to the host response to infection depends on the type of pathogen and the entry routes of the infection.
Salmonella enterica is a Gram-negative pathogen that infects a wide range of animals, including humans (6). TLR ligands for TLR4, TLR2, TLR5, and TLR9 have been found in Salmonella enterica, including lipopolysaccharide (LPS), bacterial lipoproteins, flagellin, and CpG DNA, respectively. The principal clinical syndromes associated with Salmonella infection range from self-limited gastrointestinal infections to systemic infections (7). Recently, acute liver injury has been reported in humans infected with Salmonella enterica serovar Typhi through the oral route, and this condition has been termed Salmonella hepatitis; patients usually present with hepatomegaly and a moderate elevation in transaminase levels (8, 9).
In murine models of human-specific S. Typhi infection using Salmonella enterica serovar Typhimurium infection, acute hepatitis can endanger the survival of infected mice (10). The process of S. Typhimurium infection normally proceeds as follows. After entry into a host by the intestinal route, S. Typhimurium dissemination occurs by first invading epithelial and M cells to enter the blood directly or by passing through lymphatic vessels and mesenteric lymph nodes (11). Once in the blood, S. Typhimurium exists either as an extracellular bacterium or an intracellular bacterium associated with CD18+ leukocytes. Bacteria are opsonized but not lysed by complement factors in the circulation and are rapidly cleared from the blood to reach the intracellular location sites, including liver, spleen, or bone marrow. Regardless of the infection routes of S. Typhimurium, the liver is recognized as a common site for secondary bacterial colonization due to the hematogenous spread of the bacteria, and this hepatic infection can adversely affect the survival of infected mice (10). Current studies show that pathogen invasion and replication within the liver are main causes of Salmonella hepatitis (10, 12), and the integrated macrophage-NK response is considered to be an important first line of defense against S. Typhimurium in the liver (13). Despite observing these clinical manifestations of S. Typhimurium infection in the liver, our understanding of the mechanisms driving the emergence of this particular virulence against the host remains relatively poor.
In the present study, we focused on studying the TLR-activated function of host macrophages as well as the cross talk between NK cells and S. Typhimurium-infected macrophages in the liver. We found that TLR2 and TLR9 display opposing roles in the host against S. Typhimurium immune responses.
MATERIALS AND METHODS
Mice.
Male wild-type (WT) C57BL/6 mice (6 to 8 weeks old) were purchased from Beijing HFK Bioscience, Ltd. (Beijing, China). TLR2−/− and TLR4−/− mice were kindly provided by Shao Bo Su (Sun Yat-Sen University, Guangzhou, China). TLR9−/− mice on the C57BL/6 background were obtained as a gift from Shizuo Akira (Osaka University, Osaka, Japan). Mouse handling and experiments were conducted in accordance with the guidelines for experimental animals from Shandong University (Shandong, China). All animal manipulations were performed in class II biological cabinets.
Bacterial strain and culture.
A virulent atrichia S. Typhimurium strain (ATCC 14028) was inoculated on Luria-Bertani (LB) agar plates and cultured at 37°C overnight; bacteria were then cultured in LB broth for 12 h. Bacterial cultures reaching saturation density were diluted (1:100) and cultured to the mid-logarithmic growth phase (optical density at 600 nm [OD600] = ∼0.4 to ∼0.6). Bacterial cultures were centrifuged and washed with LB broth or 1× phosphate-buffered saline (PBS) twice before use.
Antibodies.
The following monoclonal antibodies (MAbs) were used in this study: fluorescein isothiocyanate (FITC)–anti-NK1.1 (clone PK136), PerCp-Cy5.5–anti-CD3e (clone 145-2c11), phycoerythrin (PE)–anti-CD69 (clone H1.2F3), APC–anti-NKG2D (clone cx5), PE–anti-gamma interferon (IFN-γ) (clone XMG1.2), PerCpCy5.5–anti-F4/80 (clone BM8), FITC–anti-CD11b (clone M1/70), FITC–anti-TLR2 (clone 6C2), PE–anti-TLR4 (clone UT41), and FITC–anti-TLR9 (clone M9.D6) were purchased from eBioscience (San Diego, CA); anti-Qa-1b (clone 6A8.6F10.1A6) was purchased from BD Biosciences (Franklin Lakes, NJ); and PE–anti-RAE-1 (clone 186107), APC–anti-H60 (clone 205326), and PE–anti-MULT-1 (clone 237104) were purchased from R&D Systems (Minneapolis, MN). Streptavidin-conjugated FITC (BD Biosciences) was used as a secondary reagent to identify biotinylated primary antibodies. Each antibody was titrated to determine the optimal staining concentration for maximal signal. All fluorescence-activated cell sorting (FACS) data were acquired on a FACSCalibur (BD Biosciences) flow cytometer and analyzed with WinMDI 2.9 software.
S. Typhimurium infection.
For in vivo experiments, age-matched mice were used at 8 to 10 weeks old. Mice were infected orally by the intragastric (i.g.) route with 200 μl of 5 × 105 or 5 × 104 S. Typhimurium CFU in sterile LB by using a gavage needle. An equal volume of a sterile LB broth vehicle was administered as a mock-infected control. For in vitro infection, peritoneal macrophages were resuspended in RPMI 1640 containing 15% fetal bovine serum (FBS), and 1.2 × 106 cells were allowed to adhere to each well in a 12-well flat-bottom plate. Macrophages were infected by S. Typhimurium at a multiplicity of infection (MOI) of 10:1 for 1 h. And then these cells were washed with PBS, and fresh culture medium containing gentamicin antibiotic (100 μg/ml) was added for 1 h; gentamicin was reduced to 10 μg/ml for the remainder of the experiment.
Bacterial enumeration.
After homogenization, macrophages or liver tissues were lysed with 0.1% Triton X-100 in PBS to release bacteria. A dilution series in PBS was plated on LB agar plates and incubated overnight at 37°C; colonies were then counted from triplicate samples.
Serum ALT levels.
Blood samples were collected at the time of sacrifice. Samples were centrifuged at 900 × g for 15 min, and the aqueous phase was used as the serum. Serum alanine aminotransferase (ALT) levels were measured by a commercially available kit (RSbio, Shanghai, China) according to the manufacturer's instructions.
Cytokine analysis.
Liver tissue (0.1 g) from sacrificed mice was homogenized in 0.5 ml of PBS. Homogenates were spun down at 20,000 × g for 10 min at 4°C, and supernatants were collected. Cytokine levels in liver and serum were analyzed using a cytometric bead array (CBA) assay (BD Biosciences) according to the manufacturer's instructions.
Necroptosis analysis.
Peritoneal macrophages were infected with S. Typhimurium as described above for 4 h, and necroptosis of infected macrophages was detected by the annexin V-propidium iodide (PI) double-staining kit (BestBio, Shanghai, China) according to the manufacturer's instructions.
ROS and iNOS measurement.
Peritoneal macrophages were infected with S. Typhimurium as described above for 12 h. ROS levels were measured by the reactive oxygen species assay kit (Beyotime, Shanghai, China) according to the manufacturer's instructions, and iNOS levels were measured by the nitric oxide synthase assay kit (Beyotime) according to the manufacturer's instructions.
Histopathology.
Liver tissues were harvested at day 7 postinfection, fixed in 10% neutral buffered formalin, and then embedded in paraffin for sectioning. Sections were then processed for hematoxylin and eosin (H&E) staining by using the standard protocols. The sections were observed by light microscopy.
Preparation of hepatic mononuclear cells.
Hepatic mononuclear cells (MNCs) were isolated as previously described (14). Briefly, livers were removed aseptically from mice and pressed through a 200-gauge stainless steel mesh. The cells were resuspended in 40% Percoll (Sigma-Aldrich, St. Louis, MO), gently overlaid on 70% Percoll, and centrifuged at 600 × g for 30 min at room temperature. Hepatic MNCs were collected from the interphase.
MACS of NK cells.
NK cells were enriched (>90% purity) from the spleen by magnetic-activated cell sorting (MACS) (Miltenyi-Biotec, Auburn, CA) by using a mouse NK cell isolation kit according to the manufacturer's instructions.
Cytotoxicity assay.
As with the target cells, peritoneal macrophages were infected with S. Typhimurium as described above and incubated with 1 ml of 2 mM carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR) for 15 min at 37°C; these cells were then washed with PBS 3 times to stop the reaction. NK cells were isolated from WT mice and added to target cells at a 5:1 effector/target (E:T) ratio at 37°C for 12 h. After incubation, cells were washed once and labeled with an optimized dose of 0.25 μg/ml 7-aminoactinomycin D (7-AAD) (BD Biosciences) for 15 min to identify dead cells. After a final wash, cells were mixed thoroughly to interrupt cell-cell contact and resuspended at 300 μl/tube in FACS buffer for immediate analysis. Cells that were double-positive for CFSE and 7-AAD were identified as the killed target cells, and the specific cytotoxicity was calculated as follows: percent lysis = (number of CFSE and 7-AAD double-positive cells/number of CFSE single-positive cells) × 100%.
Statistical analysis.
All analyses were performed using GraphPad Prism (San Diego, CA). Survival data were analyzed using the log-rank test. Data were analyzed for statistical significance using one-way analysis of variance (ANOVA). Statistical analysis was performed using a paired Student t test. A P value of <0.05 was considered statistically significant.
RESULTS
TLR2, TLR4, and TLR9 play different roles in host resistance to S. Typhimurium infection.
To begin determining how S. Typhimurium infection triggered host innate immune responses, we first investigated which host innate immune responses were activated upon infection with a virulent atrichia S. Typhimurium strain, which contained ligands for TLR2, TLR4, and TLR9. We first evaluated the survival of S. Typhimurium-infected TLR2−/−, TLR4−/−, and TLR9−/− mice orally infected with a high lethal dose (5 × 105 CFU) of S. Typhimurium. Compared to WT and TLR4−/− mice that died around day 12 postinfection, TLR9−/− mice were significantly more susceptible to S. Typhimurium infection, as they all died within 5 days after infection; in contrast, TLR2−/− mice were significantly more resistant than WT mice (Fig. 1). These data indicate that TLR9 signaling plays a positive role in host resistance to S. Typhimurium infection and is critical for the survival of mice under S. Typhimurium infection, whereas activation of TLR2 signaling by S. Typhimurium may negatively regulate host immune responses against infection.
FIG 1.
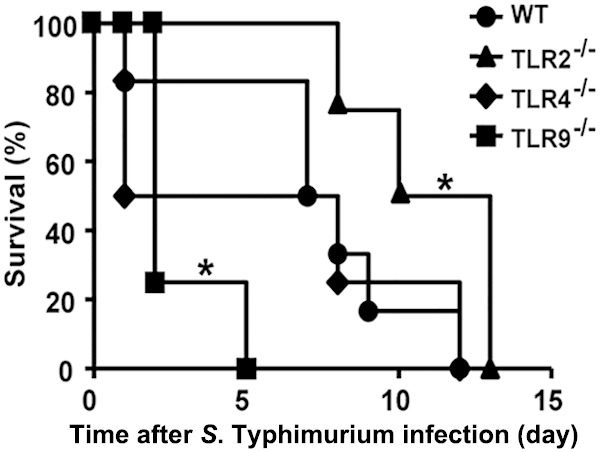
Survival of S. Typhimurium-infected WT, TLR2−/−, TLR4−/−, and TLR9−/− mice. Age-matched WT, TLR2−/−, TLR4−/−, and TLR9−/− mice were i.g. infected with 5 × 105 CFU of S. Typhimurium, and survival was monitored every day; n ≥ 6 mice in each group. *, P < 0.05 by the log-rank curve comparison test.
TLR9 protects the host against systemic inflammation induced by S. Typhimurium infection.
Inflammation is the host's attempt at self-protection against pathogen invasion, but excessive inflammatory responses can eventually lead to severe tissue damage, sometimes resulting in death. Since we established that TLR2- and TLR9-mediated innate immune responses are both regulated after S. Typhimurium infection, we next wondered whether their downstream effects were related to their ability to initiate inflammation against the pathogen in the host. To address this, we orally infected WT, TLR2−/−, and TLR9−/− mice with a low dose (5 × 104 CFU) of S. Typhimurium for 7 days and then determined serum cytokine levels. In response to low-dose S. Typhimurium infection, inflammatory mediator levels were significantly higher in TLR9−/− mice than in WT and TLR2−/− mice, especially the endogenous pyrogens interleukin 6 (IL-6), IL-18, and tumor necrosis factor alpha (TNF-α), the inflammatory cytokines IFN-γ and IL-17 (Fig. 2A), and the chemokines granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein 1 (MIP-1) (Fig. 2B). In contrast, the levels of these inflammatory mediators were similar between TLR2−/− and WT mice after S. Typhimurium infection, but the level of transforming growth factor β (TGF-β), a powerful immunosuppressive factor, was much higher in TLR2−/− mice than in WT mice. Meanwhile, the Th2 cytokine IL-10, an important regulatory cytokine in inflammation, was also elevated in infected TLR9−/− mice (Fig. 2C). These results showing that TLR9 deficiency resulted in systemic inflammation in response to S. Typhimurium infection could provide an explanation for the early death of TLR9−/− mice and suggested that TLR9 normally functioned to protect the host against a systemic cytokine storm after S. Typhimurium infection.
FIG 2.

Systemic immune responses in mice following S. Typhimurium infection. Age-matched WT (n = 6), TLR2−/−, and TLR9−/− (n ≥ 4) mice were i.g. infected with 5 × 104 CFU of S. Typhimurium (ST), and serum was harvested at 7 days postinfection. Inflammatory cytokine (A), chemokine (B), and immunosuppressive cytokine (C) levels were measured by CBA. n.d., not detected. Values are expressed in pg/ml or ng/ml serum as the means ± standard deviations (SD); statistical significance was determined as P values of <0.05 (*) and <0.01 (**) compared with infected WT mice.
TLR9 protects from severe Salmonella hepatitis after S. Typhimurium infection.
As an important complication of S. Typhimurium infection, Salmonella hepatitis impairs pathogen clearance and threatens survival. To determine the effect of TLR-mediated innate immunity on the pathogenesis of Salmonella hepatitis, we assessed histopathology, bacterial burden, and cytokine levels in the livers of S. Typhimurium-infected mice. On day 7 postinfection, TLR9−/− mice exhibited significant hepatomegaly accompanied by apparent bacterial plaque in the liver tissues; however, TLR2−/− mice showed only a slight increase in liver weight over that of the WT mice (Fig. 3A). Furthermore, H&E-stained liver sections and significantly higher ALT levels indicated severe liver injury in TLR9−/− mice compared to that in infected WT mice, whereas lymphocyte infiltration was rarely observed in the liver tissues from infected TLR2−/− mice (Fig. 3B and C). Since pathogen invasion was the key cause of Salmonella hepatitis, we determined S. Typhimurium localization and bacterial number within the liver. As shown in Fig. 3D, infected TLR9−/− mice had significantly higher bacterial numbers in livers than infected WT mice, while infected TLR2−/− mice exhibited significantly lower bacterial numbers. Furthermore, production of inflammatory mediators, including TNF-α, IL-1β, IL-6, MCP-1, and IFN-γ, was increased in liver homogenate supernatants from infected TLR9−/− mice compared to that in infected WT mice, but only marginal levels were detected in infected TLR2−/− mice (Fig. 3E). These findings showed that TLR9 deficiency aggravated the S. Typhimurium infection-mediated hepatic inflammation observed in WT mice, while TLR2 deficiency dampened this hepatic inflammation, thus indicating that TLR9 normally functioned to eliminate bacteria and therefore reduce inflammation-related immunopathology in the liver, whereas TLR2 normally functioned to promote bacterial survival within the host.
FIG 3.

Salmonella hepatitis in S. Typhimurium-infected mice. Mice were challenged i.g. with 5 × 104 CFU of S. Typhimurium (ST) for 7 days. (A) The percentage of liver in total body weight. (B) Histological samples were prepared as described in Materials and Methods, and images of H&E-stained slides were obtained under the light microscope (20× objective). Lymphocyte recruitment into liver tissues is demarcated by arrows. Data presented are representative of at least 3 independent experiments. (C) Serum ALT levels were measured by enzyme-linked immunosorbent assay (ELISA). (D) Bacterial burden was assessed by determining CFU numbers in liver tissues. Results are expressed as log10, and each individual data point represents data from a single animal. (E) Supernatant from liver homogenates was collected, and cytokine levels were measured by CBA; n ≥ 4 mice in each group. n.d., not detected. Data are expressed as the means ± SD; statistical significance was determined as P values of <0.05 (*) and <0.01 (**) compared with infected WT mice.
TLR2 and TLR9 play opposing roles in macrophage-mediated bactericidal activity.
TLR-mediated recognition, phagocytosis, and elimination of pathogens by macrophages are key processes in antimicrobial innate immunity. In order to address whether TLR2 or TLR9 activation influenced macrophage-mediated clearance of S. Typhimurium, we tested macrophage function in TLR2−/− or TLR9−/− mice. First, we evaluated how S. Typhimurium infection in vitro influenced TLR2 and TLR9 expression on WT peritoneal macrophages and found that TLR2 expression was markedly increased 12 h postinfection, whereas TLR9 expression was significantly decreased (Fig. 4A). The number of live bacteria residing in in vitro-infected TLR9−/− peritoneal macrophages was significantly higher at 4 h postinfection than that in WT macrophages, while significantly fewer live bacteria were detected in TLR2−/− macrophages (Fig. 4B) than in WT macrophages. Functionally, iNOS activity was increased in TLR2−/− macrophages (Fig. 4C) compared to that in WT macrophages, although ROS levels were similar between the 2 groups (Fig. 4D). In contrast, no significant change in iNOS activity was detected in infected TLR9−/− macrophages compared to that in WT macrophages, but a reduction in ROS levels was observed between the 2 groups (Fig. 4C and D).
FIG 4.

TLR2 and TLR9 signaling are involved in eliminating S. Typhimurium infection in macrophages. (A) TLR2 expression and TLR9 expression in peritoneal macrophages from WT mice were analyzed by FACS after in vitro stimulation with S. Typhimurium for 12 h. Results are representative of 3 independent experiments. (B) Intracellular bacterial levels in peritoneal macrophages from mice infected with S. Typhimurium for 4 h. (C and D) Peritoneal macrophages were stimulated with S. Typhimurium (MOI = 10:1) for 12 h, iNOS generation was analyzed by the nitric oxide synthase assay kit (C), and the mean fluorescence (MFI) of ROS was analyzed by the reactive oxygen species assay kit (D). (E) Peritoneal macrophages were stimulated with S. Typhimurium (MOI = 10:1) for 4 h, and macrophage necroptosis was measured by FACS using annexin V-PI double staining. For the left panel, one representative from 3 independent experiments is shown. Data are expressed as the means ± SD from at least 3 independent experiments. Statistical significance was determined as P values of <0.05 (*) and <0.01 (**) compared with infected WT mice.
S. Typhimurium expresses Salmonella pathogenicity island 1 (SPI-1) and 2 (SPI-2) genes (15) that allow it to not only survive and replicate within host macrophages but also to utilize phagocytes as a carrier to transfer bacteria from the intestine to the systemic circulation, which then delivers the bacteria into the liver (16). Once inside the liver, necroptosis caused by S. Typhimurium invasion, also known as a passive method of cell death by programmed necrosis, can promote typhoid granuloma formation and tissue necrosis in the liver that can lead to higher bacterial burden and increased inflammation. Compared with WT and TLR2−/− macrophages, TLR9−/− macrophages were more sensitive to S. Typhimurium-induced necroptosis (Fig. 4E), which is consistent with the observed higher bacterial burden after oral infection with S. Typhimurium in TLR9−/− mice than in WT mice shown above. Collectively, these results suggested that the opposing roles of TLR2 and TLR9 on macrophage-mediated clearance of S. Typhimurium might determine S. Typhimurium localization and number within the liver, which could then affect the degree of inflammation in Salmonella hepatitis.
TLR2 and TLR9 both affect the sensitivity of S. Typhimurium-infected macrophages to NK cell-mediated cytolysis.
After being delivered into the liver via the circulatory system, S. Typhimurium proliferates in Kupffer cells, and the innate immune system provides the first line of defense against invading microorganisms by inducing a variety of inflammatory and antimicrobial responses. NK cells are an important component of the innate immune system and have been shown to interact with infected macrophages, including Kupffer cells. This interaction plays a vital role in the clearance of intracellular pathogens through the natural cytotoxicity receptor NKG2D and its ligands, including RAE-1, H60, and MULT-1, expressed on intracellular pathogen-infected mononuclear phagocytes (17, 18).
To begin determining whether and how TLR2 and/or TLR9 played any role in the NK-macrophage interaction, we first tested whether and how NK cell frequency and function changed after oral S. Typhimurium infection in WT, TLR2−/−, and TLR9−/− mice. As shown in Fig. 5A, the NK cell percentage was elevated in both infected WT and TLR9−/− mice, peaking at 3 days postinfection in TLR9−/− mice and at 5 days postinfection in WT mice; in contrast, the NK cell percentage did not significantly change in TLR2−/− mice over the 7-day observation period. Further analysis showed that expression of the CD69 activation marker was significantly increased on hepatic NK cells from WT and TLR9−/− mice on day 7 after infection (Fig. 5B). However, the degranulation marker CD107a, a molecule representing activated NK cell cytotoxic activity, was significantly lower in both TLR2−/− and TLR9−/− mice than in WT mice after infection (Fig. 5C); similar results were observed for the percentage of IFN-γ+ NK cells (Fig. 5D). Thus, even though the percentage of CD69+ NK cells increased in TLR9−/− mice, the lower CD107a and IFN-γ expression in NK cells in infected TLR2−/− and TLR9−/− mice than in WT mice showed that they harbored functionally deficient NK cells against S. Typhimurium, indicating that TLR2 and TLR9 both normally played a role in promoting NK cell activation after infection.
FIG 5.
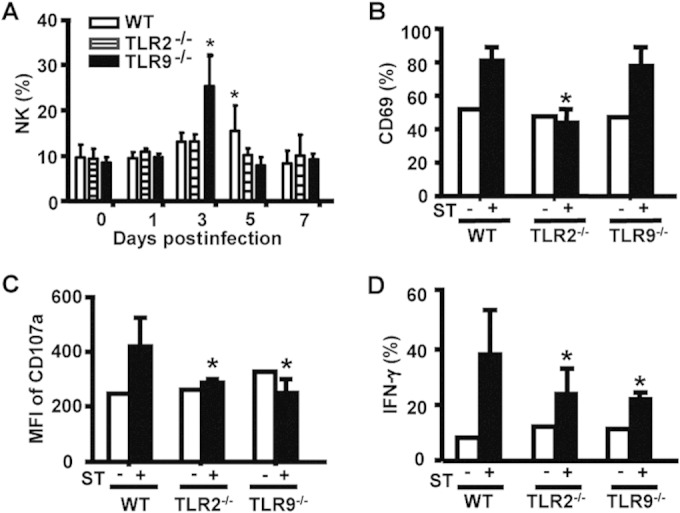
TLR2 or TLR9 deficiency reduces liver NK cell activation during S. Typhimurium infection. WT, TLR2−/−, and TLR9−/− mice were i.g. infected with 5 × 104 CFU of S. Typhimurium. (A) The percentage of NK1.1+CD3− NK cells within liver MNCs was analyzed from day 0 to day 7. CD69 expression (B) and the MFI of CD107a expression (C) on NK1.1+CD3− liver NK cells were measured by FACS analysis. (D) The percentage of IFN-γ-producing NK1.1+CD3− from mice 7 days after S. Typhimurium infection was also measured by FACS analysis. Data are expressed as the means ± SD from at least 4 mice. Statistical significance was determined as P values of <0.05 (*) compared with infected WT mice.
To next determine whether TLR2 and TLR9 played a role in the cross talk among NK cells, macrophages, and S. Typhimurium, we first evaluated the percentage of activated CD69+ NK cells in WT mice after S. Typhimurium or S. Typhimurium-infected macrophages were injected into the livers of WT mice by tail vein injection. While infection with S. Typhimurium alone increased the percentage of CD69+ NK cells above that of the noninfected control, NK cell activation by injection of S. Typhimurium-infected macrophages was much lower than that by injection of S. Typhimurium alone (Fig. 6A). In vitro experiments further demonstrated that S. Typhimurium directly activated liver MNC-derived WT, TLR2−/−, and TLR9−/− NK cells in a similar manner (Fig. 6B), indicating that NK cell activation by S. Typhimurium was independent of TLR2 or TLR9 activity. However, compared to WT NK cells cocultured with S. Typhimurium-infected WT macrophages, the CD69+ NK cell percentage was significantly decreased when WT NK cells were cocultured with S. Typhimurium-infected TLR2−/− or TLR9−/− macrophages, which was similar to that for NK cells alone; and this increase of CD69+ NK cell percentage was inhibited, as cocultured cells were separated in Transwell chambers, indicating that this macrophage-expressed TLR2- and TLR9-mediated effect on NK cell function occurred in a manner dependent upon cell-cell contact (Fig. 6C). Consistent with this inhibited NK cell activation, NK cell cytotoxicity against S. Typhimurium-infected TLR2−/− or TLR9−/− macrophages was also lower than that against S. Typhimurium-infected WT macrophages (Fig. 6D). These results showed that both TLR2 and TLR9 deficiency suppressed the sensitivity of S. Typhimurium-infected macrophages to NK cell-mediated cytolysis, indicating that both TLR2 and TLR9 played a role to promote NK cell-mediated cytolysis against S. Typhimurium-infected macrophages.
FIG 6.

TLR2 and TLR9 deficiency suppresses the susceptibility of S. Typhimurium-infected macrophages to NK cell-mediated cytolysis. (A) S. Typhimurium or S. Typhimurium-infected macrophages were injected into the livers of WT mice (n = 4) by tail vein injection, and the percentage of CD69+NK1.1+CD3− hepatic lymphocytes was evaluated after 12 h by FACS. (B) Hepatic MNCs from WT, TLR2−/−, and TLR9−/− mice were stimulated in vitro with S. Typhimurium for 12 h, and the percentage of CD69+NK1.1+CD3− NK cells was then detected by FACS. (C) Hepatic MNCs from WT mice were cocultured with WT, TLR2−/−, or TLR9−/− S. Typhimurium-infected macrophages either in the same wells (Mϕ+ST+NK) or in Transwell chambers in which the hepatic MNCs were separated from the macrophages (Mϕ+ST/NK). After 12 h, the percentage of CD69+NK1.1+CD3− lymphocytes was evaluated by FACS. (D) NK cell cytotoxicity against WT, TLR2−/−, and TLR9−/− macrophages from mice treated with S. Typhimurium for 1 h (E:T ratio = 5:1) was measured by FACS using CFSE–7-AAD double staining. (E) The percentage of NKG2D+NK1.1+CD3− liver lymphocytes from mice infected with S. Typhimurium for 7 days was measured by FACS analysis. (F) Peritoneal macrophages from WT, TLR2−/−, and TLR9−/− mice were stimulated in vitro with S. Typhimurium for 12 h, and RAE-1 and Qa-1b surface expression were evaluated by FACS. Data are expressed as the means ± SD from at least 4 mice. Statistical significance was determined as P values of <0.05 (#) compared with the mock-infected group, P values of <0.01 (▲▲) compared with the infected TLR2−/− macrophage group, and P values of <0.05 (*) and <0.01 (**) compared with the infected WT macrophage group.
Previous studies have shown that interactions between NKG2D and its ligands RAE-1, H60, and MULT-1 are involved in NK cell recognition and lysis of infected mononuclear phagocytes (17, 19). We therefore attempted to determine whether TLRs influence the expression of NKG2D on NK cells or NKG2D ligands on macrophages in order to address how the macrophages might communicate with NK cells to affect NK cell function. Flow cytometry analysis showed that the percentages of NKG2D+ NK cells from liver lymphocytes of WT, TLR2−/−, and TLR9−/− mice infected with S. Typhimurium were all decreased (Fig. 6E). While the percentage of RAE-1+ macrophages (RAE-1+ Mϕ) increased after S. Typhimurium infection in WT, TLR2−/−, and TLR9−/− macrophages in vitro, the extent of elevation after infection was much lower in TLR2−/− and TLR9−/− macrophages than in WT macrophages (Fig. 6F). In contrast, Qa-1, the ligand for the NKG2A inhibitory receptor, was downregulated in all macrophage phenotypes after S. Typhimurium infection, but no detectable differences in the magnitude of expression with infection among these groups were observed (Fig. 6F); furthermore, no statistically significant changes in macrophage expression of H60 and MULT-1 were detected between WT and either TLR2−/− or TLR9−/− macrophages (data not shown). Collectively, these results showed that TLR2 and TLR9 deficiency enhanced the resistance of S. Typhimurium-infected macrophages to NK cell lysis through inhibiting RAE-1 expression, indicating that TLR2 and TLR9 normally functioned to upregulate RAE-1 and promote NK cell lysis of S. Typhimurium-infected macrophages.
DISCUSSION
As the key sensors for pathogen-associated molecular patterns (PAMPs), TLRs play an essential role in host defense against microbes by recognizing conserved microbial molecules (18). Previous studies show that TLR activation induces APC maturation, enhances antigen presentation, and upregulates costimulatory molecules and cytokine production. However, individual TLRs can display different functions in pathogen clearance depending on differences in the virulence of a specific pathogen and the route of infection (20). Thus, for these reasons, our current understanding regarding the contributions of each TLR to host defense against S. Typhimurium infection remains relatively poor.
In the present study, we investigated the roles of TLR2, TLR9, and TLR4 in the innate immune response to atrichia S. Typhimurium infection, a Salmonella strain that lacks flagellin. Interestingly, we found that, compared to WT mice, TLR2−/− mice were more resistant, TLR9−/− mice were more susceptible (Fig. 1), and TLR4−/− mice exhibited a similar response in terms of survival after high-dose oral S. Typhimurium infection. Talbot et al. previously observed that TLR4−/− mice had an increased rate of survival following S. Typhimurium infection compared to that of WT mice (21), but this seeming contradiction to our data may be due to the different experimental design or different virulent S. Typhimurium strain used between the studies. Since septicemia is the main cause of S. Typhimurium-induced death in mice, the difference in susceptibility between TLR2−/− and TLR9−/− mice indicated that TLR2 and TLR9 may have opposing effects on the host immune response in terms of resistance to, and clearance of, S. Typhimurium.
Inflammation is the prerequisite of immune responses to pathogens. But tissue injury caused by acute inflammation also affects the survival of S. Typhimurium-infected mice (22). Indeed, extremely high levels of inflammatory mediators were observed in serum from TLR9−/− mice 7 days after infection (Fig. 2); these included IFN-γ, TNF-α, IL-6, IL-12p40, IL-17, and IL-18 proinflammatory cytokines as well as MIP-1β, G-CSF, and MCP-1, which were all potent neutrophil chemoattractants. Neutrophils are known to be the first white blood cells to enter the tissue during acute inflammation and to be particularly effective at killing extracellular S. Typhimurium (23). TNF-a, IL-12, IFN-γ, and IL-18 production are necessary for controlling S. Typhimurium growth in the acute stage of infection. However, the excessive production of these cytokines in TLR9−/− mice suggested a more severe inflammatory reaction in these mice. Although a cytokine storm in infected TLR9−/− mice may be beneficial to eliminating a higher bacterial load, this overexpression of inflammatory mediators may also cause an immunopathology that could very likely explain the hastened death observed in infected TLR9−/− mice. Interestingly, infected TLR9−/− mice also exhibited a markedly higher level of serum IL-10 than infected WT mice, which could suppress bactericidal responses and ultimately promote the macrophages in acting as hosts for S. Typhimurium replication, leading to further increased bacterial burden (24). Thus, these data reveal that TLR9 normally plays a critical role in the control of anti-S. Typhimurium-induced excessive hepatic inflammation.
TLR2 is known as an activating signal on phagocytes. In contrast to TLR9−/− mice, TLR2−/− and WT mice exhibited only slightly increased inflammatory cytokine and chemokine production after S. Typhimurium infection (Fig. 2). We also observed that TLR2−/− mice displayed much higher TGF-β levels than WT and TLR9−/− mice in response to S. Typhimurium infection. TGF-β is well known as a key regulator of inflammation and also functions as an anti-inflammatory cytokine through upregulating IL-10 (25). Consistent with our findings, TGF-β treatment in Salmonella infection has been shown to reduce bacterial numbers in the organs of infected mice by inducing IL-1α, IFN-γ, and NO production (26). Macrophages are a major source of TGF-β upon stimulation by microorganisms, and TLR2 and TLR9 expressed on macrophages might be involved in the regulation of TGF-β production. Thus, these data reveal that TLR2 normally promotes hepatic inflammatory reaction to S. Typhimurium infection. Taking the TLR2 and TLR9 data together, these findings demonstrate that the opposing effects of TLR2 and TLR9 in host immune response during S. Typhimurium infection lie in their different roles in regulating hepatic inflammation.
The excessive production of inflammatory cytokines in S. Typhimurium-infected mice was likely due to uncontrolled pathogen infection. The typical S. Typhimurium course of infection occurs in a host by first ingesting the Salmonella into the intestine, where the bacteria can then directly enter the blood (11). Once in the blood, S. Typhimurium can travel extracellularly or in CD18+ leukocytes to secondary sites. The liver in particular is a common site of secondary bacterial colonization, which can have an effect on the survival of infected mice depending on the severity of the ensuing Salmonella hepatitis (10). Here, TLR9−/− mice showed much more severe Salmonella hepatitis and higher bacterial burden than WT mice 7 days postinfection; however, TLR2−/− mice presented only slightly elevated readouts of infection and lower bacterial burdens than WT mice (Fig. 3). These results suggest that TLR9 activation may be critical for controlling S. Typhimurium infection in the liver, while TLR2 activation may exacerbate pathogen invasion.
Macrophages are a key component of the innate immune system. After engulfing pathogens, macrophages are activated and produce massive cytokines and chemokines, resulting in an inflammation reaction. The pathogenesis of severe hepatic damage involved in Salmonella hepatitis may be multifactorial and include contributions from bacterial endotoxin (LPS), local inflammation, and host immune reactions. Since LPS released by virulent S. Typhimurium strains within Kupffer cells is the key determinant of hepatic damage (12), we speculate that the differing numbers of Salmonella CFU residing in the livers of WT and TLR-deficient mice will affect the degree of liver injury. To test this hypothesis, we evaluated TLR2 and TLR9 expression levels on WT peritoneal macrophages after in vitro infection with S. Typhimurium. While TLR2 expression increased on the macrophage surface, both surface and intracellular TLR9 expression decreased after engulfing S. Typhimurium (Fig. 4). This expression data suggest the possibility that TLR2 expression may be unfavorable for S. Typhimurium clearance, an idea supported by the data showing that TLR2−/− macrophages exhibit a significantly lower bacterial load and higher iNOS activity than WT and TLR9−/− macrophages. Thus, S. Typhimurium may utilize the TLR2 signaling pathway as a subversion technique to avoid the endocellular digestion process, and more iNOS in infected TLR2−/− macrophages may be due to its high TLR4 expression (data not shown), which is known to be involved in iNOS production in macrophages (27). In contrast to TLR2 function, TLR9 functions as an excellent antimicrobial mechanism in macrophages; indeed, our data show that TLR9 activation is essential for ROS production, as previously shown in DCs (28), and also protects against necroptosis, which will help to prevent bacterium spread in the host.
NK cells are another important component of the innate immune system, playing a central role in response to intracellular bacterial infection. Besides degranulation and IFN-γ secretion (29, 30), NK cells can also lyse S. Typhimurium-infected monocytes as a major mechanism to remove bacteria from the liver (31). Here, we observed that both TLR2 deficiency and TLR9 deficiency decreased NK cell degranulation (CD107a) and IFN-γ expression in response to S. Typhimurium infection (Fig. 5), suggesting that TLR2 and TLR9 normally function to promote NK cell function and activation. Our data also showed that the percentage of activated CD69+ NK cells actually increased in TLR9−/− mice, but these cells might be nonfunctional NK cells based on their low expression of CD107a and IFN-γ, although this idea remains to be formally tested. Investigation into the mechanism underlying how TLR2 and TLR9 were involved in regulating NK cell function after S. Typhimurium infection showed that although S. Typhimurium alone directly activated WT, TLR2−/−, and TLR9−/− NK cells in vitro, TLR2 or TLR9 deficiency in S. Typhimurium-infected macrophages inhibited NK cell activation in a cell-to-cell-dependent manner (Fig. 6A to C). This result suggested that the cross talk occurring between NK cells and S. Typhimurium-infected macrophages involved TLR2 and TLR9 functions. NK cell cytotoxicity is dependent on the balance between activating and inhibitory receptors expressed in NK cells. NKG2D is an important activating receptor for NK cell-mediated cytolysis, and murine NKG2D interacts with RAE-1, H60, and MULT-1 ligands. The TLR signaling pathway activation is known to regulate RAE-1 expression (32). Here, although RAE-1 surface expression increased after infection in WT, TLR2−/−, and TLR9−/− macrophages, the magnitude of RAE-1 upregulation was attenuated in TLR2−/− and TLR9−/− macrophages compared to that in WT macrophages and was accompanied by increased resistance of these macrophages to NK cell-mediated cytolysis (Fig. 6). These data indicate that TLR2 and TLR9 likely participate in NK-macrophage cross talk after S. Typhimurium infection by regulating RAE-1 expression. In addition, we found that S. Typhimurium may also regulate NK cell cytolysis as a bacterial self-protection mechanism independent of TLR activity by downregulating NKG2D on NK cells.
In summary (Fig. 7), these findings collectively suggest that TLR2 and TLR9 likely play different roles in the immune response to S. Typhimurium infection. Although we found that both TLR2 and TLR9 promote NK cell cytolytic function against S. Typhimurium-infected macrophages, our data reveal that the roles of TLR2 and TLR9 in macrophage-mediated clearance of S. Typhimurium are opposite. TLR9 activation is critical for controlling S. Typhimurium infection in the liver; indeed, TLR9 expression is propitious to clearance of S. Typhimurium in the host, where TLR9 deficiency leads to macrophage dysfunction, which then contributes to a cytokine storm, system inflammation, severe Salmonella hepatitis, and shortened survival. In contrast to TLR9, our data reveal that TLR2 activation exacerbates pathogen invasion, where TLR2 deficiency augments the resistance to S. Typhimurium by enhancing the bactericidal capability of macrophages, reducing S. Typhimurium load, and suppressing inflammation. These results also suggest that TLR2 antagonists or TLR9 agonists may serve as potential anti-S. Typhimurium therapeutic agents.
FIG 7.
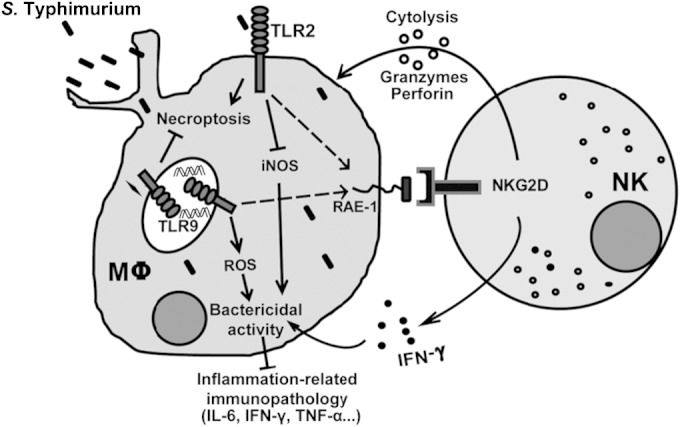
Working model for TLR2 and TLR9 contribution to the protection from S. Typhimurium infection. In response to S. Typhimurium infection, TLR2 and TLR9 normally function to upregulate RAE-1 and promote NK cell-mediated lysis of S. Typhimurium-infected macrophages. However, in macrophage-mediated clearance of S. Typhimurium, TLR9 promotes bactericidal activity and reduces inflammation-related immunopathology, whereas TLR2 normally functions to promote bacterial survival within macrophage and results in systemic inflammation.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81373222) and the National Basic Research Program of China (973 Program, grant no. 2006CB504303).
REFERENCES
- 1.Broz P, Ohlson MB, Monack DM. 2012. Innate immune response to Salmonella Typhimurium, a model enteric pathogen. Gut Microbes 3:62–70. doi: 10.4161/gmic.19141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coburn B, Grassl GA, Finlay BB. 2007. Salmonella, the host and disease: a brief review. Immunol Cell Biol 85:112–118. doi: 10.1038/sj.icb.7100007. [DOI] [PubMed] [Google Scholar]
- 3.Gowda DC. 2007. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol 23:596–604. doi: 10.1016/j.pt.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Jung C, Meinzer U, Montcuquet N, Thachil E, Chateau D, Thiebaut R, Roy M, Alnabhani Z, Berrebi D, Dussaillant M, Pedruzzi E, Thenet S, Cerf-Bensussan N, Hugot JP, Barreau F. 2012. Yersinia pseudotuberculosis disrupts intestinal barrier integrity through hematopoietic TLR-2 signaling. J Clin Invest 122:2239–2251. doi: 10.1172/JCI58147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, Buzoni-Gatel D, Kasper LH. 2006. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol 176:7589–7597. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- 6.Ohl ME, Miller SI. 2001. Salmonella: a model for bacterial pathogenesis. Annu Rev Med 52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Zhang C, Zhou Z, Zhang J, Tian Z. 2012. Small intestinal intraepithelial lymphocytes expressing CD8 and T cell receptor gammadelta are involved in bacterial clearance during Salmonella enterica serovar Typhimurium infection. Infect Immun 80:565–574. doi: 10.1128/IAI.05078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pramoolsinsap C, Viranuvatti V. 1998. Salmonella hepatitis. J Gastroenterol Hepatol 13:745–750. doi: 10.1111/j.1440-1746.1998.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 9.El-Newihi HM, Alamy ME, Reynolds TB. 1996. Salmonella hepatitis: analysis of 27 cases and comparison with acute viral hepatitis. Hepatology 24:516–519. doi: 10.1002/hep.510240308. [DOI] [PubMed] [Google Scholar]
- 10.Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, Baumler AJ. 2001. Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect 3:1335–1344. doi: 10.1016/S1286-4579(01)01495-2. [DOI] [PubMed] [Google Scholar]
- 11.Mastroeni P, Grant A, Restif O, Maskell D. 2009. A dynamic view of the spread and intracellular distribution of Salmonella enterica. Nat Rev Microbiol 7:73–80. doi: 10.1038/nrmicro2034. [DOI] [PubMed] [Google Scholar]
- 12.Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, Fang FC. 2004. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J Immunol 172:6202–6208. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 13.Tian Z, Chen Y, Gao B. 2013. Natural killer cells in liver disease. Hepatology 57(4):1654–1662. doi: 10.1002/hep.26115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Sun R, Wei H, Dong Z, Gao B, Tian Z. 2006. Poly I:C prevents T cell-mediated hepatitis via an NK-dependent mechanism. J Hepatol 44:446–454. doi: 10.1016/j.jhep.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Finlay BB, Brumell JH. 2000. Salmonella interactions with host cells: in vitro to in vivo. Philos Trans R Soc Lond B Biol Sci 355:623–631. doi: 10.1098/rstb.2000.0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, Brown W, Le M, Berggren R, Parks WT, Fang FC. 1999. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature 401:804–808. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- 17.Vankayalapati R, Wizel B, Weis SE, Safi H, Lakey DL, Mandelboim O, Samten B, Porgador A, Barnes PF. 2002. The NKp46 receptor contributes to NK cell lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol 168:3451–3457. doi: 10.4049/jimmunol.168.7.3451. [DOI] [PubMed] [Google Scholar]
- 18.Albiger B, Dahlberg S, Henriques-Normark B, Normark S. 2007. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J Intern Med 261:511–528. doi: 10.1111/j.1365-2796.2007.01821.x. [DOI] [PubMed] [Google Scholar]
- 19.Vankayalapati R, Garg A, Porgador A, Griffith DE, Klucar P, Safi H, Girard WM, Cosman D, Spies T, Barnes PF. 2005. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol 175:4611–4617. doi: 10.4049/jimmunol.175.7.4611. [DOI] [PubMed] [Google Scholar]
- 20.Netea MG, Van der Meer JW, Kullberg BJ. 2004. Toll-like receptors as an escape mechanism from the host defense. Trends Microbiol 12:484–488. doi: 10.1016/j.tim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Talbot S, Totemeyer S, Yamamoto M, Akira S, Hughes K, Gray D, Barr T, Mastroeni P, Maskell DJ, Bryant CE. 2009. Toll-like receptor 4 signalling through MyD88 is essential to control Salmonella enterica serovar Typhimurium infection, but not for the initiation of bacterial clearance. Immunology 128:472–483. doi: 10.1111/j.1365-2567.2009.03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakoneczna I, Hsu HS. 1980. The comparative histopathology of primary and secondary lesions in murine salmonellosis. Br J Exp Pathol 61:76–84. [PMC free article] [PubMed] [Google Scholar]
- 23.Conlan JW. 1996. Neutrophils prevent extracellular colonization of the liver microvasculature by Salmonella typhimurium. Infect Immun 64:1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KS, Jeong ES, Heo SH, Seo JH, Jeong DG, Choi YK. 2011. IL-10 suppresses bactericidal response of macrophages against Salmonella Typhimurium. J Microbiol 49:1050–1053. doi: 10.1007/s12275-011-1043-z. [DOI] [PubMed] [Google Scholar]
- 25.Li Q, Sun B, Dastgheib K, Chan CC. 1996. Suppressive effect of transforming growth factor beta1 on the recurrence of experimental melanin protein-induced uveitis: upregulation of ocular interleukin-10. Clin Immunol Immunopathol 81:55–61. doi: 10.1006/clin.1996.0157. [DOI] [PubMed] [Google Scholar]
- 26.Galdiero M, Marcatili A, Cipollaro de l'Ero G, Nuzzo I, Bentivoglio C, Romano Carratelli C. 1999. Effect of transforming growth factor beta on experimental Salmonella typhimurium infection in mice. Infect Immun 67:1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vazquez-Torres A, Fang FC. 2001. Oxygen-dependent anti-Salmonella activity of macrophages. Trends Microbiol 9:29–33. doi: 10.1016/S0966-842X(00)01897-7. [DOI] [PubMed] [Google Scholar]
- 28.Lahiri A, Das P, Vani J, Shaila MS, Chakravortty D. 2010. TLR 9 activation in dendritic cells enhances Salmonella killing and antigen presentation via involvement of the reactive oxygen species. PLoS One 5:e13772. doi: 10.1371/journal.pone.0013772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowdery JS, Chace JH, Yi AK, Krieg AM. 1996. Bacterial DNA induces NK cells to produce IFN-gamma in vivo and increases the toxicity of lipopolysaccharides. J Immunol 156:4570–4575. [PubMed] [Google Scholar]
- 30.Lapaque N, Walzer T, Meresse S, Vivier E, Trowsdale J. 2009. Interactions between human NK cells and macrophages in response to Salmonella infection. J Immunol 182:4339–4348. doi: 10.4049/jimmunol.0803329. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z, Zhang C, Zhang J, Tian Z. 2012. Macrophages help NK cells to attack tumor cells by stimulatory NKG2D ligand but protect themselves from NK killing by inhibitory ligand Qa-1. PLoS One 7:e36928. doi: 10.1371/journal.pone.0036928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamerman JA, Ogasawara K, Lanier LL. 2004. Cutting edge: Toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J Immunol 172:2001–2005. doi: 10.4049/jimmunol.172.4.2001. [DOI] [PubMed] [Google Scholar]