

Abstract
Periapical disease, an inflammatory disease mainly caused by dental caries, is one of the most prevalent infectious diseases of humans, affecting both children and adults. The infection travels through the root, leading to inflammation, bone destruction, and severe pain for the patient. Therefore, the development of a new class of anti-periapical disease therapies is necessary and critical for treatment and prevention. A small molecule, odanacatib (ODN), which is a cathepsin K (Ctsk) inhibitor, was investigated to determine its ability to treat this disease in a mouse model of periapical disease. While Ctsk was originally found in osteoclasts as an osteoclast-specific lysosomal protease, we were surprised to find that ODN can suppress the bacterium-induced immune response as well as bone destruction in the lesion area. X rays and microcomputed tomography (micro-CT) showed that ODN treatment had significant bone protection effects at different time points. Immunohistochemical and immunofluorescent staining show that ODN treatment dramatically decreased F4/80+ macrophages and CD3+ T cells in the lesion areas 42 days after infection. Consistent with these findings, quantitative real-time PCR (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA) analysis showed low levels of proinflammatory mRNAs (for tumor necrosis factor alpha, interleukin 6, and interleukin 23α) and corresponding cytokine expression in the ODN-treated disease group. The levels of mRNA for Toll-like receptors 4, 5, and 9 also largely decreased in the ODN-treated disease group. Our results demonstrated that ODN can inhibit endodontic disease development, bone erosion, and immune response. These results indicate that application of this small molecule offers a new opportunity to design effective therapies that could prevent periapical inflammation and revolutionize current treatment options.
INTRODUCTION
Periapical lesions result from microbial infection of the dental pulp tissue by members of the autogenous oral microflora, which induce inflammation in the pulp tissue (1). This inflammatory process, much like periodontitis, increases in magnitude in the apical region of the root canal system and subsequently in the periapical area, leading to periapical bone resorption as the infection spreads throughout the canal system toward the apical foramen and into the adjacent bone (2). Furthermore, periapical lesions that present with radiographic bone lesions are preceded by necrotic pulp tissues. Unlike periodontal disease inflammation (3, 4), endodontic lesions exhibit differences in the character of the immune response (5).
Despite many studies aimed at discovering ways to alleviate the effects of oral disease, there is still an urgent need to improve the health of millions with periapical disease who suffer from oral-bacterial-infection-induced periapical inflammation, oral bone erosion, and the potential loss of teeth. The discovery of the critical roles of cathepsin K (Ctsk) in osteoclastic bone resorption is the fruit of decades of investigation on bone biology and disease (6, 7). The Ctsk gene is the only gene for which an essential role in bone resorption has been clearly demonstrated in both mice and humans, and current research data show that Ctsk is also indispensable in the immune system (8). Recent studies demonstrated that cathepsins are required for expression of Toll-like receptor 9 in dendritic cells, which plays an essential role in innate recognition of microbial products and activation of defense responses (9). Our research led to the surprising and pivotal realization that Ctsk also has significant functions in the immune system (10, 11), so we termed the Ctsk gene an “osteoimmune gene.” The study of bone and the immune system, which is termed osteoimmunology, represents a new perspective which takes into account the multiple advances made in the study of biological events in bone and the immune system in recent years (12, 13).
The success of the novel treatment of inflammatory diseases like periodontitis and rheumatoid arthritis (RA) has impressively demonstrated that a clinical benefit can be gained from therapeutic intervention regarding specific proteins by using small molecules (14). Odanacatib (ODN) is a potent, orally active selective inhibitor of Ctsk being developed for the treatment of postmenopausal osteoporosis. Treatment with ODN decreases bone resorption by selectively inhibiting proteolysis of matrix protein by Ctsk without affecting other osteoclast activities or osteoclast viability (15). However, there is still no research on the application of Ctsk inhibitors to the treatment of oral inflammatory diseases, considering its dual functions in bone and immune system. In addition, based on current knowledge gained from recent studies, there are still questions as to what functions Ctsk has in activating immune cells, such as macrophages and dendritic cells (antigen-presenting cells [APCs]), and what leads to T cell activation and autoimmunity, causing the subsequent production of inflammatory cytokines. Therefore, based on the cited research and our previous results, the current investigation was conducted with respect to the likelihood that Ctsk has a significant role in the activation of macrophages and Toll-like receptor (TLR)-mediated innate and adaptive immune responses in the pathogenesis of periapical lesions by using a specific Ctsk small-molecule inhibitor, ODN.
MATERIALS AND METHODS
Animals.
One hundred five 7- to 8-week-old male wild-type (WT) BALB/cJ mice, purchased from the Jackson Laboratory, were used for introducing a periapical mouse model. Mice were divided into 4 groups of seven mice each at each time point (7, 21, and 42 days): (i) normal group with Ctsk inhibitor (no bacterial infection) (3.606 mg/kg/week); (ii) normal group without Ctsk inhibitor (no bacterial infection); (iii) bacterial infection group treated with Ctsk inhibitor (3.606 mg/kg/week or 0.7212 mg/kg/week); (iv) bacterial infection group without Ctsk inhibitor. The animals were maintained in the University of Alabama at Birmingham animal facility and were given distilled water and allowed to feed freely. All experimental protocols were approved by the NIH and the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and completed within 16 weeks after birth. Approval for the animal protocol related to this study (animal protocol number 131209236) was renewed by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee (IACUC) on 27 August 2013.
Administration of cathepsin K (Ctsk) inhibitor.
Male mice in the normal and periapical disease groups were treated with ODN and compared with the untreated normal and disease groups. Treated mice were administered orally 3.606 mg/kg/week or 0.7212 mg/kg/week (5-fold-lower dose) of pharmacological-grade ODN (Selleckchem; USA) in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) from 1 week before the establishment of the disease model to the end of sample harvest. The proper ODN dose for mice was chosen to match the conventional treatment of humans (16). The control mice were administered DMSO orally.
Pulp exposure and bacterial infection.
Exposure of the left and right first molar pulp was performed as described previously (10, 17). In brief, mice were anesthetized with peritoneal injection of 62.5 mg/kg ketamine and 12.5 mg/kg xylazine according to body weight. The dental pulps of the mandibular first molars were exposed with a 1/4 round carbide bur powered by a variable-speed electric rotary hand piece (Osada Electric, Los Angeles, CA) under a surgical microscope (model MC-M92; Seiler, St. Louis, MO). After the roof of the pulp chamber was removed, the size of the exposure was approximately 0.5 to 1.0 mm in diameter. We used stainless steel number 8 hand files (Dentsply/Maillefer, Johnson City, TN) and stainless steel number 15 rotary files (Dentsply/Maillefer, Johnson City, TN) to establish canal patency. Eradication of infecting microorganisms was not completed in this therapy.
Bacterial culture and infection procedure protocols were conducted as described previously (10, 17). In brief, exposed pulps were infected with a mixture of four common human endodontic pathogens, including Prevotella intermedia (ATCC 25611; American Type Culture Collection [ATCC], Manassas, VA), Fusobacterium nucleatum (ATCC 25586), Peptostreptococcus micros (ATCC 33270), and Streptococcus intermedius (ATCC 27335). All four species of bacteria were cultured under strict anaerobic conditions (80% N2, 10% H2, and 10% CO2), inoculated into broth, and cultivated over 7 days. Microbes were harvested, resuspended, and evaluated for cell concentration of each species via optical density reading. The wavelength used for cell number counting was 600 nm (one OD unit equals 6.67 × 108 particles); the cell density per ml can be obtained by calculation. Then the cell density of each species was adjusted to 1010 per ml for bacterial inoculation. The four organisms were then mixed for a total of 1010 cells of each bacterial species/ml phosphate-buffered saline (PBS) in 3% methylcellulose. Ten microliters of the polymicrobial solution was placed inside the access opening of each molar and carried to the periapical tissues using a number 8 endodontic file. After exposed pulps were infected with the mixture of four common human endodontic pathogens, access openings were left open to the oral environment for 24 h. All exposure sites were then filled with self-curing composite temporary filling.
Harvest and preparation of samples.
Animals were sacrificed by CO2 inhalation on days 7, 21, and 42 after the initial infection. The mandibles were removed and hemisected. After removal of soft tissue, the jaw samples from the left side were fixed in 4% formaldehyde for 24 h and then stored in 70% ethanol prior to X-ray exposure, micro-computed tomography (micro-CT), and measurement of bone loss. The jaw samples from the right side were fixed in 4% paraformaldehyde and prepared for histological analysis. The right sections were prepared for histology analysis according to a standard protocol, with modification for samples that were prepared for paraffin sectioning. In brief, samples intended for paraffin sectioning were fixed in 4% formaldehyde for 24 h, washed with PBS, and decalcified in 10% EDTA for 25 days (EDTA was replenished each day). For the samples used for RNA and protein extraction, the periapical bone tissues surrounding the mesial and distal root were extracted from the mandible together with surrounding bone in a block specimen using a surgical microscope for total RNA and cytokine enzyme-linked immunosorbent assays (ELISA). For the RNA extraction, periapical tissues were rinsed in chilled PBS, weighed (3 to 5 mg/tissue), immediately put into RNAlater ICE (Invitrogen, USA) overnight at 4°C, and then stored at −80°C until used for RNA extraction. For ELISA, the periapical tissues were rinsed in chilled PBS, weighed (3 to 5 mg/tissue), and immediately frozen at −80°C until used for protein extraction.
Micro-CT analysis.
Micro-computed tomography (micro-CT) scans were evaluated for bone loss as described previously, with modifications (10, 17). Briefly, the two-dimensional picture that showed the largest target root canal was selected as the baseline image. For each sample, an approximate total of 101 microtomographic slices with an increment of 12 μm were acquired in front of and behind the baseline image. From the three-dimensional stack of micro-CT images, a “pivot” section was the periapical area, including the root area and furcation area, where the inflammation may cause the bone resorption around the apex of the tooth. The segmentation parameter is 0.7/1/210.
Histological analysis.
Samples were fixed with 4% paraformaldehyde for 24 h, decalcified by 10% EDTA for 25 days with a new solution each day, dehydrated by 50%, 70%, 95%, and 100% alcohol for 2 h each, washed in paraffin acetone for 30 min in a 60°C incubator, washed in chloroform for 30 min in a 60°C incubator, infiltrated with paraffin in a 60°C incubator three times for 1 h each time, and embedded in paraffin. To detect bone-resorbing osteoclasts, tartrate-resistant acid phosphatase (TRAP) staining was performed. Tissue sections were deparaffinized and hydrated through xylenes and graded alcohol series, preincubated with 50 mM sodium acetate and 40 mM potassium sodium tartrate buffer for 20 min, and incubated with TRAP substrate solution.
Immunofluorescence analysis.
We performed immunofluorescence analysis as we described previously (18), with the exceptions that we used rat polyclonal anti-CD3 (Abcam, Cambridge, MA) as the primary antibodies and observations were performed by epifluorescence in a Zeiss Axioplan microscope (Carl Zeiss Microscopy, LLC, USA). Nuclei were visualized with 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich, USA). The experiments were carried out in triplicate on three independent occasions.
Immunohistochemistry analysis.
We performed immunohistochemistry using corresponding sections of mandibular tissue from the normal group (with or without inhibitor) and the disease group (with or without inhibitor); then the slides were analyzed by immunohistochemistry for expression and localization of proteins of the macrophage marker F4/80 (rat monoclonal antibody, 1:200) (eBioscience, San Diego, CA), TLR4 (goat polyclonal antibody, 1:200), TLR5 (rabbit polyclonal antibody, 1:200) (Santa Cruz, USA), and TLR9 (rabbit monoclonal antibody, 1:500) (Sigma-Aldrich, USA). Vector stain ABC kit anti-rat, -goat, and -rabbit IgG peroxidase polymer detection systems along with a 3,3′-diaminobenzidine kit (Vector Laboratories, Burlingame, CA) as a substrate were used for the peroxidase-mediated reaction. The experiments were carried out in triplicate on three independent occasions.
RNA extraction and quantitative real-time PCR (qRT-PCR).
For RNA extraction, the prepared samples were transferred to the tube prefilled with beads (Nextadvance Company, USA) and homogenized using a Bullet blender (Nextadvance Company, USA). The RNA extraction was performed with the standard procedure using TRIzol reagent (Invitrogen, USA). The extracted RNA was used for reverse transcription using a Vilo master kit (Invitrogen). Real-time quantitative PCR (qPCR) was performed as described previously (10, 17) using primers purchased from Invitrogen as listed (see Table S1 in the supplemental material). Briefly, cDNA fragments were amplified with Sybr green fast advanced master mix (Applied Biosystems, Foster City, CA) and detected by a Step-One real-time PCR system (Applied Biosystems). The mRNA expression level of the β-actin housekeeping gene was used as an endogenous control and enabled calculation of specific mRNA expression levels as a ratio to the β-actin level. Experiments were repeated at least three times.
Protein extraction and cytokines for enzyme-linked immunosorbent assay (ELISA).
For protein extraction, the frozen periapical tissue samples were transferred to a tube prefilled with beads (Nextadvance Company, USA) and homogenized using a Bullet blender (Nextadvance Company, USA). The tissue fragments were dispersed in 300 μl of lysis buffer. The mixture was centrifuged at 12,000 rpm for 10 min, and the supernatant was collected and stored at −80°C until assay. ELISA was used as described previously (19, 20) to evaluate the effect of inhibition of Ctsk on the levels of tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and IL-23α in inflammatory periapical tissues extracted from the normal group (with or without inhibitor) and the disease group (with or without inhibitor). Briefly, assays for cytokines in extracts employed commercially available ELISA kits that are obtained from the following sources: TNF-α, eBioscience; IL-6, eBioscience; and IL-23α, BioLegend. All assays were conducted in accordance with the manufacturer's instructions. Results were expressed as pg cytokine/ml tissue.
Statistical analysis and data quantification analysis.
Experimental data are reported as means ± standard deviations (SD) for triplicate independent samples. All experiments were performed in triplicate on three independent occasions. The figures show representative data. Data were analyzed with the two-tailed Student t test. The Mann-Whitney U test was used for the nonparametric test. P values of <0.05 and U values of >1.96 were considered significant. Data quantification analyses were performed using the NIH Image J program as described previously (10, 17).
RESULTS
Inhibition of Ctsk exhibits bone-protective effects of the endodontic lesion area at different time points.
Our previous study on adeno-associated virus (AAV)-mediated Ctsk inhibition in our periapical lesion mouse model revealed that Ctsk is important for bone resorption and inflammation in periapical lesions but is not useful for exploring the possible mechanisms of these events (10). Thus, it is important to determine the changes in the trends of the cells (number change) and the inflammation (more severe or not) in the periapical lesion area by inhibition of Ctsk and its possible mechanism at different time points. To test the possible role during the progress of periapical lesions mediated by Ctsk, we applied the periapical mouse model established by our previous study (10, 17). Samples from the normal group with or without inhibitor (only images of day 42 are shown in Fig. 1) and from the bacterium-infected group with or without inhibitor (3.606 mg/kg/week) were analyzed by X ray and micro-CT at days 7, 21, and 42 after infection (Fig. 1). X-ray imaging of the first molar tooth of the normal group with and without inhibitor at different time points showed that there was no area with reduced bone density (see Fig. S1 in the supplemental material). The X ray and the micro-CT test showed a low-bone-density area at the furcation and the periapical area in the bacterial-infection group without inhibitor at 7, 21, and 42 days (Fig. 1B to D). As the time increased, the low density of bone regions in the disease group without inhibitor widened (red arrows). In contrast, there was no obvious reduced bone density area in the infected disease group with inhibitor (3.606 mg/kg/week) at different time points (Fig. 1B to D). To further quantify the bone resorption in the periapical lesion area, we used quantification of micro-CT to determine the differences between groups (Fig. 1E). The quantification analysis showed a significant difference in the disease groups with and without inhibitor at 7, 21, and 42 days (Fig. 1E). The Ctsk inhibitor (3.606 mg/kg/week or 0.7212 mg/kg/week [5-fold lower]) seemed to have a dose-dependent bone protective effect, as shown by micro-CT analysis (see Fig. S2 in the supplemental material). We also conducted histological analysis by hematoxylin-and-eosin (H&E) staining for the periapical lesion area for the different groups (Fig. 2). The H&E stain showed that the remaining bone area in mesial, distal, and furcation areas in the disease group with inhibitor (3.606 mg/kg/week) was much greater than that in the disease group without inhibitor at 7, 21, and 42 days (Fig. 2A and B). We also found that the distribution of mononuclear inflammatory cells was evident in the disease group without inhibitor at different time points (red arrows in Fig. 2A). Although the bone area decreased in the disease group without inhibitor, there was no significant change at the first 21 days. However, the remaining bone area in the disease group without inhibitor was still significantly less than that in the inhibitor-treated group at 42 days (Fig. 2C). There was no significant change in the normal group with or without inhibitor (see Fig. S3A and C in the supplemental material). Above all, the results showed that Ctsk is critical for bone resorption during the progression of periapical lesions.
FIG 1.
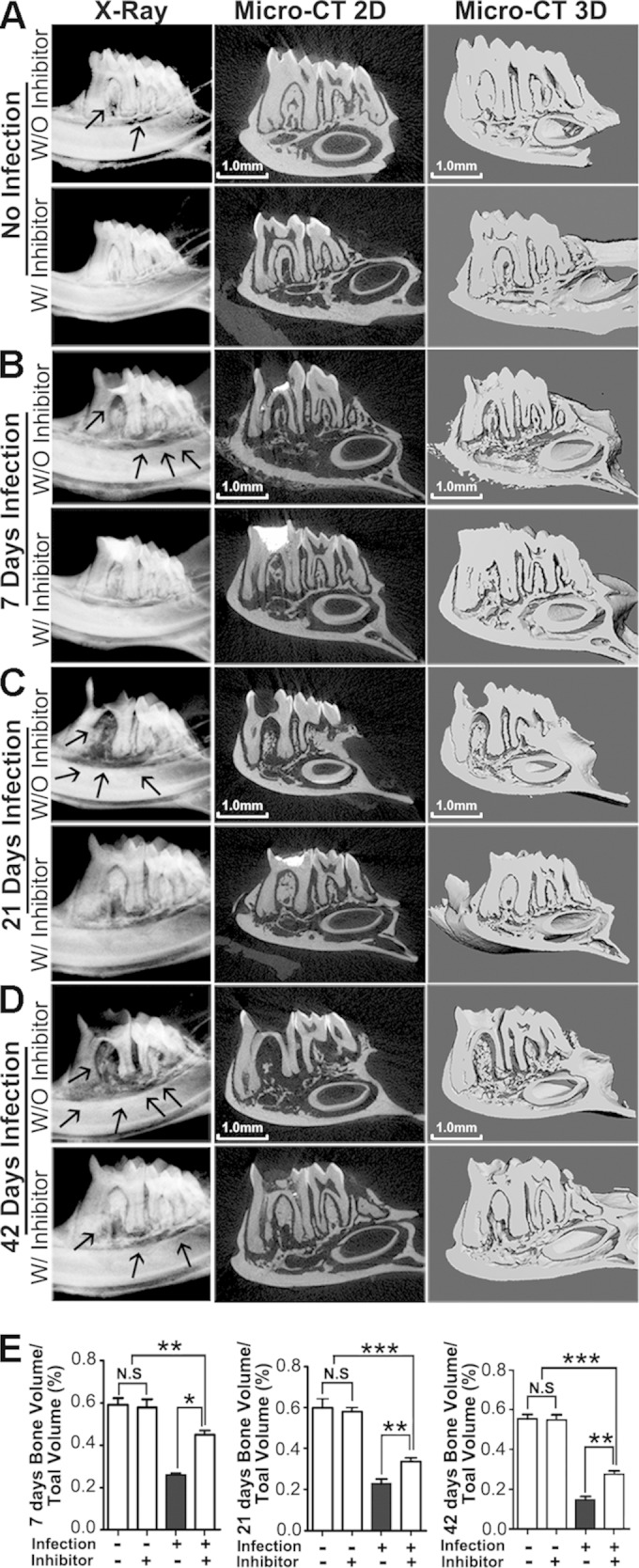
Inhibition of Ctsk by specific inhibitor reduced infection-stimulated endodontic bone resorption by micro-computed tomography (micro-CT) analysis. (A) X-ray and micro-CT analysis of endodontic lesion areas of the normal groups over the natural course of infection with and without inhibitor (3.606 mg/kg/week) at 42 days. (B to D) X-ray and micro-CT analysis of end odontic lesion areas of the bacterium-infected groups with and without inhibitor (3.606 mg/kg/week) at 7, 21, and 42 days. There was an obvious reduced bone density area compared to the inhibitor-treated disease group at different time points (arrows). The X-ray and micro-CT analysis showed increased low-bone-density areas in the furcation and periapical areas at 42 days compared with 7 days and 21 days. (E) Quantification of bone volume/tissue volume measured for periapical lesion areas in different groups. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Each experiment was repeated three times (n = 3). N.S, no significance. The left columns of panels A to D are X-ray images, the middle columns are typical slice two-dimensional micro-CT images, and the right columns are three-dimensional reconstituted images.
FIG 2.

Inhibition of Ctsk by specific inhibitor showed bone protection effects at different time points by histology analysis. (A) H&E staining of the periapical mesial and distal root sections and furcation area sections from the bacterium-infected groups with and without inhibitor (3.606 mg/kg/week) at 7, 21, and 42 days. Red arrows indicate the inflammatory monocyte infiltration. The dotted red outline indicates the remaining bone in the lesion area. (B) Quantification of remaining bone area in these three areas at 7, 21, and 42 days. (C) Time course analysis of the bone resorption in the bacterium-infected groups with and without inhibitor (3.606 mg/kg/week). *, P < 0.05; **, P < 0.01; ***, P < 0.001. N.S, no significance. Each experiment was repeated three times (n = 3).
Inhibition of Ctsk exhibits decreased osteoclast numbers at endodontic and furcation areas at different time points.
By applying TRAP staining, we found that the number of osteoclasts decreased in the disease group with inhibitor (3.606 mg/kg/week) compared to the disease group without inhibitor at different time points (Fig. 3A and B). We also noticed that the number of osteoclasts increased significantly in the disease group without inhibitor compared to the number in the inhibitor group at 7 days (Fig. 3C), and the osteoclast numbers remained high during the progress of periapical lesion formation. There was no significant difference of osteoclast number in the normal group with or without inhibitors at 42 days (see Fig. S3B and D in the supplemental material).
FIG 3.

Inhibition of Ctsk by the specific inhibitor showed reduced TRAP+ cells in periapical lesions at different time points by histology analysis. (A) TRAP staining of the periapical mesial and distal root sections and furcation area sections from the bacterium-infected group with and without inhibitor (3.606 mg/kg/week) at different time points (7, 21, and 42 days). (B) Quantification of TRAP+ cell numbers in these three areas at 7, 21, and 42 days. (C) Time course analysis of TRAP+ cell numbers in the bacterium-infected groups with and without inhibitor (3.606 mg/kg/week). The TRAP+ cell number increased in the bacterium-infected group without inhibitor at different time points, and the TRAP+ cell number in the infected group with inhibitor (3.606 mg/kg/week) was significantly lower than that in the infected group without inhibitor. *, P < 0.05; ***, P < 0.001. N.S, no significance. Each experiment was repeated three times (n = 3).
Ctsk may have effects on immune response and have functions in immune cells at the periapical lesion area.
The macrophage is important for the host immune response during the periapical bacterial infection (2). In order to determine whether inhibition of Ctsk has effects on the expression of macrophages, we conducted immunohistochemistry (IHC) analysis for F4/80, a specific marker of macrophages (Fig. 4). The results showed that the F4/80-positive macrophages increased significantly in the disease group without inhibitor (Fig. 4C). We also conducted immunofluorescence analysis for the periapical lesion area for the CD3-positive T cells in different groups (Fig. 5). We then merged each set of images for comparison and location (Fig. 5A). These images showed significantly fewer visible CD3-positive T cells (Fig. 5A, white arrows) in the disease group with inhibitor (3.606 mg/kg/week) than in the disease group without inhibitor, which indicated that inhibition of Ctsk decreased the number of T cells in the periapical lesion area (Fig. 5C).
FIG 4.

Inhibition of Ctsk by the specific inhibitor showed reduced F4/80+ cells in periapical lesions at 42 days by immunohistological analysis. (A) Immunohistochemistry stains of F4/80+ (brown) macrophages in periapical lesions in the normal group and the bacterium-infected group with and without inhibitor (3.606 mg/kg/week) at 42 days. Normal serum served as the negative control. (B) Higher-magnification view of the images from the infection disease group at 42 days. Red arrows indicate F4/80+ macrophages. (C) Quantification of F4/80+ macrophages in the alveolar area of the normal group and the bacterium-infected group with and without inhibitor (3.606 mg/kg/week) at 42 days. **, P < 0.01; ***, P < 0.001. Each experiment was repeated three times (n = 3).
FIG 5.

Inhibition of Ctsk by the specific inhibitor decreased the number of T cells in periapical lesions at 42 days. (A) Immunofluorescence staining of CD3+ (red) T cells in periapical lesion area in the normal group and the bacterium-infected group with or without inhibitor (3.606 mg/kg/week) at 42 days. White arrows show visible CD3-positive T cells. (B) Normal serum served as a negative control in the same area (without primary antibody). (C) Quantification of CD3+ cells demonstrated that inhibition of Ctsk significantly reduced expression of CD3+ T cells in bacterium-infected group with inhibitor (3.606 mg/kg/week) at 42 days. ***, P < 0.001. Each experiment was repeated three times (n = 3).
Inhibition of Ctsk reduced the expression of proinflammatory and osteoclast marker genes as well as TLRs and cytokines in the periapical lesion at different time points.
To evaluate the effect of Ctsk inhibition on the mRNA levels of the genes for TNF-α, IL-6, IL-23α, and Ctsk, which are related to inflammatory and osteoclast status in periapical tissues, qRT-PCR was used as described previously (10, 17, 19). The relative mRNA expression levels showed that the expression of Ctsk was inhibited at different time points in the inhibitor (3.606 mg/kg/week)-treated disease group but not in the untreated disease group (Fig. 6A). Proinflammatory-gene expression also increased in the disease group at 7, 21, and 42 days (Fig. 6A). The results of TLR expression analysis showed that TLR9 expression increased significantly starting from day 21 in the disease group (Fig. 6A). The expression of TLR4 and TLR5 was also detected at different time points, since TLR4 and TLR5 are responsible for recognition of lipopolysaccharide (LPS) and flagellin of bacteria pathogens (21). Although TLR4 increased in the disease group without inhibitor at 7 and 21 days, there was no significant difference between the disease group and the inhibitor-treated group at 42 days (Fig. 6A). The IHC stain confirmed the qRT-PCR result (see Fig. S4 in the supplemental material). The expression of TLR5 decreased significantly in the inhibitor-treated disease group starting from 21 days (Fig. 6A). IHC staining of TLR5 showed that TLR5 expression decreased significantly in the inhibitor-treated disease group at 42 days (see Fig. S5 in the supplemental material), which was consistent with the qRT-PCR result. Although the gene expression levels of IL-6 and IL-23α decreased in the untreated disease group and the disease group treated with inhibitor (3.606 mg/kg/week) at 42 days, cytokine expression was still higher than that in the normal group. The ELISA result showed that TNF-α, IL-6, and IL-23α reached the highest levels in the disease group without inhibitor at 42 days (Fig. 6B). Since Ctsk has been shown to possibly play a critical role in the TLR9-related pathway (9), we also conducted IHC analysis of TLR9 to explore whether Ctsk affects the TLR9 expression in periapical disease at 42 days after infection. The results showed that TLR9 was expressed abundantly in the periapical lesion area in the disease group without inhibitor but not in the inhibitor-treated group (Fig. 7).
FIG 6.

Inhibition of Ctsk by the specific inhibitor reduced the expression of proinflammatory genes as well as TLR and cytokine genes in the periapical lesion at different time points. (A) qRT-PCR of osteoclast genes (i.e., Ctsk) and proinflammatory genes (i.e., TNF-α, IL-6, and IL-23α) as well as TLR genes (i.e., TLR4, -5, and -9) in the periapical lesion from the normal group and the bacterium-infected group with and without inhibitor (3.606 mg/kg/week) at 7, 21, and 42 days. β-actin was used as an endogenous control. (B) Expression of TNF-α, IL-6, and IL-23α in the periapical lesion at 7, 21, and 42 days detected by ELISA. Significance was compared between disease groups with and without inhibitor at different time points. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Each experiment was repeated three times (n = 4).
FIG 7.

Inhibition of Ctsk by the specific inhibitor shows reduced expression of TLR9 in periapical lesions at 42 days by immunohistological analysis. (A) Immunohistochemistry stains of TLR9+ (brown) cell areas in the normal group and the bacterium-infected group with and without inhibitor (3.606 mg/kg/week) at 42 days. Normal serum served as a negative control. (B) Higher-magnification view of the images from the infection disease group at 42 days. Red arrows indicate TLR9+ cells. (C) Quantification of TLR9+ cells in furcation, mesial, and distal periapical area of the normal group and the bacterium-infected group with and without inhibitor (3.606 mg/kg/week) at 42 days. *, P < 0.05; **, P < 0.01; ***, P < 0.001. N.S, no significance. Each experiment was repeated three times (n = 3).
DISCUSSION
Periapical lesions result from pulp necrosis induced by bacterial infection. The infection produces an inflammatory reaction first within the pulp and subsequently in the periapical region after egress of microorganisms, their by-products, and/or altered pulp tissues from the infected root canal. The periapical lesion is characterized by the presence of numerous inflammatory cells, which then lead to immune inflammation of periapical tissue. Generally, the body's immune defense system gradually removes the pathogens after the removal of bacterial infections, but during the process of obliterating foreign microorganisms, inflammatory factors produced by the body can also damage the surrounding normal tissue and cause periapical bone tissue absorption. If this process continues, teeth will eventually become loose or fall out. Bone resorption is mediated by bone-resorptive cytokines produced by the inflammatory response. Consequently, the question of how to control an excessive inflammatory response in the inflammatory process while inhibiting corresponding bone resorption is particularly important.
Cathepsin K, a lysosomal cysteine protease, is abundantly expressed in osteoclasts. This fact, as well as its enzymatic properties, implies that it is a key factor in normal bone remodeling and in pathological processes, such as osteoporosis, osteoarthritis, and inflammatory response-mediated bone resorption disease such as periapical disease (10, 11). Recent studies showed that Ctsk also has important functions in immune responses (9, 22, 23). Our previous study demonstrated that knocking down Ctsk by AAV gene therapy can significantly reduce bone resorption and inflammation (10). However, the main drawback of using AAV as a vector is that the small size of the genome significantly limits the amount of genetic material it can carry (24). Small molecules overcome this by means of several advantages: a greater range of treatable diseases, lower cost with greater ease of manufacturing, and oral availability, which corresponds to patient preference (14, 25). ODN is a highly selective, potent, and reversible inhibitor of Ctsk and inhibits osteoclast-mediated bone resorption in vitro but does not reduce the number of osteoclasts and appears to reduce bone resorption while preserving bone formation (26, 27). ODN is currently being developed as an orally bioavailable therapeutic for the treatment of postmenopausal osteoporosis. In a clinical trial, 36 months of once-weekly ODN treatment increased lumbar spine and total hip bone mineral density (BMD) and reduced bone turnover markers in postmenopausal women with low BMD (15).
Since Ctsk has been shown to have functions in both osteoclasts and immune cells, such as macrophages and dendritic cells (9, 11, 28), whether inhibition of Ctsk can affect not only osteoclasts but also immune responses is still unclear. This study proposes a novel role for Ctsk in endodontic inflammation lesions. Patients with endodontic disease suffer from both inflammation-induced tissue damage and bone loss (2); a single target that can be treated to dramatically improve both conditions is ideal. In the current study, we tested the inhibition of Ctsk using the specific small-molecule inhibitor ODN by oral administration in a periapical lesion mouse model to determine whether Ctsk plays dual roles in periapical disease to reduce both inflammation and bone resorption at different time points.
Due to the nature and progression of endodontic infection, changing microenvironmental conditions select for more anaerobic and virulent species of bacteria (29). We induced periapical disease by inoculating the root canal system of the mandibular first molar with a mixture of four common endodontic pathogens: Prevotella intermedia (ATCC 25611), Fusobacterium nucleatum (ATCC 25586), Peptostreptococcus micros (ATCC 33270), and Streptococcus intermedius (ATCC 27335), as described previously (10, 17). Our results showed that bone resorption at different time points was much more advanced in the periapical periodontitis group without inhibitor than in the periapical periodontitis group with inhibitor, and meanwhile, we found that bone resorption increased over time in different groups, but this trend was more obvious in the periapical disease group without inhibitor (Fig. 1). Further quantitative analysis by micro-CT confirmed the bone-protective effects at 7, 21, and 42 days by inhibition of Ctsk (Fig. 1, arrows). To determine the trends of bone resorption and the infiltration of inflammatory cells in the periapical lesion area, we also conducted H&E staining to detect the changes of bone resorption in different groups at 7, 21, and 42 days after infection (Fig. 2), which was consistent with the results of X-ray analysis and micro-CT. We also wanted to explore the proper dose used for treatment of periapical disease in mice, so we chose two different doses, 3.606 mg/kg/week and 0.7212 mg/kg/week, which were calculated from the dose used for clinical trials in humans by considering the difference in body surface area between humans and mice (16). The result showed that the 3.606-mg/kg/week dose had a better bone protection effect at 42 days after infection (see Fig. S2 in the supplemental material). These results showed that the bone resorption was more notable in the periapical disease group without inhibitor than in the inhibitor-treated disease group at different time points. Inflammatory-cell infiltration in the periapical disease group without inhibitor was increasingly more serious at 7, 21, and 42 days; a large number of blue-stained monocytes were distributed in the apical, mesial/distal, and furcation areas (Fig. 2A, red arrows). In the inhibitor-treated periapical disease group, the trabecular bone area was basically unchanged and infiltration of inflammatory cells was significantly reduced. Above all, inhibition of Ctsk had significant bone protection effects and prevented inflammatory-cell infiltration.
To investigate the mechanism of the effect of Ctsk inhibition in vivo, TRAP staining and immunohistochemistry (IHC) staining were carried out. Studies showed that periapical inflammatory cells play an important role in the development of periapical periodontitis, as they secrete a variety of cytokines (IL-1α, TNF-α, IL-6, and IL-11) and are involved in the adjustment of the activation and apoptosis of corresponding effector cells, such as osteoclasts, thus controlling bone resorption (30–32). In our previous study, we showed that Ctsk knockdown can abolish the extracellular acidification and then inhibit bone resorption, while osteoclast differentiation is only slightly affected in vitro (10, 11). ODN has also been shown not to affect the osteoclast numbers in vivo (33). Therefore, the number of osteoclasts of various groups in vivo at different time points was determined in this experiment to clarify the possible mechanism. We found that the number of osteoclasts did not change in the normal control group (see Fig. S3 in the supplemental material) but was significantly reduced at different time points in the lesion area of the inhibitor-treated periapical periodontitis group compared to that of the untreated periapical disease group (Fig. 3). This indicated that the immune response may also be affected by inhibition of Ctsk, since preosteoclast activation is due to RANKL, which is secreted by osteoblasts, T cells, and B cells, and these cells can be activated by proinflammatory cytokines and chemokines, such as IL-1, IL-6, TNF-α, and IL-17 (34). Studies indicated that activated macrophages are the source of bone-resorbing cytokines in periapical disease (35), which can be activated by T-lymphocytes or by bacterial endotoxin, as part of innate immunity. In order to fully demonstrate the function of Ctsk in the immune response to periapical disease, the present study also examined whether inhibition of Ctsk affects the expression of macrophages by detecting the macrophage marker F4/80 (Fig. 4). The results showed that the number of macrophages increased significantly in the periapical disease group without inhibitor at 42 days but not in the inhibitor-treated group (Fig. 4), which indicated that Ctsk may play an important role in both osteoclasts and immune responses (Fig. 3 and 4).
Since macrophage expression decreased in the inhibitor-treated disease group, and to further investigate the effect of Ctsk function in adaptive immune response, immune fluorescence analysis was carried out. We determined the number of CD3-positive T cells in the periapical disease area from different groups at 42 days (Fig. 5). The results showed that the CD3-positive T cells decreased significantly in the inhibitor-treated infection group, which indicated that the adaptive immune response was impaired by inhibition of Ctsk. As the effector cell of inflammation, T cells can produce a variety of effector molecules (36, 37). Therefore, the number of T cells is an important indicator of the severity of the local inflammatory response. As a T-cell surface marker, CD3 is commonly used to detect T cell populations, including αβ T cells and γδ T cells. The results showed that the number of CD3+ T cells was significantly reduced in the inhibitor-treated periapical disease group than in the disease group (Fig. 5A). T cells showed aggregated distribution in the periapical periodontitis group, while the inhibitor-treated periapical periodontitis group showed only minor T cell distribution, which indicated that the immune system-mediated inflammatory reaction is suppressed in Ctsk-inhibited disease groups. This result could explain the reduction of the number of osteoclasts from another aspect (Fig. 3). T cells and macrophages decreased, which might result in a lower proinflammatory-cytokine production, which might finally cause lower osteoclast activation and differentiation.
In order to confirm this hypothesis, the relative mRNA level was quantified by Sybr green-based qRT-PCR by using the primer set listed in Table S1 in the supplemental material. Our results showed that the Ctsk gene reached the highest level at 42 days in the periapical disease group in vivo but not in the inhibitor-treated disease group, which showed no obvious change of Ctsk expression (Fig. 6A). To further investigate the mechanism of the protective effect of inhibition of Ctsk in the mouse model of periapical disease, we tested the expression level of proinflammatory-cytokine genes, including TNF-α, IL-6, and IL-23α. The TNF-α and IL-6 expression levels increased significantly in the periapical disease group at day 7 compared to the inhibitor-treated disease group (Fig. 6A). The expression of IL-23α reached the highest level at day 21. The expression levels of cytokines, including TNF-α, IL-6, and IL-23, all increased in the periapical disease group at different time points. Although the proinflammatory cytokines also increased in the inhibitor-treated disease group, the level was much lower than that in the disease group (Fig. 6B). Cytokines such as TNF-α, IL-6, and IL-23α are soluble mediators and are released from immunocompetent cells in the periapical inflammatory processes. These cytokines can stimulate bone resorption in periapical lesion areas (38). Proinflammatory cytokines, such as TNF-α, have been reported to have a key role in regulating the inflammatory response (39). IL-6 is produced at the site of inflammation and also plays a key role in the acute-phase response, which is also shown by our qRT-PCR results (Fig. 6A) (40). As receptors recognize several major bacterial pathogens, Toll-like receptors (TLRs) are important in immune responses. Currently, 12 TLR family members have been found in humans and other mammals, but not all TLRs are expressed in mammals. For example, TLR11 and -12 are expressed in mice but not in humans (41). TLR4, -5, and -9 can specifically identify different types of bacterial antigens, which play an important role in bacterial infection. The study from Beklen et al. showed that except for TLR7 and TLR8, expression levels of TLR1 to TLR10 in the human gingival surface are higher in periodontitis patients than healthy people (42), suggesting that TLRs play a significant role in the activation and development of periodontitis. Therefore, TLR4, -5, and -9 were selected as the research subject in this experiment to detect the changes in the inhibition of Ctsk in the periapical disease model. The relationship between Ctsk and TLRs has not been fully clarified yet. Recent studies demonstrate that Ctsk has a critical role in TLRs functions (9, 43, 44). Asagiri et al. demonstrated that in rheumatoid arthritis, Ctsk might have an effect on dendritic-cell (DC) cytokine expression through TLR9 in vitro (9). Hirabara et al. showed that the Ctsk inhibitor might inhibit TLR4-dependent Ctsk expression in human fibroblasts in vitro (43). Ctsk has also been shown to be involved in development of psoriasis-like skin lesions through TLR7 activation (44). In the current study, we showed that TLR5 and -9 decreased in the inhibitor-treated disease group compared to the disease group at different time points (Fig. 6A). The IHC staining of TLR5 and -9 confirmed the qRT-PCR result (Fig. 7; also, see Fig. S5 in supplemental material). TLR4 expression increased in the disease group at 7 and 21 days; however, it decreased at 42 days. There was no significant difference at 42 days between the disease group and the inhibitor-treated disease group (Fig. 6; also, see Fig. S4 in supplemental material).
In summary, we investigated the possible mechanism of the effect of Ctsk, which simultaneously targets osteoclasts and immune cells. The inhibition of Ctsk by the specific small molecule ODN reduced bone resorption by inhibition of the osteoclast function and inflammation simultaneously and in turn decreased osteoclast activation and differentiation, which exhibits a promising possibility for therapy for periapical inflammatory disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christie Paulson for her excellent assistance with the manuscript. We appreciate the assistance of the Center for Metabolic Bone Disease at the University of Alabama at Birmingham (P30 AR046031). We are also grateful for the assistance of the Small Animal Phenotyping Core (P30 DK079626), Metabolism Core, and Neuroscience Molecular Detection Core Laboratory at the University of Alabama at Birmingham (P30 NS0474666).
This work was supported by NIH grant RC1-DE-020533-01 (Y.P.L.).
We have no potential conflicts of interest with respect to the authorship and/or publication of this article.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01713-14.
REFERENCES
- 1.Wang QQ, Zhang CF, Chu CH, Zhu XF. 2012. Prevalence of Enterococcus faecalis in saliva and filled root canals of teeth associated with apical periodontitis. Int J Oral Sci 4:19–23. doi: 10.1038/ijos.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stashenko P, Teles R, D'Souza R. 1998. Periapical inflammatory responses and their modulation. Crit Rev Oral Biol Med 9:498–521. doi: 10.1177/10454411980090040701. [DOI] [PubMed] [Google Scholar]
- 3.Jiang H, Chen W, Zhu G, Zhang L, Tucker B, Hao L, Feng S, Ci H, Ma J, Wang L, Stashenko P, Li YP. 2013. RNAi-mediated silencing of Atp6i and Atp6i haploinsufficiency prevents both bone loss and inflammation in a mouse model of periodontal disease. PLoS One 8:e58599. doi: 10.1371/journal.pone.0058599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang S, Hao L, McConnell M, Zhou X, Wang M, Zhang Y, Mountz JD, Reddy M, Eleazer PD, Li YP, Chen W. 2013. Inhibition of Rgs10 expression prevents immune cell infiltration in bacteria-induced inflammatory lesions and osteoclast-mediated bone destruction. Bone Res 1:267–281. doi: 10.4248/BR201303005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eleazer PD, Farber PA, Seltzer S. 1975. Lack of lymphocyte stimulation by root canal products. J Endod 1:388–394. doi: 10.1016/S0099-2399(75)80157-9. [DOI] [PubMed] [Google Scholar]
- 6.Duong LT. 2013. Inhibition of cathepsin K: blocking osteoclast bone resorption and more. IBMS BoneKEy 10:396. doi: 10.1038/bonekey.2013.130. [DOI] [Google Scholar]
- 7.Troen BR. 2004. The role of cathepsin K in normal bone resorption. Drug News Perspect 17:19–28. doi: 10.1358/dnp.2004.17.1.829022. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto F, Saitoh S-i, Fukui R, Kobayashi T, Tanimura N, Konno K, Kusumoto Y, Akashi-Takamura S, Miyake K. 2008. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun 367:693–699. doi: 10.1016/j.bbrc.2007.12.130. [DOI] [PubMed] [Google Scholar]
- 9.Asagiri M, Hirai T, Kunigami T, Kamano S, Gober H-J, Okamoto K, Nishikawa K, Latz E, Golenbock DT, Aoki K, Ohya K, Imai Y, Morishita Y, Miyazono K, Kato S, Saftig P, Takayanagi H. 2008. Cathepsin K-dependent Toll-like receptor 9 signaling revealed in experimental arthritis. Science 319:624–627. doi: 10.1126/science.1150110. [DOI] [PubMed] [Google Scholar]
- 10.Gao B, Chen W, Hao L, Zhu G, Feng S, Ci H, Zhou X, Stashenko P, Li YP. 2013. Inhibiting periapical lesions through AAV-RNAi silencing of cathepsin K. J Dent Res 92:180–186. doi: 10.1177/0022034512468757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YP, Chen W. 1999. Characterization of mouse cathepsin K gene, the gene promoter, and the gene expression. J Bone Mineral Res 14:487–499. doi: 10.1359/jbmr.1999.14.4.487. [DOI] [PubMed] [Google Scholar]
- 12.Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, Choi Y. 2006. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol 24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- 13.Takayanagi H. 2007. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 14.Fleischmann R. 2012. Novel small-molecular therapeutics for rheumatoid arthritis. Curr Opin Rheumatol 24:335–341. doi: 10.1097/BOR.0b013e32835190ef. [DOI] [PubMed] [Google Scholar]
- 15.Eisman JA, Bone HG, Hosking DJ, McClung MR, Reid IR, Rizzoli R, Resch H, Verbruggen N, Hustad CM, DaSilva C, Petrovic R, Santora AC, Ince BA, Lombardi A. 2011. Odanacatib in the treatment of postmenopausal women with low bone mineral density: three-year continued therapy and resolution of effect. J Bone Mineral Res 26:242–251. doi: 10.1002/jbmr.212. [DOI] [PubMed] [Google Scholar]
- 16.Stoch SA, Zajic S, Stone J, Miller DL, Van Dyck K, Gutierrez MJ, De Decker M, Liu L, Liu Q, Scott BB, Panebianco D, Jin B, Duong LT, Gottesdiener K, Wagner JA. 2009. Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double-blind, randomized, placebo-controlled phase I studies. Clin Pharmacol Ther 86:175–182. doi: 10.1038/clpt.2009.60. [DOI] [PubMed] [Google Scholar]
- 17.Ma J, Chen W, Zhang L, Tucker B, Zhu G, Sasaki H, Hao L, Wang L, Ci H, Jiang H, Stashenko P, Li Y-P. 2013. RNA interference-mediated silencing of Atp6i prevents both periapical bone erosion and inflammation in the mouse model of endodontic disease. Infect Immun 81:1021–1030. doi: 10.1128/IAI.00756-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng S, Deng L, Chen W, Shao J, Xu G, Li YP. 2009. Atp6v1c1 is an essential component of the osteoclast proton pump and in F-actin ring formation in osteoclasts. Biochem J 417:195–203. doi: 10.1042/BJ20081073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki H, Hou L, Belani A, Wang CY, Uchiyama T, Muller R, Stashenko P. 2000. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J Immunol 165:3626–3630. doi: 10.4049/jimmunol.165.7.3626. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki H, Okamatsu Y, Kawai T, Kent R, Taubman M, Stashenko P. 2004. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J Periodontal Res 39:432–441. doi: 10.1111/j.1600-0765.2004.00760.x. [DOI] [PubMed] [Google Scholar]
- 21.O'Neill LAJ, Golenbock D, Bowie AG. 2013. The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol 13:453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 22.Conus SSH. 2010. Cathepsins and their involvement in immune responses. Swiss Med Wkly 140:w13042. doi: 10.4414/smw.2010.13042. [DOI] [PubMed] [Google Scholar]
- 23.Krieg AM, Lipford GB. 2008. Immunology: the toll of cathepsin K deficiency. Science 319:576–577. doi: 10.1126/science.1154207. [DOI] [PubMed] [Google Scholar]
- 24.Owens RA. 2002. Second generation adeno-associated virus type 2-based gene therapy systems with the potential for preferential integration into AAVS1. Curr Gene Ther 2:145–159. doi: 10.2174/1566523024605627. [DOI] [PubMed] [Google Scholar]
- 25.Spring DR. 2005. Chemical genetics to chemical genomics: small molecules offer big insights. Chem Soc Rev 34:472–482. doi: 10.1039/b312875j. [DOI] [PubMed] [Google Scholar]
- 26.Yasuda Y, Kaleta J, Bromme D. 2005. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev 57:973–993. doi: 10.1016/j.addr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. 2007. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des 13:387–403. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- 28.Herroon MK, Rajagurubandara E, Rudy DL, Chalasani A, Hardaway AL, Podgorski I. 2013. Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene 32:1580–1593. doi: 10.1038/onc.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moller AJ. 1966. Microbiological examination of root canals and periapical tissues of human teeth. Methodological studies. Odontol Tidskr 74(Suppl):1–380. [PubMed] [Google Scholar]
- 30.Strand V, Kavanaugh AF. 2004. The role of interleukin-1 in bone resorption in rheumatoid arthritis. Rheumatology 43(Suppl 3):iii10–iii16. doi: 10.1093/rheumatology/keh202. [DOI] [PubMed] [Google Scholar]
- 31.de la Mata J, Uy HL, Guise TA, Story B, Boyce BF, Mundy GR, Roodman GD. 1995. Interleukin-6 enhances hypercalcemia and bone resorption mediated by parathyroid hormone-related protein in vivo. J Clin Invest 95:2846–2852. doi: 10.1172/JCI117990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schett G. 2011. Effects of inflammatory and anti-inflammatory cytokines on the bone. Eur J Clin Invest 41:1361–1366. doi: 10.1111/j.1365-2362.2011.02545.x. [DOI] [PubMed] [Google Scholar]
- 33.Cusick T, Chen CM, Pennypacker BL, Pickarski M, Kimmel DB, Scott BB, Duong LT. 2012. Odanacatib treatment increases hip bone mass and cortical thickness by preserving endocortical bone formation and stimulating periosteal bone formation in the ovariectomized adult rhesus monkey. J Bone Mineral Res 27:524–537. doi: 10.1002/jbmr.1477. [DOI] [PubMed] [Google Scholar]
- 34.Cochran DL. 2008. Inflammation and bone loss in periodontal disease. J Periodontol 79:1569–1576. doi: 10.1902/jop.2008.080233. [DOI] [PubMed] [Google Scholar]
- 35.Metzger Z. 2000. Macrophages in periapical lesions. Endod Dent Traumatol 16:1–8. doi: 10.1034/j.1600-9657.2000.016001001.x. [DOI] [PubMed] [Google Scholar]
- 36.Ueda H, Howson JMM, Esposito L, Heward J, Snook Chamberlain G, Rainbow DB, Hunter KMD, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RCJ, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SCL. 2003. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 37.Marçal JRB, Samuel RO, Fernandes D, de Araujo MS, Napimoga MH, Pereira SAL, Clemente-Napimoga JT, Alves PM, Mattar R, Rodrigues V Jr, Rodrigues DBR. 2010. T-helper cell type 17/regulatory T-cell immunoregulatory balance in human radicular cysts and periapical granulomas. J Endod 36:995–999. doi: 10.1016/j.joen.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 38.Ataoglu T, Ungor M, Serpek B, Haliloglu S, Ataoglu H, Ari H. 2002. Interleukin-1beta and tumour necrosis factor-alpha levels in periapical exudates. Int Endod J 35:181–185. doi: 10.1046/j.1365-2591.2002.00467.x. [DOI] [PubMed] [Google Scholar]
- 39.Beutler B, Cerami A. 1989. The biology of cachectin/TNF—a primary mediator of the host response. Annu Rev Immunol 7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 40.Gabay C. 2006. Interleukin-6 and chronic inflammation. Arthritis Res Ther 8(Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beutler B. 2004. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 42.Beklen A, Hukkanen M, Richardson R, Konttinen YT. 2008. Immunohistochemical localization of Toll-like receptors 1-10 in periodontitis. Oral Microbiol Immunol 23:425–431. doi: 10.1111/j.1399-302X.2008.00448.x. [DOI] [PubMed] [Google Scholar]
- 43.Hirabara S, Kojima T, Takahashi N, Hanabayashi M, Ishiguro N. 2013. Hyaluronan inhibits TLR-4 dependent cathepsin K and matrix metalloproteinase 1 expression in human fibroblasts. Biochem Biophys Res Commun 430:519–522. doi: 10.1016/j.bbrc.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Hirai T, Kanda T, Sato K, Takaishi M, Nakajima K, Yamamoto M, Kamijima R, Digiovanni J, Sano S. 2013. Cathepsin K is involved in development of psoriasis-like skin lesions through TLR-dependent Th17 activation. J Immunol 190:4805–4811. doi: 10.4049/jimmunol.1200901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.