

Abstract
Cryptococcosis due to a highly virulent fungus, Cryptococcus gattii, emerged as an infectious disease on Vancouver Island in Canada and surrounding areas in 1999, causing deaths among immunocompetent individuals. Previous studies indicated that C. gattii strain R265 isolated from the Canadian outbreak had immune avoidance or immune suppression capabilities. However, protective immunity against C. gattii has not been identified. In this study, we used a gain-of-function approach to investigate the protective immunity against C. gattii infection using a dendritic cell (DC)-based vaccine. Bone marrow-derived dendritic cells (BMDCs) efficiently engulfed acapsular C. gattii (Δcap60 strain), which resulted in their expression of costimulatory molecules and inflammatory cytokines. This was not observed for BMDCs that were cultured with encapsulated strains. When Δcap60 strain-pulsed BMDCs were transferred to mice prior to intratracheal R265 infection, significant amelioration of pathology, fungal burden, and the survival rate resulted compared with those in controls. Multinucleated giant cells (MGCs) that engulfed fungal cells were significantly increased in the lungs of immunized mice. Interleukin 17A (IL-17A)-, gamma interferon (IFN-γ)-, and tumor necrosis factor alpha (TNF-α)-producing lymphocytes were significantly increased in the spleens and lungs of immunized mice. The protective effect of this DC vaccine was significantly reduced in IFN-γ knockout mice. These results demonstrated that an increase in cytokine-producing lymphocytes and the development of MGCs that engulfed fungal cells were associated with the protection against pulmonary infection with highly virulent C. gattii and suggested that IFN-γ may have been an important mediator for this vaccine-induced protection.
INTRODUCTION
Inhalation of the airborne fungal pathogens Cryptococcus neoformans and Cryptococcus gattii causes life-threatening infectious diseases despite treatment with antifungal drugs. These two species are genetically close, although they have some distinct features. C. neoformans typically causes fatal infections, such as meningitis, in immunocompromised hosts, whereas C. gattii causes similar infections in immunocompetent hosts. Although cryptococcosis caused by C. gattii is endemic in tropical areas, such as Australia and Papua New Guinea, outbreaks of C. gattii, including fatalities among healthy individuals, were reported on Vancouver Island and surrounding areas beginning in 1999 (1, 2). In response to this, the Centers for Disease Control and Prevention (CDC) of the United States and British Columbia organized a public health working group to promote awareness of this outbreak (3–5).
Using mouse pulmonary infection models, two groups independently showed that C. gattii strain R265, which was clinically isolated during the Canadian outbreak, was more virulent than C. neoformans strain H99, which is frequently studied (6, 7). Although the mechanisms for its hypervirulence remain unknown, there is evidence that C. gattii induces a less severe inflammatory response than that induced by C. neoformans infection. Histological and flow cytometry analyses showed reduced migration of inflammatory cells into the lungs of mice infected with R265 compared with those infected with H99 (7–9). Additionally, a smaller amount of inflammatory cytokines was found in the lungs of mice infected with C. gattii (9) and in the cerebrospinal fluid of humans infected with C. gattii (10, 11). These findings suggest that C. gattii has a superior ability to suppress or evade the inflammatory response.
Previous studies indicated that one of the capsular components of C. gattii may have been involved in immune avoidance or immune suppression and was required for the complete virulence of C. gattii (12, 13). Because C. gattii can induce immune avoidance or suppression, an analysis of loss-of-function using gene knockout (KO) mice is not applicable for studying any protective immune responses against C. gattii, and the protective immunity against highly virulent C. gattii has not been well characterized. Thus, it is important to unravel any protective immunity against C. gattii to garner insights for the prevention, diagnosis, and treatment of cryptococcosis with highly virulent C. gattii.
Dendritic cells (DCs) play a central role in inducing effector T cells (14) and can also be utilized as antigen delivery systems for vaccinations for cancers or infections (15, 16). Adoptive transfer of DCs pulsed with fungal cells or with fungal RNA has also been utilized as a means to assess T cell-mediated immunity against pathogenic fungi (17–19). In this study, we implemented a gain-of-function approach to investigate protective immunity against C. gattii. We tested whether a DC-based vaccine could augment protective immunity against the highly virulent C. gattii strain R265 by evaluating the pathology, fungal burden, and survival rate after pulmonary infection in mice. Further, we examined cytokine-producing cells in the mouse spleen and lung. Moreover, we assessed the role of gamma interferon (IFN-γ) in the protective effect exerted by this DC-based vaccine using IFN-γ knockout mice.
MATERIALS AND METHODS
Ethics.
All our animal experiments were in compliance with the guidelines and policies of the Principles of Morality for Animal Experiments of the National Institute of Infectious Disease, Japan (approval numbers 213072, 21608, 214044, 114004, and 114029).
Mice.
C57BL/6J mice were purchased from Japan SLC, Inc. IFN-γ knockout (KO) mice (C57BL/6 background) were purchased from the Jackson Laboratory. Mice used in experiments were 6 to 8 weeks old and were maintained under specific-pathogen-free conditions at the National Institute of Infectious Diseases of Japan.
C. gattii.
C. gattii strain R265 (genotype VGII, mating type α, and serotype B) was kindly provided by Kyung J. Kwon-Chung (National Institutes of Health, Bethesda, MD); in 2001, this strain was clinically isolated from bronchial washings of infected patients during the Vancouver Island outbreak (20). To construct the acapsular C. gattii Δcap60 strain, CAP60 (GenBank accession number CGB_A6290C) of C. gattii strain PNG18 (genotype VGI, mating type α, and serotype B) was disrupted by gene replacement using the nourseothricin resistance gene NAT1, which was also clinically isolated from an infected person in Papua New Guinea (21). The disruption strategy used is shown in Fig. S1 in the supplemental material. The primers and sequences that were used for strain construction are listed in Table S1 in the supplemental material.
To prepare a gene replacement cassette, DNA fragments, such as for a marker gene and homologous regions, were amplified by PCR. The marker fragment NAT, including the ACT1 promoter, NAT1, and the TRP1 terminator, were amplified using the primers NAT-F and NAT-R derived from plasmid pCH233 used as a template (22). Homologous upstream and downstream regions (≈1 kb) of CAP60 were amplified with the primers CAP60up-F and CAP60up-R for the upstream region and CAP60down-F and CAP60down-R for the downstream region using C. gattii genomic DNA as a template. The primer pairs CAP60up-R and NAT-R and CAP60down-F and NAT-F contained complementary sequences that allowed them to anneal with the marker fragment NAT. Three fragments, (i) 5′ upstream region, (ii) 3′ downstream region, and (iii) NAT fragment, were equally mixed and used as a template for PCR with the primers CAP60up-F and CAP60down-R, which harbored a single gene disruption cassette. This cassette was introduced into C. gattii strain PNG18 using a helium-driven biolistic system (Bio-Rad) as described previously (23). The transformants were screened on a yeast extract-peptone-dextrose (YPD) agar plate (1% [wt/vol] yeast extract, 2% [wt/vol] Bacto peptone, and 2% [wt/vol] dextrose) that contained 100 μg/ml of nourseothricin. Homologous integration events were confirmed by PCR using the primers shown in Fig. S1. The CAP60 open reading frame (ORF), the region from the ATG codon to the TAG stop codon, was completely deleted in the Δcap60 strain.
Plasmid pJAF12-CAP60 was constructed to integrate the CAP60 ORF with a 1-kb flanking region into the genomic DNA of the Δcap60 strain. Primers CAP60up-NotI-F and CAP60down-SacII-R were used to amplify the CAP60 locus, and the amplified fragment was digested with NotI and SacII. The digested fragment was cloned into the NotI and SacII sites of pJAF12 (24). Plasmid pJAF12-CAP60 was introduced in the Δcap60 strain as described above, and the transformants were screened on a YPD agar plate that contained 100 μg/ml of nourseothricin and 200 μg/ml of Geneticin (G418). Homologous integration events were confirmed by PCR with the primers shown in Fig. S1 in the supplemental material.
To prepare heat-killed fungal cells, C. gattii was cultured in YPD medium with shaking overnight at 30°C. Cells were harvested and washed with sterile Dulbecco's phosphate-buffered saline (DPBS; Invitrogen), resuspended in DPBS, and boiled for 1 h. Heat-killed C. gattii cells were not washed further. The morphologies of heat-killed C. gattii were not altered based on microscopic observations. A suspension of heat-killed C. gattii cells was spread onto YPD agar and cultured at 30°C for 7 days to check whether all C. gattii cells were dead.
A conventional India ink preparation was used to observe capsule formation. C. gattii cells were grown in YPD medium at 30°C with shaking overnight. Then, 100 μl of a culture suspension was centrifuged and the harvested cells were resuspended in 20 μl of an ink solution, which included equal amounts of 4% (wt/vol) paraformaldehyde and India ink. Then, 5 μl of an ink-cell suspension was placed on a slide glass with a coverslip. Cells were observed by differential interference contrast microscopy (Inverted microscope IX81; Olympus).
BMDCs.
Bone marrow (BM) cells were harvested from the femurs and tibias of female C57BL/6J mice. Red blood cells were lysed with lysis buffer (9 volumes of 0.83% [wt/vol] NH4Cl and 1 volume of 200 mM Tris-HCl, pH 7.6). BM cells (3 × 106 cells/10 ml per dish) were cultured in RPMI 1640 (complete) medium (Sigma) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 1% (vol/vol) streptomycin-penicillin solution (Sigma; 10,000 U of penicillin and 10 mg/ml of streptomycin), 44 μM 2-mercaptoethanol, and 10 ng/ml of mouse granulocyte-macrophage colony-stimulating factor (mGM-CSF; PeproTech, Inc.) at 37°C under 5% CO2. Sterile petri dishes (not treated for cell culture) were used for cell culture. On day 3, 6 ml of the culture medium was removed, and 7 ml of fresh complete medium was added to the dishes. On day 5, 5 ml of fresh complete medium was added to the dishes. On day 6, nonadherent cells were collected and used as BM-derived dendritic cells (BMDCs).
Lung leukocytes.
Mouse lungs were perfused and rinsed with saline prior to being minced with scissors. Lung pieces were enzymatically digested at 37°C for 60 min with 5 ml/lung of a digestion solution (RPMI 1640 medium, 5% FBS, 2 mg/ml of collagenase D [Roche], and 10 μg/ml of DNase I [Sigma]) in 15-ml conical tubes and a tube rotator. After digestion, 250 μl of 100 mM EDTA solution was added to the tubes to stop digestion (final concentration, 5 mM EDTA). The lung pieces were then homogenized with a 70-μm cell strainer (BD-Falcon), and cell suspensions were harvested. A cell pellet was resuspended in 2 ml of 30% Percoll (GE Healthcare) and layered onto 4 ml of 44% and 70% Percoll. All Percoll solutions contained 2 mM EDTA to prevent cell aggregation. After centrifugation at 1,000 × g without acceleration and deceleration for 20 min at room temperature, cells at the 44-70% interface were collected and washed twice with complete RPMI 1640 medium. Cells (2 × 105) were attached to a glass slide using a Cytofuge-12 (Statspin, Inc.) and stained with Diff-Quik (Sysmex Corporation) for Wright-Giemsa staining, followed by determination of the proportion of each cell type under a microscope. The numbers of lymphocytes, macrophages, and polymorphonuclear cells (PMNs) were determined by multiplying the total leukocyte count by the proportion of each cell type.
Phagocytosis assay.
Prior to doing a phagocytosis assay, heat-killed C. gattii cells (1 × 109 cells/ml) were stained with acridine orange (stock concentration, 0.1 mg/ml; working concentration, 0.01 mg/ml) at room temperature for 1 h in the dark. Stained cells were washed with DPBS and resuspended in RPMI 1640 medium supplemented with 10% (vol/vol) heat-inactivated FBS. We confirmed that capsular strains as well as the wild-type strain were stained. A suspension of stained cells was stored at 4°C overnight. Harvested BMDCs (1 × 106 cells/ml) were incubated with prestained C. gattii cells (5 × 106 cells/ml; multiplicity of infection [MOI] = 5) in complete RPMI 1640 medium that contained mGM-CSF in 24-well culture plates. After incubation, 500 μl of 4% (vol/vol) paraformaldehyde (Fixation Buffer; Biolegend) was added and cells were fixed at room temperature for 5 min, followed by a washing with DPBS. Fungal cells that were not engulfed by BMDCs were stained with calcofluor white (Sigma; 1/10 dilution) at room temperature for 5 min. Culture wells were rinsed with DPBS before fluorescence microscopic analysis (inverted microscope IX81; Olympus). A mercury apo lamp, Olympus filter cube WU (BP330–385, DM400, BA420), and Olympus filter cube GFP (BP460–480, DM485, BA495–540) were used for the fluorescence imaging. At least five fields of view with ≈100 BMDCs/field were randomly selected. At least three fields were used to determine a phagocytosis rate, defined as the number of BMDCs engulfing fungal cells/total BMDCs × 100%. ImageJ software (National Institutes of Health, USA) was used for cell counting and color image merging.
Activation marker expression on BMDCs and cytokine-producing T cells.
BMDCs (2 × 106 cells/ml) were incubated in complete RPMI 1640 medium that contained mGM-CSF and heat-killed C. gattii cells (2 × 104 cells/ml; MOI = 0.1) or 100 ng/ml of lipopolysaccharide (LPS) from Escherichia coli O111:B4 (Sigma) for 24 h. Cells were then collected and stained for flow cytometry analysis. All antibodies and buffer used for cell staining were from Biolegend. Fc receptors were blocked with an anti-CD16/32 antibody (clone 93), after which cells were stained with the following antibodies: anti-CD11b (clone M1/70), anti-CD11c (clone N418), anti-CD86 (clone GL-1), anti-CD40 (clone MR1), and anti-I-Ab (clone AF6-120.1).
To assess cytokine-producing T cells, spleen cells (2 × 106 cells/ml) were cultured in complete RPMI 1640 medium with 100 ng/ml of anti-CD28 monoclonal antibody (MAb) (clone 37.51) and 100 ng/ml of anti-CD49d MAb (clone 9C10) in the presence of heat-killed Δcap60 cells (MOI = 0.1) for 5 to 6 days. Lung leukocytes were incubated in complete RPMI 1640 medium with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA) and 1 μM ionomycin for 3 h. To stop cytokine release, brefeldin A (final concentration, 5 μg/ml; Biolegend) and monensin (final concentration, 2 μM; Sigma; stock concentration, 72 mM in methanol) were added for the last 1.5 to 4 h of culture. After blocking of Fc receptors, cell surface molecules on harvested cells were stained with the following antibodies: anti-CD4 (clone GK1.5), anti-CD3 (clone 145-2C11), and anti-Thy1.2 (clone 30-H12). Intracellular cytokines were stained according to the manufacturer's instructions (Biolegend) with the following antibodies: anti-IFN-γ (clone XMG1.2), anti-interleukin 17A (anti-IL-17A) (clone TC11-18H10.1), and anti-tumor necrosis factor alpha (anti-TNF-α) (clone MP6-XT22). For RORγt staining, a FOXP3/transcription factor staining buffer set and anti-human/mouse RORγt (clone AFKJS-9) were used according to the manufacturer's instructions (eBioscience Inc.). Isotype-matched IgG was used for control staining for IFN-γ, IL-17A, TNF-α, and RORγt. Data were acquired with a BD FACSCaliburor BD FACSCantoII flow cytometer (BD Bioscience) and analyzed using FlowJo software (TreeStar Inc.).
Cytokine determinations.
BMDCs (1 × 106 cells/ml) were incubated with heat-killed C. gattii cells for 24 h. After centrifugation, culture supernatants were collected and cytokine levels were determined by enzyme-linked immunosorbent assay (ELISA). Cytokines in lung homogenates prepared from C. gattii-infected mice were also measured by ELISA. A MaxiSorp plate and a DuoSet ELISA kit (R&D Systems) or BD OptEIA ELISA sets (BD Bioscience) were used according to the manufacturers' instructions.
Vaccination.
BMDCs (1 × 106 cells/ml) were incubated in complete RPMI 1640 medium that contained mGM-CSF and heat-killed C. gattii cells (1 × 107 cells/ml; MOI = 10) for 24 h in petri dishes. After incubation, nonadherent cells were collected and washed twice with PBS. BMDCs that were pulsed with heat-killed C. gattii cells (5 × 105 cells/mouse) were injected via a tail vein, both at 14 days and 1 day before intratracheal infection with C. gattii R265.
Infection study.
C. gattii R265 was cultured in YPD at 30°C with shaking overnight. Cells were then washed and resuspended in PBS. A mouse was anesthetized with isoflurane, after which 50 μl of a fungal suspension (3 × 103 CFU) was intratracheally injected using a 24-gauge indwelling needle (TOP Corporation, Japan). Mice were euthanized by carbon dioxide inhalation, and their lungs were harvested to determine their weight and fungal burden. Lungs were homogenized using a stainless steel mesh in 5 ml of PBS. Homogenates were diluted and spread onto YPD plates. These plates were incubated at 30°C for 24 h, after which colonies were counted.
Histological analysis.
Histological analyses were as previously described (8). To prepare specimens, three isolated lungs were fixed in 10% formalin, dehydrated, and embedded in paraffin. Paraffin blocks were cut into 4-μm sections and stained with hematoxylin and eosin, alcian blue, or Elastica van Gieson stain for light microscopy analysis. The cross point intervals of alveoli were measured to assess alveolar spaces and the levels of alveolar destruction as described previously (25). Multinucleated giant cells (MGCs) were examined to assess macrophage recruitment into lungs after infection. The numbers of MGCs per unit area (square millimeters), the numbers of nuclei within each MGC, and the nuclear density in MGCs were determined as described previously (26).
Statistical analysis.
GraphPad Prism5 (GraphPad Software, Inc.) was used for statistical analyses. P values of <0.05 were considered significant.
RESULTS
BMDCs are activated by acapsular C. gattii.
To design an effective DC-based vaccine, we first evaluated the phagocytosis efficiency, costimulatory molecule expression, and cytokine production by BMDCs that were cultured with C. gattii cells. Because the capsular component of C. neoformans is known to suppress several immune responses by BMDCs (27, 28), we constructed an acapsular Δcap60 strain (see Fig. S1 in the supplemental material) and compared this with encapsulated strains (Fig. 1). In a phagocytosis assay, 80% of BMDCs engulfed several Δcap60 cells within 24 h, while only 10% of BMDCs engulfed one or two cells of the encapsulated strains (Fig. 1B; see also Fig. S2 in the supplemental material). Major histocompatibility complex class II (MHC-II) molecules (I-Ab) are essential for antigen presentation to CD4 T cells, and CD86 and CD40 are important costimulatory molecules for T cell stimulation. The percentages of BMDCs that expressed these molecules were significantly increased when these cells were stimulated with the acapsular Δcap60 strain (Fig. 1C; see also Fig. S3 in the supplemental material). Additionally, BMDCs that were stimulated with the Δcap60 strain produced more inflammatory cytokines, including IL-12p40 and TNF-α, than BMDCs that were stimulated with encapsulated strains (Fig. 1D). These data showed that BMDCs could be activated by the acapsular Δcap60 strain and suggested that BMDCs pulsed with this strain could be used for a DC-based vaccine to induce T cell responses against C. gattii.
FIG 1.

The C. gattii acapsular Δcap60 strain efficiently activates BMDCs. (A) Capsule formation was assessed using the conventional India ink method. Parental strain PNG18, the Δcap60 strain, and the revertant (CAP60C) were grown in YPD medium at 30°C overnight. BMDCs were incubated with heat-killed C. gattii cells for 24 h, and the phagocytosis rate (B), costimulatory molecule expression (C), and cytokine production (D) were evaluated by fluorescence microscopy, flow cytometry, and ELISA, respectively. For flow cytometry analysis, gates were set for CD11c+ CD11b+ cells. Representative data (means ± SDs) from 2 or 3 independent experiments are shown. *, P < 0.05 versus PNG18 by unpaired t test; #, P < 0.05 versus PNG18 by Mann-Whitney U test.
Transferring CAP60Δ/DCs ameliorates pulmonary infection with highly virulent C. gattii.
We tested for a protective effect after vaccination with Δcap60 strain-pulsed DCs (CAP60Δ/DCs) in a mouse model of pulmonary C. gattii infection. Transferring CAP60Δ/DCs significantly suppressed fungal growth in mouse lungs and improved mouse survival rates after pulmonary infection with the highly virulent strain R265 (Fig. 2). Although C. neoformans acapsular mutants have been successfully used as vaccines (29), vaccination using heat-killed R265 or heat-killed Δcap60 strain without DCs had no protective effect (see Fig. S4 in the supplemental material). In these experiments, we could not evaluate fungal burdens in the brain or spleen because of the limited number of fungal cells that had disseminated to the brain and spleen (data not shown). A previous study also showed that C. gattii R265 only minimally disseminated from the lungs to other organs in a murine pulmonary infection model (7).
FIG 2.

Transferring CAP60Δ/DCs ameliorates pulmonary infection with highly virulent C. gattii strain R265. CAP60Δ/DCs or unloaded DCs were injected into tail veins of mice at 14 days and 1 day prior to intratracheal infection with 3 × 103 CFU of strain R265. (A and B) Fungal burdens (n = 5) in the lungs were determined at day 14 (A) and day 3 (B). Pooled data from two independent experiments are plotted, and median values are shown by short horizontal lines. *, P < 0.05 by analysis of variance with Dunn's post hoc test (A) or P < 0.05 by Mann-Whitney U test (B). (C) Survival curves (n = 8) were compared by log rank test. Representative data from two independent experiments are shown. *, P < 0.01. Median survival times were 28 days in the case of nontransfer and 238 days in the case of CAP60Δ/DC transfer.
To determine how our CAP60Δ/DC vaccine suppressed fungal growth, we histologically assessed mouse lung sections after pulmonary infection with highly virulent C. gattii. Lungs were significantly lighter in those mice immunized with our CAP60Δ/DC vaccine than in unvaccinated mice at day 13 postinfection (see Fig. S5 in the supplemental material). In unvaccinated mice, the increased numbers of fungal cells destroyed alveolar structures by enlarging alveolar spaces. In contrast, fewer fungal cells, increased numbers of leukocytes, and reduced alveolar destruction were observed in the lungs of mice that had been immunized with Δcap60 strain-loaded DCs (Fig. 3; see also Fig. S5 and Table S2 in the supplemental material). The majority of these leukocytes were mononuclear cells, including lymphocytes and macrophages, which developed into multinucleated giant cells (MGCs) that engulfed the fungal cells in the lungs of mice immunized with our CAP60Δ/DC vaccine. Large MGCs with a number of nuclei and an eosinophilic cytoplasm that represented the accumulated macrophages and well-matured cytoplasmic organelles, respectively, were significantly increased in immunized mice (Fig. 3C). Although transferring antigen-unloaded DCs also induced leukocyte accumulation and the development of MGCs in the lungs to some extent, MGCs with lesser eosinophilic cytoplasm and engulfing fewer fungal cells were observed in those mice that received antigen-unloaded DCs compared with those in immunized mice (Fig. 3). Increased numbers of leukocytes and multinucleated cells were also observed in the lungs of vaccinated mice at day 7 posttransfer (see Fig. S5). Taken together, these data suggested that the development of MGCs that engulfed fungal cells had suppressed the fungal growth in the lungs of immunized mice.
FIG 3.

Multinucleated giant cells (MGCs) engulf fungal cells in the lungs of immunized mice. Lung sections obtained from three mice, at 13 days after infection with strain R265, were stained with hematoxylin-eosin (HE) or alcian blue (AB). C. gattii cells were stained with alcian blue. Each section was observed at magnifications of ×40 (A), ×200 (B), and ×1,000 (C).
Transferring CAP60Δ/DCs induces cytokine-producing lymphocytes in spleen and lungs.
The development of MGCs that engulf fungal cells in granulomas is associated with protection from cryptococcosis due to C. neoforomans, which is also associated with an increase in cytokine-producing T cells, such as Th1 and Th17 cells (30–33). Because DC-based vaccines reportedly augmented cytokine-producing effector T cells (17, 34), we assessed cytokine-producing T cells in the spleen and lungs after vaccination. IFN-γ+ and TNF-α+ CD4 T cells that responded to antigen restimulation were significantly increased in the spleens of vaccinated mice at day 14 after infection (Fig. 4). However, we could not detect any IL-17A-producing T cells (data not shown). Because the numbers of total splenocytes and proportions of CD4+ cells among splenocytes were equal between unvaccinated and vaccinated mice at day 14 postinfection, the increased proportion of these cells indicated an increase in the numbers of this cell population.
FIG 4.

Transferring CAP60Δ/DCs induces cytokine-producing CD4 T cells in spleen. Splenocytes were obtained from three mice at 14 days after infection and cultured with (+Ag) or without (−Ag) antigen and with or without heat-killed Δcap60 cells (MOI = 0.1) for 5 to 6 days. For flow cytometry analysis, gates were set for CD4+ cells. Representative flow cytometry profiles (A) and histograms for statistical analysis (B) are shown. Pooled data from two separate experiments were used to prepare histograms (means ± SEMs). *, P < 0.05 by analysis of variance with Dunnett's post hoc test.
Also, we utilized short-pulse stimulation using phorbol 12-myristate 13-acetate (PMA) and ionomycin to determine the numbers of cytokine-producing T cells in the lungs of vaccinated mice. IL-17A+ RORγt+ T cells and innate lymphoid cells (ILCs) were significantly increased in the lungs of vaccinated mice at day 1 postinfection (Fig. 5). CD4 T cells that produced IL-17A, IFN-γ, or TNF-α were also increased in the lungs of vaccinated mice at day 7 after transfer of CAP60Δ/DCs (see Fig. S6 in the supplemental material). At day 14 after infection, the amounts of IFN-γ, IL-17A, TNF-α, and CXCL1 (KC) were significantly increased in the lungs of immunized mice, and comparable amounts of IL-1β and smaller amounts of CCL2 (MCP-1) were detected (see Fig. S6). Because numerous lymphocytes and macrophages had already accumulated in the lungs of immunized mice, the amounts of MCP-1 may have decreased by day 14 after infection. These data suggested that increased T cell cytokine responses in vaccinated mice may have contributed to the development of MGCs that engulfed fungal cells after pulmonary infection with C. gattii.
FIG 5.
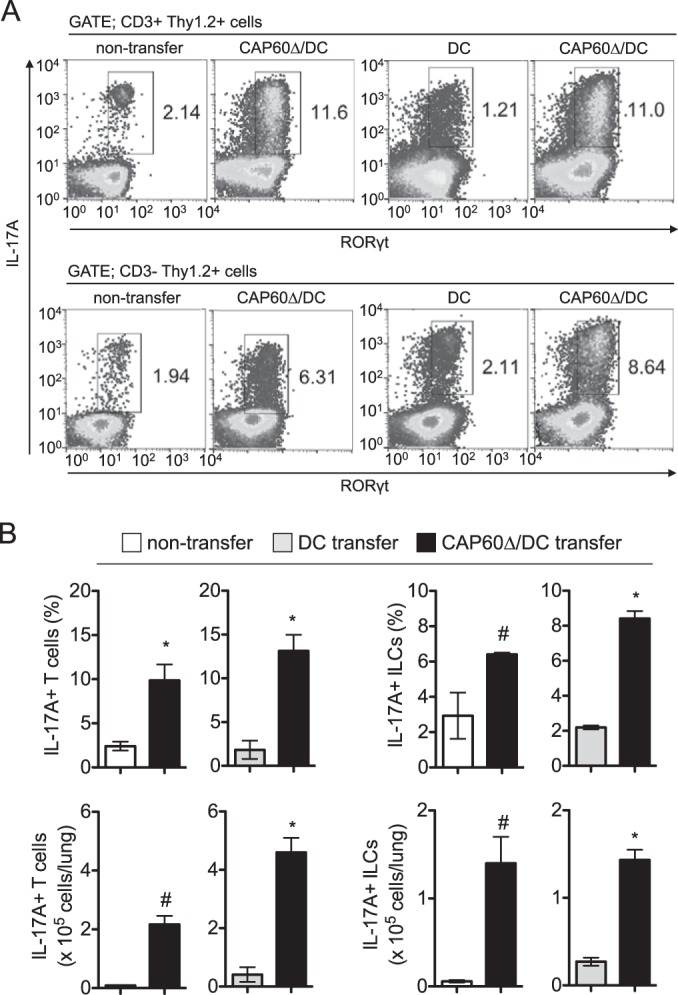
Transferring CAP60Δ/DCs induces IL-17A-producing cells in lungs. Lung leukocytes were obtained from three mice at 1 day after infection and were stimulated with PMA and ionomycin for 3 h. For flow cytometry analysis, gates were set for CD3+ Thy1.2+ cells or CD3− Thy1.2+ cells. Representative flow cytometry profiles (A) and histograms for statistical analysis (B) from two independent experiments are shown. Results in histograms are means ± SDs. *, P < 0.05 versus control by unpaired t test; #, P < 0.05 versus control by unpaired t test with Welch's correction.
IFN-γ may be an important mediator for vaccine-induced protection against pulmonary infection with highly virulent C. gattii.
Previous studies showed that IFN-γ-producing T cells had a protective role in pulmonary infection with C. neoformans (35, 36). However, surprisingly, the fungal burden was significantly reduced in the lungs of IFN-γ knockout (KO) mice after the pulmonary infection with C. gattii compared with that in C57BL/6J wild-type (WT) mice (Fig. 6). Thus, we investigated a role for IFN-γ in DC vaccine-induced protection by determining the reduction rate of the fungal burden in the lungs of vaccinated mice. The DC-based vaccine induced an 8,900-fold reduction and a 2,700-fold reduction in the fungal burdens in the lungs of WT mice and IFN-γ KO mice, respectively. The protective effect of our DC vaccine was significantly reduced in IFN-γ knockout mice. This suggested that IFN-γ may be an important mediator for DC vaccine-induced protection against pulmonary infection with highly virulent C. gattii.
FIG 6.
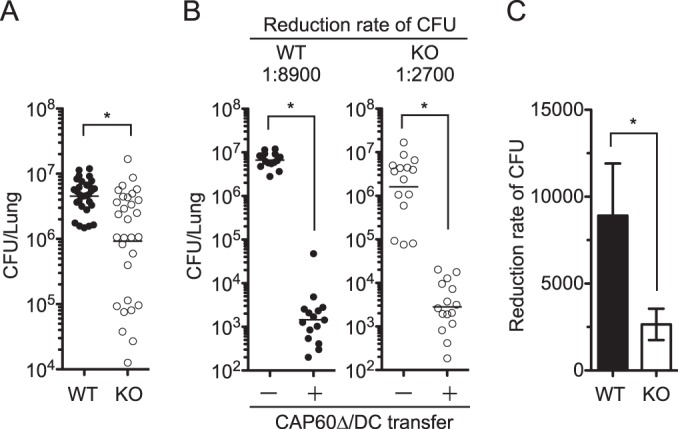
DC-based vaccine amelioration of the fungal burden is partially reduced in IFN-γ KO mice. (A) Fungal burdens in the lungs of C57BL/6J (WT) mice and IFN-γ KO mice were determined on days 9 to 14 postinfection. Data from six independent experiments were combined. (B and C) Vaccination and infection were performed as described in the legend to Fig. 2, and the fungal burden in lungs was determined on day 14 postinfection. Data from three independent experiments were combined. Vaccine effects between WT and KO mice were compared based on the reduced fungal burden rate (C). Panels A and B show dot plots with geometric means, and panel C shows means ± SEMs. *, P < 0.05 by Mann-Whitney U test.
DISCUSSION
In this study, we developed a new DC-based vaccine to investigate protective immunity against highly virulent C. gattii. Our results showed that an increase in cytokine-producing CD4 T cells and the development of MGCs that engulfed fungal cells were associated with protection against pulmonary infection with highly virulent C. gattii.
Recently, Chaturvedi et al. reported a protein-based vaccine against highly virulent C. gattii. They identified several protein antigen candidates using two-dimensional PAGE (2D-PAGE) and immunoblotting with sera from vaccinated mice after infection (37). They immunized mice three times at 4-week intervals without any adjuvant and then challenged mice intranasally with 1 × 104 CFU of C. gattii R265 at 10 days after the final immunization. Although the protein vaccine significantly ameliorated the fungal burden in lungs and improved the survival rate after this challenge, this protein-based vaccine induced no more than about a 5-fold reduction in the fungal burden in lungs on day 14 postinfection (37). In our study, Δcap60 strain-loaded DCs were transferred at day 14 and day 1 prior to infection, and this DC-based vaccine provided for an 8,900-fold reduction in the fungal burden in mouse lungs at day 14 postinfection (Fig. 6). This DC-based vaccine still effectively suppressed fungal growth even when mice were infected with C. gattii at 2 months after the final transfer of CAP60Δ/DCs (unpublished data). Interestingly, vaccination with DCs loaded with the Δcap60 strain (VGI) effectively suppressed the growth of R265 (VGII). Thus, our data suggested that our CAP60Δ/DC vaccine could induce protection against C. gattii in a cross-strain manner and that this DC-based vaccine was useful for studying protective immunity against highly virulent C. gattii.
Huston et al. showed that human DCs derived from CD14+ peripheral blood mononuclear cells (PBMCs) could engulf and kill C. gattii strain R265 but that DCs failed to mature in the presence of C. gattii (38). They also showed that TNF-α could restore DC maturation to induce T cell responses in the presence of C. gattii, which suggested the potential of DC-based therapies to improve the outcomes of patients with C. gattii infections. Another study showed that encapsulated C. neoformans strain B3501 (serotype D) could induce human DC maturation based on DC expression of CD40, CD86, and MHC-II (28). In our study, murine BMDCs did not mature in the presence of encapsulated C. gattii strains, because they did not efficiently engulf encapsulated C. gattii strains (Fig. 1). One study showed that murine BMDCs could mature after upregulated CD86 and MHC-II expression and cytokine production in the presence of encapsulated C. neoformans strain 1841 (serotype D) (27). Thus, both human DCs and murine BMDCs can become mature in the presence of encapsulated C. neoformans, but they did not mature in the presence of encapsulated C. gattii, as the phagocytosis rate for C. gattii cells seemed to be different between human DCs and murine BMDCs.
This failure of DC maturation may have negatively affected protection against pulmonary infection with C. gattii. In fact, transferring antigen-unloaded DCs prior to infection induced about a 10-fold reduction in the fungal burden in the lungs at day 14 postinfection (Fig. 2). Because BMDCs were cultured in the presence of GM-CSF, even antigen-unloaded DCs secreted a small amount (∼300 pg/ml) of IL-12p40 (Fig. 1). Thus, transferring unloaded DCs seemed to induce a small number of IFN-γ-producing CD4 T cells in the spleen (Fig. 4). However, the protective effect was limited in those mice that received unloaded DCs compared with the effect in those that received Δcap60 strain-pulsed DCs, as transferring unloaded DCs induced fewer antigen-specific T cells. These findings suggested that the complete activation of DC and T cell responses was required for protection against pulmonary infection with highly virulent C. gattii.
In a strict sense, vaccination using CAP60Δ/DCs only delayed the progression of infection, as the fungal burden in the lungs at day 14 after infection was not lower than the inoculated burden (Fig. 2). In fact, even though mice were immunized with CAP60Δ/DCs, most of these mice ultimately died (Fig. 2C). This implied that highly virulent C. gattii could cause a persistent infection or could change to a latent state during a protective immune response. Persistent infection with C. neoformans was shown in a previous report, as C. neoformans could be detected in the lungs of immunocompetent rats at 18 months after an intratracheal infection, along with reduced nitric oxide synthase in pulmonary granulomas that harbored this pathogen (39). C. gattii may also cause a persistent infection by similar mechanisms. To overcome persistent C. gattii infection, the DC-based vaccine used in this study needs to be refined. Previous studies showed that treating antigen-loaded DCs with rapamycin, an mTOR inhibitor, enhanced T cell responses and augmented the vaccine's efficacy against tuberculosis or tumors. Because rapamycin can enhance DC autophagy, which can enhance antigen processing and presentation and also improve DC life span (34, 40), it may be useful to augment the protective effect of our DC-based vaccine against C. gattii infection.
Previous studies showed that T cells, particularly IFN-γ-producing T cells, were essential for protection against C. neoformans infection in mice (35, 36). IFN-γ production induced by C. neoformans infection enhanced the migration and killing capacity of macrophages (32, 41). Although transferring CAP60Δ/DCs strongly induced an IFN-γ response that was partially required for protection against C. gattii, this DC-based vaccine was still effective for inhibiting fungal growth in IFN-γ KO mice (Fig. 6). This suggested that both IFN-γ-dependent and IFN-γ-independent responses were involved in vaccine-induced protection against pulmonary infection with C. gattii.
The role of IL-17A in cryptococcosis is controversial. Several reports showed that IL-17A-producing CD4 T cells were increased in the lungs after pulmonary infection with C. neoformans (33, 42). Two reports indicated that IL-17A was dispensable for protection against pulmonary infection with C. neoformans (42, 43), while another report showed that IL-17A enhanced host defenses against pulmonary C. neoformans infection (33). It has also been shown that IL-17A was required for leukocyte accumulation, including numerous granulocytes, at the early phase of infection and for the development of MGCs that engulfed fungal cells at the late phase of C. neoformans infection. Thus, fungal clearance was impaired in IL-17A KO mice (33).
In our study, IL-17A+ T cells and IL-17A+ innate lymphoid cells (ILCs) increased in the lungs of immunized mice at day 1 postinfection (Fig. 5), and the fungal growth in mouse lungs was significantly suppressed at day 3 postinfection (Fig. 2). Additionally, the amount of IL-17A was also increased in the lungs of immunized mice at day 14 after infection (see Fig. S6 in the supplemental material). In agreement with previous findings that an IL-17A response induced neutrophil recruitment, the number of neutrophils increased in the lungs of immunized mice (see Fig. S5). Collectively, these results implied that IL-17A might contribute to vaccine-induced protection against pulmonary infection with highly virulent C. gattii. Further studies will be needed to determine the main factors of DC-based vaccine-induced immunity for protection against pulmonary infection with highly virulent C. gattii.
In summary, we demonstrated that transferring CAP60Δ/DCs strongly induced cytokine-producing CD4 T cells and MGCs that engulfed fungal cells and that this was associated with protection against pulmonary infection with highly virulent C. gattii. We propose that this DC-based vaccine is useful for (i) determining which antigens can induce cytokine-producing T cells and (ii) assessing those memory T cell responses that contribute to protection against highly virulent C. gattii. This may lead to the development of new means to control lethal C. gattii infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Health Science Research Grants for Research on Emerging and Re-emerging Infectious Diseases (H25-Shinkou-Shitei-001, H25-Shinkou-Shitei-002, H25-shinkou-Wakate-005, H25-Shinkou-Ippan-006, and H26-Shinkoujitsuyouka-Ippan-010) from the Ministry of Health, Labor and Welfare of Japan, by a grant from the Strategic Research Foundation Grant-aided Project for Private Schools at Heisei 23rd, KAKENHI (26860774), from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by a grant from the Life Science Foundation of Japan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02827-14.
REFERENCES
- 1.Chen S, Sorrell T, Nimmo G, Speed B, Currie B, Ellis D, Marriott D, Pfeiffer T, Parr D, Byth K. 2000. Epidemiology and host- and variety-dependent characteristics of infection due to Cryptococcus neoformans in Australia and New Zealand. Australasian Cryptococcal Study Group. Clin Infect Dis 31:499–508. doi: 10.1086/313992. [DOI] [PubMed] [Google Scholar]
- 2.Galanis E, MacDougall L, Kidd S, Morshed M, British Columbia Cryptococcus gattii Working Group . 2010. Epidemiology of Cryptococcus gattii, British Columbia, Canada, 1999–2007. Emerging Infect Dis 16:251–257. doi: 10.3201/eid1602.090900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith RM, Mba-Jonas A, Tourdjman M, Schimek T, DeBess E, Marsden-Haug N, Harris JR. 2014. Treatment and outcomes among patients with Cryptococcus gattii infections in the United States Pacific Northwest. PLoS One 9:e88875. doi: 10.1371/journal.pone.0088875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.BCCDC. 2011. Environmental pathogens, Cryptococcus gattii, p 112–113. British Columbia Annual Summary of Reportable Diseases 2011. BC Centre for Disease Control, Vancouver, BC, Canada: http://www.bccdc.ca/util/about/annreport/default.htm. [Google Scholar]
- 5.CDC. 2010. Emergence of Cryptococcus gattii, Pacific Northwest, 2004–2010. MMWR Morb Mortal Wkly Rep 59:865–868. [PubMed] [Google Scholar]
- 6.Fraser JA, Giles SS, Wenink EC, Geunes-Boyer SG, Wright JR, Diezmann S, Allen A, Stajich JE, Dietrich FS, Perfect JR, Heitman J. 2005. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437:1360–1364. doi: 10.1038/nature04220. [DOI] [PubMed] [Google Scholar]
- 7.Ngamskulrungroj P, Chang Y, Sionov E, Kwon-Chung KJ. 2012. The primary target organ of Cryptococcus gattii is different from that of Cryptococcus neoformans in a murine model. mBio 3(3):e00103-12. doi: 10.1128/mBio.00103-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okubo Y, Wakayama M, Ohno H, Yamamoto S, Tochigi N, Tanabe K, Kaneko Y, Yamagoe S, Umeyama T, Shinozaki M, Nemoto T, Nakayama H, Sasai D, Ishiwatari T, Shimodaira K, Yamamoto Y, Kamei K, Miyazaki Y, Shibuya K. 2013. Histopathological study of murine pulmonary cryptococcosis induced by Cryptococcus gattii and Cryptococcus neoformans. Jpn J Infect Dis 66:216–221. doi: 10.7883/yoken.66.216. [DOI] [PubMed] [Google Scholar]
- 9.Cheng P-Y, Sham A, Kronstad JW. 2009. Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect Immun 77:4284–4294. doi: 10.1128/IAI.00628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Einsiedel L, Gordon DL, Dyer JR. 2004. Paradoxical inflammatory reaction during treatment of Cryptococcus neoformans var. gattii meningitis in an HIV-seronegative woman. Clin Infect Dis 39:e78–e82. doi: 10.1086/424746. [DOI] [PubMed] [Google Scholar]
- 11.Brouwer AE, Siddiqui AA, Kester MI, Sigaloff KCE, Rajanuwong A, Wannapasni S, Chierakul W, Harrison TS. 2007. Immune dysfunction in HIV-seronegative, Cryptococcus gattii meningitis. J Infect 54:e165–e168. doi: 10.1016/j.jinf.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Springer DJ, Ren P, Raina R, Dong Y, Behr MJ, McEwen BF, Bowser SS, Samsonoff WA, Chaturvedi S, Chaturvedi V. 2010. Extracellular fibrils of pathogenic yeast Cryptococcus gattii are important for ecological niche, murine virulence and human neutrophil interactions. PLoS One 5:e10978. doi: 10.1371/journal.pone.0010978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yauch LE, Lam JS, Levitz SM. 2006. Direct inhibition of T-cell responses by the Cryptococcus capsular polysaccharide glucuronoxylomannan. PLoS Pathog 2:e120. doi: 10.1371/journal.ppat.0020120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinman RM, Witmer MD. 1978. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci U S A 75:5132–5136. doi: 10.1073/pnas.75.10.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palucka K, Banchereau J. 2013. Dendritic-cell-based therapeutic cancer vaccines. Immunity 39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.García F, Climent N, Assoumou L, Gil C, González N, Alcamí J, León A, Romeu J, Dalmau J, Martínez-Picado J, Lifson J, Autran B, Costagliola D, Clotet B, Gatell JM, Plana M, Gallart T, DCV2/MANON07- AIDS Vaccine Research Objective Study Group . 2011. A therapeutic dendritic cell-based vaccine for HIV-1 infection. J Infect Dis 203:473–478. doi: 10.1093/infdis/jiq077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.d'Ostiani CF, Del Sero G, Bacci A, Montagnoli C, Spreca A, Mencacci A, Ricciardi-Castagnoli P, Romani L. 2000. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. Implications for initiation of T helper cell immunity in vitro and in vivo. J Exp Med 191:1661–1674. doi: 10.1084/jem.191.10.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Nkwanyuo G, Pitzurra L, Velardi A, Romani L. 2003. A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood 102:3807–3814. doi: 10.1182/blood-2003-03-0748. [DOI] [PubMed] [Google Scholar]
- 19.Roy RM, Klein BS. 2012. Dendritic cells in antifungal immunity and vaccine design. Cell Host Microbe 11:436–446. doi: 10.1016/j.chom.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kidd SE, Hagen F, Tscharke RL, Huynh M, Bartlett KH, Fyfe M, Macdougall L, Boekhout T, Kwon-Chung KJ, Meyer W. 2004. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc Natl Acad Sci U S A 101:17258–17263. doi: 10.1073/pnas.0402981101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell LT, Currie BJ, Krockenberger M, Malik R, Meyer W, Heitman J, Carter D. 2005. Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii. Eukaryot Cell 4:1403–1409. doi: 10.1128/EC.4.8.1403-1409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fraser JA, Subaran RL, Nichols CB, Heitman J. 2003. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2:1036–1045. doi: 10.1128/EC.2.5.1036-1045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol 175:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahn Y-S, Hicks JK, Giles SS, Cox GM, Heitman J. 2004. Adenylyl cyclase-associated protein AcaI regulates virulence and differentiation of Cryptococcus neoformans via the cyclic AMP-protein kinase A cascade. Eukaryot Cell 3:1476–1491. doi: 10.1128/EC.3.6.1476-1491.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon DM, Tsai LW, Ingenito EP, Starcher BC, Mariani TJ. 2010. PPARgamma deficiency results in reduced lung elastic recoil and abnormalities in airspace distribution. Respir Res 11:69. doi: 10.1186/1465-9921-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvalho NB, Oliveira FS, Durães FV, de Almeida LA, Flórido M, Prata LO, Caliari MV, Appelberg R, Oliveira SC. 2011. Toll-like receptor 9 is required for full host resistance to Mycobacterium avium infection but plays no role in induction of Th1 responses. Infect Immun 79:1638–1646. doi: 10.1128/IAI.01030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siegemund S, Alber G. 2008. Cryptococcus neoformans activates bone marrow-derived conventional dendritic cells rather than plasmacytoid dendritic cells and down-regulates macrophages. FEMS Immunol Med Microbiol 52:417–427. doi: 10.1111/j.1574-695X.2008.00391.x. [DOI] [PubMed] [Google Scholar]
- 28.Vecchiarelli A, Pietrella D, Lupo P, Bistoni F, McFadden DC, Casadevall A. 2003. The polysaccharide capsule of Cryptococcus neoformans interferes with human dendritic cell maturation and activation. J Leukoc Biol 74:370–378. doi: 10.1189/jlb.1002476. [DOI] [PubMed] [Google Scholar]
- 29.Fromtling RA, Kaplan AM, Shadomy HJ. 1983. Immunization of mice with stable, acapsular, yeast-like mutants of Cryptococcus neoformans. Sabouraudia 21:113–119. doi: 10.1080/00362178385380181. [DOI] [PubMed] [Google Scholar]
- 30.Hill JO. 1992. CD4+ T cells cause multinucleated giant cells to form around Cryptococcus neoformans and confine the yeast within the primary site of infection in the respiratory tract. J Exp Med 175:1685–1695. doi: 10.1084/jem.175.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wozniak KL, Ravi S, Macias S, Young ML, Olszewski MA, Steele C, Wormley FL. 2009. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS One 4:e6854. doi: 10.1371/journal.pone.0006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawakami K, Qureshi MH, Zhang T, Koguchi Y, Shibuya K, Naoe S, Saito A. 1999. Interferon-gamma (IFN-gamma)-dependent protection and synthesis of chemoattractants for mononuclear leucocytes caused by IL-12 in the lungs of mice infected with Cryptococcus neoformans. Clin Exp Immunol 117:113–122. doi: 10.1046/j.1365-2249.1999.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ. 2014. Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production. Infect Immun 82:937–948. doi: 10.1128/IAI.01477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Eissa NT. 2009. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 15:267–276. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 35.Huffnagle GB, Lipscomb MF, Lovchik JA, Hoag KA, Street NE. 1994. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J Leukoc Biol 55:35–42. [DOI] [PubMed] [Google Scholar]
- 36.Kawakami K. 2004. Regulation by innate immune T lymphocytes in the host defense against pulmonary infection with Cryptococcus neoformans. Jpn J Infect Dis 57:137–145. [PubMed] [Google Scholar]
- 37.Chaturvedi AK, Hameed RS, Wozniak KL, Hole CR, Leopold Wager CM, Weintraub ST, Lopez-Ribot JL, Wormley FL. 2014. Vaccine-mediated immune responses to experimental pulmonary Cryptococcus gattii infection in mice. PLoS One 9:e104316. doi: 10.1371/journal.pone.0104316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huston SM, Li SS, Stack D, Timm-McCann M, Jones GJ, Islam A, Berenger BM, Xiang RF, Colarusso P, Mody CH. 2013. Cryptococcus gattii is killed by dendritic cells, but evades adaptive immunity by failing to induce dendritic cell maturation. J Immunol 191:249–261. doi: 10.4049/jimmunol.1202707. [DOI] [PubMed] [Google Scholar]
- 39.Goldman DL, Lee SC, Mednick AJ, Montella L, Casadevall A. 2000. Persistent Cryptococcus neoformans pulmonary infection in the rat is associated with intracellular parasitism, decreased inducible nitric oxide synthase expression, and altered antibody responsiveness to cryptococcal polysaccharide. Infect Immun 68:832–838. doi: 10.1128/IAI.68.2.832-838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, Pearce EJ. 2012. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol 189:2151–2158. doi: 10.4049/jimmunol.1103741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flesch IE, Schwamberger G, Kaufmann SH. 1989. Fungicidal activity of IFN-gamma-activated macrophages. Extracellular killing of Cryptococcus neoformans. J Immunol 142:3219–3224. [PubMed] [Google Scholar]
- 42.Yamamoto H, Nakamura Y, Sato K, Takahashi Y, Nomura T, Miyasaka T, Ishii K, Hara H, Yamamoto N, Kanno E, Iwakura Y, Kawakami K. 2014. Defect of CARD9 leads to impaired accumulation of gamma interferon-producing memory phenotype T cells in lungs and increased susceptibility to pulmonary infection with Cryptococcus neoformans. Infect Immun 82:1606–1615. doi: 10.1128/IAI.01089-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardison SE, Wozniak KL, Kolls JK, Wormley FL. 2010. Interleukin-17 is not required for classical macrophage activation in a pulmonary mouse model of Cryptococcus neoformans infection. Infect Immun 78:5341–5351. doi: 10.1128/IAI.00845-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.