

Abstract
The cytolethal distending toxin (Cdt) is produced from a number of bacteria capable of causing infection and inflammatory disease. Our previous studies with Actinobacillus actinomycetemcomitans Cdt demonstrate not only that the active toxin subunit functions as a phosphatidylinositol-3,4,5-triphosphate (PIP3) phosphatase but also that macrophages exposed to the toxin were stimulated to produce proinflammatory cytokines. We now demonstrate that the Cdt-induced proinflammatory response involves the activation of the NLRP3 inflammasome. Specific inhibitors and short hairpin RNA (shRNA) were employed to demonstrate requirements for NLRP3 and ASC as well as caspase-1. Furthermore, Cdt-mediated inflammasome activation is dependent upon upstream signals, including reactive oxygen species (ROS) generation and Cdt-induced increases in extracellular ATP levels. Increases in extracellular ATP levels contribute to the activation of the P2X7 purinergic receptor, leading to K+ efflux. The relationship between the abilities of the active toxin subunit CdtB to function as a lipid phosphatase, activate the NLRP3 inflammasome, and induce a proinflammatory cytokine response is discussed. These studies provide new insight into the virulence potential of Cdt in mediating the pathogenesis of disease caused by Cdt-producing organisms such as Aggregatibacter actinomycetemcomitans.
INTRODUCTION
The cytolethal distending toxins (Cdts) are a family of heat-labile protein cytotoxins produced by several bacterial species, including Campylobacter jejuni, Shigella species, Haemophilus ducreyi, Aggregatibacter actinomycetemcomitans, and diarrheal disease-causing enteropathogens such as some Escherichia coli isolates (1–7). There is clear evidence that Cdts are encoded by three genes, designated cdtA, cdtB, and cdtC, which are arranged as an apparent operon (7–12). These three genes specify three polypeptides, designated CdtA, CdtB, and CdtC, with apparent molecular masses of 28, 32, and 20 kDa, respectively, that form a heterotrimeric holotoxin. Several cell lines and cell types have been shown to be sensitive to Cdt-induced cell cycle arrest and cell death via apoptosis; these include human lymphoid cells, fibroblasts, human embryonic intestinal epithelial cells, a human colon carcinoma cell line, and human keratinocytes, among others (7, 11, 12). There is considerable agreement that the heterotrimeric holotoxin functions as an AB2 toxin, where CdtB is the active (A) unit and the complex of CdtA and CdtC comprises the binding (B) unit (13–15). In this regard, we have shown that CdtA and CdtC are required for the toxin to associate with lipid microdomains within lymphocyte membranes and that Cdt-mediated toxicity is dependent upon the integrity of these lipid domains (16). Furthermore, we have demonstrated that the CdtC subunit contains a cholesterol recognition sequence that is required for interactions with membrane cholesterol (16, 17).
The active Cdt subunit CdtB exhibits enzymatic activity, enabling it to degrade the signaling lipid phosphatidylinositol-3,4,5-triphosphate (PIP3), a key component of the phosphatidylinositol 3-kinase (PI-3K) signaling pathway (18, 19). Mutation analysis of CdtB clearly indicates that the ability of CdtB to induce cell cycle arrest and cell death in lymphocytes correlates with PIP3 phosphatase activity. Furthermore, we have demonstrated that treatment of lymphocytes with Cdt leads to perturbations in PI-3K signaling, including PIP3 depletion and decreased phosphorylation of both Akt and glycogen synthase kinase 3β (GSK3β), resulting in concomitant changes in their respective enzymatic activities: decreased Akt and increased GSK3β kinase activity. Moreover, differential susceptibility to the toxin of lymphoid cell lines with defects in this signaling pathway supports the notion that CdtB-mediated hydrolysis of PIP3 is key to the molecular mechanism by which the toxin acts to impair lymphocyte proliferation and survival.
Recently, we reported that human macrophages were not susceptible to Cdt-induced apoptosis (19). Nonetheless, the toxin was capable of binding to macrophages and perturbing PI-3K signaling, resulting in decreased phosphorylation of Akt and GSK3β; these changes were also accompanied by concomitant alterations in kinase activity, as observed with lymphocytes. While Cdt did not induce apoptosis, treatment of macrophages with toxin resulted in proinflammatory cytokine production, including increased levels of expression and release of interleukin-1β (IL-1β), tumor necrosis factor alpha (TNF-α), and IL-6. Furthermore, treatment of cells with either Toll-like receptor 2 (TLR-2), TLR-3, or TLR4 agonists in the presence of Cdt resulted in augmented proinflammatory responses over those observed with the agonist alone. These effects were found to be dependent upon the ability of Cdt to function as a lipid phosphatase and thereby deplete cells of PIP3, which in turn leads to GSK3β activation. Of particular importance to these observations, GSK3β activation has been linked to NF-κB activation, which in turn induces increased expression levels of proinflammatory cytokines, including IL-1β, TNF-α, IL-18, and IL-6, among others (20–25). In contrast, the activation of PI-3K signaling has been linked to the phosphorylation and inactivation of GSK3β, with preferential expression of anti-inflammatory cytokines such as IL-10 and IL-1 receptor antagonist (IL-1RA) (26–28). We now report that the Cdt-mediated induction of the proinflammatory cytokine response by macrophages is also dependent upon the Cdt-mediated activation of caspase-1, which in turn is dependent upon the activation of the NLRP3 inflammasome.
MATERIALS AND METHODS
Cells.
The human acute monocytic leukemia cell line THP-1 was obtained from the ATCC; cells were maintained in RPMI 1640 containing 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 20 μM 2-mercaptoethanol, and 2% penicillin-streptomycin at 37°C with 5% CO2 in a humidified incubator. Preparation of THP-1 cells stably expressing short hairpin RNA (shRNA) against caspase-1 (shCasp1), NLRP3 (shNLRP3), and ASC (shASC) as well as nontarget controls was previously described (29–31); these cells have been well characterized, and expression levels were quantified to be reduced >75% for shNLRP3, >95% for shASC, and >90% for shCasp1 cells, with no effect on the nontarget controls (32). THP-1 cells were differentiated into macrophages by incubating cells in the presence of 50 ng/ml phorbol myristate acetate (PMA) for 48 h, at which time the cells were washed and incubated an additional 24 h in medium prior to use.
Expression and purification of Cdt holotoxin.
The construction and expression of the plasmid containing the cdt genes for the holotoxin (pUCAacdtABChis) were previously reported (33). The plasmids were constructed so that the cdt genes were under the control of the lac promoter and transformed into E. coli DH5α. Cultures of transformed E. coli cells were grown in 1 liter of LB broth and induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 2 h, and bacterial cells were harvested, washed, and resuspended in 50 mM Tris (pH 8.0). The cells were frozen overnight, thawed, and sonicated. The histidine-tagged holotoxin was isolated by nickel affinity chromatography as previously described (33); the isolated toxin was analyzed by SDS-PAGE and found to contain the three Cdt subunits CdtA (18 kDa), CdtB (32 kDa), and CdtC (20 kDa) (see Fig. S1A in the supplemental material).
Analysis of cytokine release and caspase-1 activation.
Cytokine production was measured in THP-1-derived macrophages (2 × 105 cells) incubated for 5 h (IL-1β and TNF-α) or 48 h (IL-18) in the presence or absence of various amounts of Cdt (0 to 200 ng/ml). Culture supernatants were collected and analyzed for IL-1β (Quantikine Elisa kit; R and D Systems), TNF-α (Peprotech), and IL-18 (MBL) by enzyme-linked immunosorbent assays (ELISAs) using commercially available kits, according to the manufacturers' instructions. In each instance, the amount of cytokine present in the supernatant was determined by using a standard curve.
Caspase-1 activity in cells treated with 200 ng/ml Cdt for 2 and 4 h was determined as described above. Cells were harvested and lysed, and the extracts were assessed for caspase-1 activity by using a colorimetric assay (Abcam) based upon the cleavage and release of the chromophore p-nitroanilide (p-NA) from the labeled substrate Z-Tyr-Val-Ala-Asp(OMe) (YVAD)–p-NA. Additionally, caspase-1 release into culture supernatants was assessed by an ELISA using the anti-p20 caspase-1 Quantikine Elisa kit (R and D Systems).
Western blot analysis.
Replicate wells of THP-1-derived macrophages (4 × 106 cells/well) were incubated with Cdt as described above. The cells were washed and treated with 20 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate, and protease and phosphatase inhibitors (Pierce); replicate wells were pooled; and the protein concentration was determined. Samples (30 μg) were separated on 12% SDS-PAGE gels and then transferred onto nitrocellulose. The membrane was blocked with BLOTTO and then incubated with anti-pro-IL-1β or actin antibody (Santa Cruz Biotechnology, Inc.) for 18 h at 4°C (8). Membranes were washed and incubated with donkey anti-rabbit immunoglobulin (1:1,000 dilution; GE Healthcare) conjugated to horseradish peroxidase. The Western blots were developed by using chemiluminescence and analyzed by digital densitometry (Kodak Image Systems), as previously described (34).
ATP analysis.
Macrophages (4 × 106) were incubated in medium or with Cdt (0 to 200 ng/ml) for 4 h. Supernatants were harvested and assayed for ATP content by using a commercially available kit (ATP determination kit; Molecular Probes). Concentrations of ATP were determined by using a standard curve.
Statistical analysis.
Means ± standard errors of the means were calculated for replicate experiments. Significance was determined by using Student's t test, differences between multiple treatments were compared by analysis of variance (ANOVA) paired with Tukey's honestly significant difference (HSD) posttest, and a P value of <0.05 was considered to be statistically significant.
RESULTS
We previously demonstrated that human macrophages were not susceptible to the cytotoxic effects of Cdt, which typically involve cell death resulting from the activation of the apoptotic cascade. Moreover, we now confirm that Cdt also does not alter cell viability, as assessed by propidium iodide exclusion (see Fig. S2 in the supplemental material). Nonetheless, the toxin is able to bind to macrophages, deliver CdtB to intracellular compartments, and perturb PI-3K signaling (19). Moreover, we have shown that a Cdt-mediated blockade of the PI-3K signaling pathway led to the expression and release of proinflammatory cytokines; these effects are consistent with the putative regulatory role for this signaling pathway in regulating cytokine production. It is well established that the synthesis of cytokines such as IL-1β requires further processing prior to release; this involves enzymatic cleavage leading to formation of the biologically active mature molecule. In the case of IL-1β and IL-18, maturation involves caspase-1 activation, which in turn is dependent upon inflammasome activation. To determine whether Cdt was able to promote caspase-1 activation, THP-1-derived macrophages were exposed to 200 ng/ml of toxin for 2 and 4 h. Cell extracts were assessed for active caspase-1; as shown in Fig. 1A, toxin-treated cells exhibited 2- to 3-fold increases in caspase-1 activity at 2 and 4 h, respectively. Caspase-1 activation was further demonstrated by Western blotting, which demonstrated 2.5-fold and >3-fold increases in the amount of mature caspase-1 (p20) generated in the presence of 250 and 500 ng/ml Cdt, respectively (Fig. 1A, inset). Additionally, the supernatants of cells treated with Cdt were assessed for the presence of caspase-1, as the mature/active caspase is typically secreted along with cytokines. Culture supernatants were analyzed for the secretion of caspase-1 by an ELISA; as shown in Fig. 1B, THP-1-derived macrophages secreted caspase-1 in a dose-dependent manner. Macrophages released 4.8 ± 2.3 pg/ml in the presence of 8 ng/ml Cdt; caspase-1 secretion increased to 23.3 ± 6.8 pg/ml in the presence of 200 ng/ml Cdt. Caspase-1 was not detectable in supernatants obtained from untreated cells.
FIG 1.

Cdt-induced proinflammatory cytokine release from macrophages is dependent upon caspase-1 activation. (A) THP-1-derived macrophages were treated with 200 ng/ml Cdt for 2 and 4 h. Cell extracts were assessed for caspase-1 activity as described in Materials and Methods. The inset shows Western blot analysis of extracts obtained from THP-1-derived macrophages treated with 0 to 500 ng/ml Cdt; the mature form of caspase-1 (p20 subunit) is shown, along with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. Levels of expression are shown below the blot as a percentage of levels in untreated control cells. OD, optical density. (B) THP-1-derived macrophages were treated with Cdt (0 to 200 ng/ml) for 5 h. Supernatants were then analyzed for the presence of caspase-1 (anti-p20) by an ELISA. Results are the means ± standard errors of the means for three experiments, each performed in triplicate. (C) Effects of caspase-1 inhibition on release of proinflammatory cytokines. Macrophages were exposed to various amounts of the caspase inhibitor Z-WEHD-FMK for 60 min; 50 ng/ml Cdt was then added to the cells for 5 h. Culture supernatants were analyzed for IL-1β (circles), IL-18 (diamonds), and TNF-α (triangles) by an ELISA. Results are plotted as the mean ± the standard error of the mean pg/ml cytokine released versus the Z-WEHD-FMK concentration. Levels of cytokines release from untreated control cells were 9.8 ± 7.8 pg/ml (IL-1β), 38.1 ± 14.0 pg/ml (IL-18), and 120.9 ± 2.6 pg/ml (TNF-α). Asterisks indicate statistical significance (P ≤ 0.05) compared to untreated control cells (A and B) or compared to cells treated with toxin alone (C). (D) Cells were assessed for the expression of pro-IL-1β after 5 h of exposure to Cdt following solubilization, fractionation by SDS-PAGE, and analysis by Western blotting. Actin is shown as a loading control. Western blots are representative of results from three experiments.
Two approaches were initially employed to determine if caspase-1 activation is required for cytokine maturation and release following exposure to Cdt. Macrophages were first treated with various amounts (0.1 to 100 μM) of the cell-permeable caspase-1 inhibitor Z-Trp-Glu(OMe)-His-Asp(OMe)-fluoromethylketone (Z-WEHD-FMK) and then exposed to Cdt (50 ng/ml), and the supernatants were analyzed 5 h later for IL-1β, IL-18, and TNF-α by an ELISA. As shown in Fig. 1C, cells treated with Cdt alone showed significant IL-1β release (802 ± 107 pg/ml) relative to that of control cells, which were incubated in medium alone (9.8 ± 7.8 pg/ml). It should be noted that the induction of IL-1β production is dependent upon active Cdt, as inactive mutants (19) and heat-inactivated toxin failed to induce the release of mature IL-1β (see Fig. S2B in the supplemental material); furthermore, polymyxin B did not alter the ability of Cdt to induce cytokine production (see Fig. S2B in the supplemental material). Macrophages pretreated with the caspase-1 inhibitor exhibited dose-dependent reductions in IL-1β release of 608 ± 127, 293 ± 60, 151 ± 32, and 72 ± 27 pg/ml in the presence of 0.1, 1, 10, and 100 μM inhibitor, respectively (Fig. 1C). We have previously shown that Cdt induces increased cytokine expression and release of mature IL-1β as a result of a Cdt-mediated PI-3K blockade due to the ability of CdtB to function as a lipid phosphatase (19). Therefore, we confirmed that the caspase-1 inhibitor affected only IL-1β maturation and not the Cdt-induced expression of pro-IL-1β. Figure 1D demonstrates that the caspase-1 inhibitor did not impair Cdt-induced increases in the synthesis of pro-IL-1β; the proform of the cytokine was upregulated in cells treated with Cdt alone, and its levels remained elevated in the presence of the toxin and caspase-1 inhibitor (Fig. 1D). Likewise, cells exposed to Cdt for 48 h were induced to release 325 ± 8 pg/ml IL-18, whereas untreated control cells released 38 ± 14 pg/ml (Fig. 1C). Pretreatment with 0.1 μM caspase-1 inhibitor reduced IL-18 release to baseline values (36 ± 23 pg/ml). In contrast to IL-1β and IL-18, the caspase-1 inhibitor partially blocked TNF-α release (Fig. 1D). Cells exposed to Cdt alone secreted 8,325 ± 276 pg/ml TNF-α; in the presence of 100 μM inhibitor, the highest concentration employed, release was reduced by 56%, to 3,720 ± 109 pg/ml.
The requirement for caspase-1 activation in the Cdt-induced proinflammatory response was further evaluated with THP-1 cells that were depleted of the enzyme by using shRNA (Fig. 2). It should be noted that the characterization of these cells, including quantification of reduced expression levels relative to those in nontarget control cells, was previously reported (see Materials and Methods) (29–32). Cdt induced a dose-dependent release of both IL-1β and IL-18 in macrophages derived from both wild-type and shRNA control (shCtl) THP-1 cells (Fig. 2A and B). For example, exposure of shCtl-derived macrophages to 40 and 200 ng/ml Cdt resulted in 586 ± 82 and 794 ± 138 pg/ml IL-1β as well as 97 ± 10 and 145 ± 8 pg/ml IL-18, respectively. Untreated cells released 100 ± 4 pg/ml IL-1β and 14 ± 9 pg/ml IL-18. In contrast, cytokine release was significantly reduced in Cdt-treated macrophages derived from cells deficient in caspase-1 (shCasp1); in the presence of 40 and 200 ng/ml Cdt, these cells secreted 48 ± 26 and 43 ± 15 pg/ml IL-1β as well as 58 ± 14 and 82 ± 11 pg/ml IL-18, respectively. As expected, these cells were significantly impaired in their ability to release caspase-1 (Fig. 2C).
FIG 2.

Cdt-induced cytokine release from THP-1-derived macrophages involves activation of the NLRP3 inflammasome. THP-1 cells were stably transfected with an shRNA that targets caspase-1, NLRP3, or ASC or with nontargeted control shRNA. (A to C) The effect of protein knockdown in cells exposed to 0 to 200 ng/ml Cdt was determined, and culture supernatants were analyzed for IL-1β (A), IL-18 (B), and caspase-1 (p20) (C) release by an ELISA. Results are the means ± standard errors of the means for three experiments performed in triplicate; asterisks indicate a statistically significant difference from the shRNA control P ≤ 0.05). (D) Cells were also analyzed by Western blotting for the presence of pro-IL-1β. Actin is shown as a loading control. WT, wild type.
Generation of the active form of caspase-1 is dependent upon the activation of the inflammasome, a multiprotein complex consisting of a nucleotide-binding oligomerization domain-like receptor (NLR) and an adaptor molecule, such as ASC, along with procaspase-1 (35, 36). We focused on the NLRP3 inflammasome and determined the requirement for NLRP3 and ASC by also depleting THP-1 cells of these components with specific shRNAs; these cells have been well characterized for reductions in expression levels (see Materials and Methods) (29, 32). Macrophages derived from NLRP3-deficient cells produced significantly reduced levels of cytokines relative to the levels observed in shRNA control cells (Fig. 2A and B). In the presence of 40 and 200 ng/ml Cdt, macrophages derived from the NLRP3-depleted cells released 108 ± 16 and 172 ± 57 pg/ml IL-1β as well as 56 ± 8 and 88 ± 13 pg/ml IL-18, respectively. Similarly, macrophages derived from ASC-depleted THP-1 cells released reduced levels of IL-1β and IL-18; exposure to 40 and 200 ng/ml Cdt resulted in the release of 172 ± 61 and 229 ± 92 pg/ml IL-1β along with 54 ± 7 and 77 ± 11 pg/ml IL-18, respectively. In order to confirm that the reduced expression of inflammasome components also resulted in a concomitant effect on Cdt-induced caspase activation, we monitored cells for the secretion of caspase-1 (p20). As shown in Fig. 2C, macrophages derived from both wild-type THP-1 and shCtl cells and exposed to 200 ng/ml Cdt released 77.4 ± 4.7 and 61.0 ± 2.3 pg/ml caspase-1, respectively, while untreated control cells released 6 ± 1 pg/ml (wild type) and 4 ± 1 pg/ml (shCtl). Macrophages derived from THP-1 cells depleted of NLRP3 or ASC exhibited significant reductions in caspase-1 release in the presence of 200 ng/ml Cdt, 24 ± 1.6 and 19 ± 1.5 pg/ml, respectively. Moreover, we confirmed that Cdt-induced expression of pro-IL-1β was not affected by the disruption of the inflammasome and reduced caspase-1 activation (Fig. 2D).
Activation of the NLRP3 inflammasome has been linked to a number of upstream events, which include, among others, reactive oxygen species (ROS) generation, K+ efflux, and increases in extracellular ATP (37–40). To determine if Cdt-induced activation of the inflammasome involves ROS, we employed the antioxidant N-acetylcysteine (NAC) and the NADPH oxidase inhibitor diphenylene iodonium (DPI). Pretreatment of THP-1-derived macrophages with 10 mM NAC followed by Cdt (50 ng/ml) resulted in reductions in the secretion of both IL-1β (174 ± 43 pg/ml) and IL-18 (103 ± 10 pg/ml) compared with cells treated with toxin alone, which released 384 ± 43 pg/ml IL-1β and 325 ± 8 pg/ml IL-18 (Fig. 3A). Furthermore, cells pretreated with 250 nM DPI also exhibited reductions in the secretion of both IL-1β (64 ± 35 pg/ml) and IL-18 (131 ± 16pg/ml). In addition to inhibiting the release of mature cytokines, these agents also reduced inflammasome activation, which was reflected by a decline in the release of caspase-1 (p20); as shown in Fig. 3B, cells pretreated with NAC and DPI released 17 ± 3 and 13 ± 1 pg/ml caspase-1 (p20), respectively, versus 53 ± 6 pg/ml in cells treated with Cdt alone. Moreover, these ROS inhibitors had no effect on the Cdt-induced synthesis of pro-IL-1β (Fig. 3C).
FIG 3.
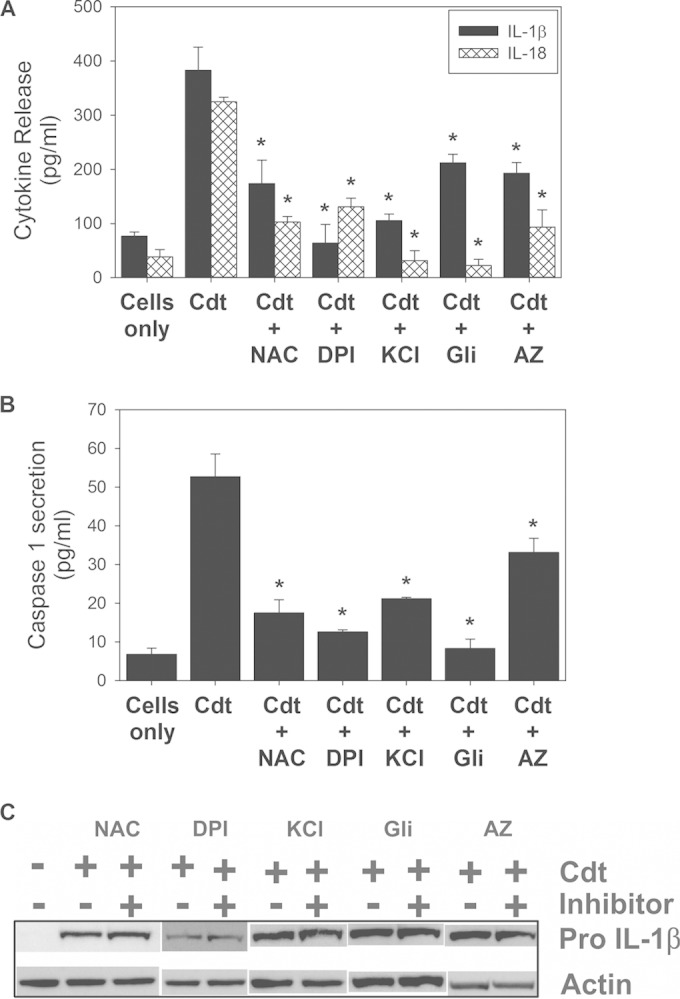
Cdt-induced inflammasome activation involves ROS, K+ efflux, and extracellular ATP. NAC (10 mM) and DPI (250 nM) were used to determine the requirement for ROS, elevated extracellular levels of K+ (70 mM) and glibenclamide (Gli) (50 μg/ml) were used to determine the requirement for K+ efflux, and AZ11645373 (AZ) (1 μM) was used to determine the requirement for an ATP-PTXR7 interaction. (A and B) Effects of these agents on Cdt (50 ng/ml)-induced production of IL-1β and IL-18 (A) and on caspase-1 release (B). Results are the means ± standard errors of the means for three experiments, each performed in triplicate; asterisks indicate statistical significance compared to the Cdt-only control (P ≤ 0.05). (C) Cells were also analyzed for the presence of pro-IL-1β by Western blotting. Actin is shown as a loading control.
K+ efflux was assessed by using two inhibitory approaches, increasing the extracellular levels of K+ with 70 mM KCl and with 50 μg/ml glibenclamide, a selective inhibitor of ATP-dependent potassium channels (Fig. 3A). THP-1-derived macrophages were treated with Cdt (50 ng/ml) in the presence of 70 mM KCl. Levels of secretion of both IL-1β and IL-18 were reduced to 105 ± 12 pg/ml and 31 ± 18 pg/ml, respectively, compared to 383 ± 43 pg/ml (IL-1β) and 325 ± 8 pg/ml (IL-18) in control cells treated with the toxin alone. Additionally, an inhibitor of ATP-dependent K+ channels, glibenclamide, reduced cytokine release as well, to 212 ± 16 pg/ml (IL-1β) and 22 ± 11 pg/ml (IL-18). In addition to their effects on cytokine release, these inhibitors also blocked the release of caspase-1 (p20), to 21 ± 0.4 pg/ml (KCl) and 8 ± 2 pg/ml (glibenclamide) (Fig. 3B); no effect on Cdt-induced pro-IL-1β levels was observed (Fig. 3C).
ATP has been implicated in the activation of the NLRP3 inflammasome via binding to the P2X7 purinergic receptor, which in turn results in ion permeability, including K+ efflux from the cytosol (31, 37, 39). The requirement for an ATP-P2X7 interaction was first assessed by using the P2X7 antagonist AZ11645373. As shown in Fig. 3A, pretreatment of macrophages with AZ11645373 resulted in reductions in cytokine release of 193 ± 19 pg/ml IL-1β and 93 ± 32 pg/ml IL-18. In addition to cytokine release, AZ11645373 treatment resulted in reduced caspase-1 release. Culture supernatants containing both the inhibitor and Cdt (50 ng/ml) contained 33 ± 4 pg/ml caspase-1 (Fig. 3B), compared to 53 ± 6 pg/ml in the Cdt control; no effect on Cdt-induced pro-IL-1β levels was observed (Fig. 3C). Similar results were observed for another P2X7 inhibitor, oxidized ATP (data not shown).
We next determined if Cdt treatment altered extracellular levels of ATP. As shown in Fig. 4A, supernatants obtained from cells exposed to 100 ng/ml Cdt contained 0.48 ± 0.05 μM ATP, while supernatants from control cells contained 0.25 ± 0.03 μM; these values increased to 0.61 ± 0.09 μM in the presence of 200 ng/ml Cdt. In order to demonstrate that extracellular ATP is required for inflammasome activation, we utilized the enzyme apyrase. We first demonstrated that the addition of apyrase to THP-1-derived macrophage cultures reduces the extracellular ATP burden (Fig. 4B). Cells treated with Cdt (200 ng/ml) exhibited an increase in the concentration of extracellular ATP to 0.76 ± 0.1 μM over that observed with untreated cells (0.39 ± 0.05 μM); in the presence of apyrase (1 U will liberate 1.0 μmol of inorganic phosphate from ATP or ADP per min at pH 6.5 at 30°C), ATP levels were reduced in a dose-dependent manner, to 0.34 ± 0.02 μM (1 mU), 0.12 ± 0.02 μM (10 mU), and 0.003 ± 0.006 μM (100 mU). The supernatants were also assessed for IL-1β and caspase-1 (p20). As shown in Fig. 4C, treatment of cells with 200 ng/ml Cdt resulted in an increase in IL-1β secretion to 474 ± 9.4 pg/ml, in contrast to 50 ± 16.9 pg/ml in untreated cells. The addition of 1 mU apyrase to Cdt-treated cells resulted in reduced IL-1β release, to 70 ± 9.4 pg/ml; treatment with 100 mU apyrase further reduced the cytokine level to 44.1 ± 5.6 pg/ml. Apyrase had a similar effect on caspase-1 release, which was not detected at any enzyme concentration used, while supernatants from Cdt-treated cells contained 87 ± 2.2 pg/ml.
FIG 4.
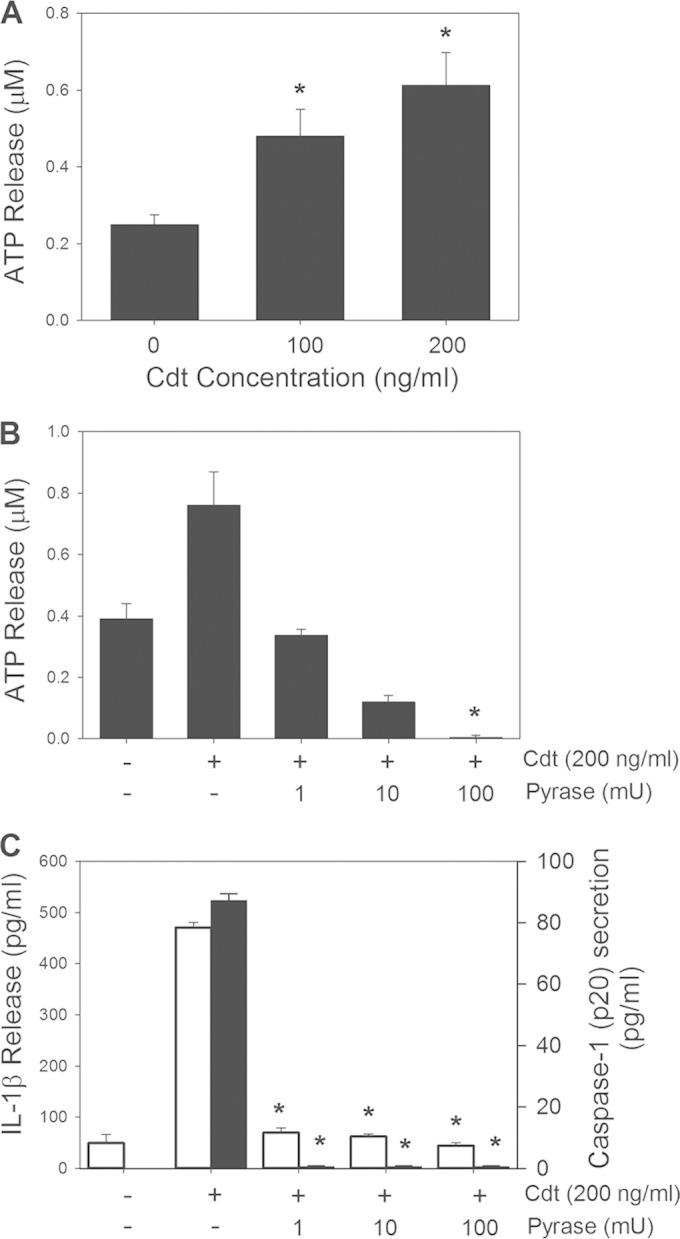
The proinflammatory response induced by Cdt is dependent upon extracellular ATP. (A) Macrophages were treated with 0 to 200 ng/ml Cdt for 2 h, and supernatants were then analyzed for ATP. (B and C) Cells were also analyzed for the effect of apyrase (Pyrase) on extracellular ATP levels (B) as well as IL-1β release (open bars) and caspase-1 release (solid bars) (C). Results represent the means ± standard errors of the means for three experiments, each performed in triplicate; asterisks indicate statistical significance compared to control cells (P ≤ 0.05).
We previously demonstrated that the proinflammatory cytokine response induced by Cdt was mediated at the molecular level by the action of the active Cdt subunit CdtB, functioning as a PIP3 phosphatase. PIP3 depletion led to a blockade of the PI-3K signaling pathway, decreased phosphorylation of GSK3β, and a concomitant activation of this kinase. In those studies, GSK3β inhibitors were effectively used to block the release of proinflammatory cytokines. Therefore, in a final series of experiments, we employed three kinase inhibitors to determine if Cdt-induced activation of GSK3β was responsible for both the expression of pro-IL-1β as well as increases in extracellular ATP levels. As shown in Fig. 5A, macrophages preexposed to each of the GSK3β inhibitors, 10 μM GSK inhibitor X, 50 μM GSK inhibitor XII, and 1 μM GSK inhibitor XV, exhibited a reduction in the pro-IL-1β expression level relative to that in cells treated with the toxin alone. The GSK inhibitors were also assessed for their ability to alter Cdt-induced increases in extracellular ATP levels. Supernatants obtained from cells exposed to Cdt alone contained 0.57 ± 0.03 μM ATP, versus 0.35 ± 0.04 μM in control cells. In contrast, cells pretreated with each of the inhibitors exhibited significant reductions in the levels of extracellular ATP: 0.34 ± 0.04 μM (GSK inhibitor X), 0.33 ± 0.02 μM (GSK inhibitor XII), and 0.35 ± 0.02 μM (GSK inhibitor XV).
FIG 5.
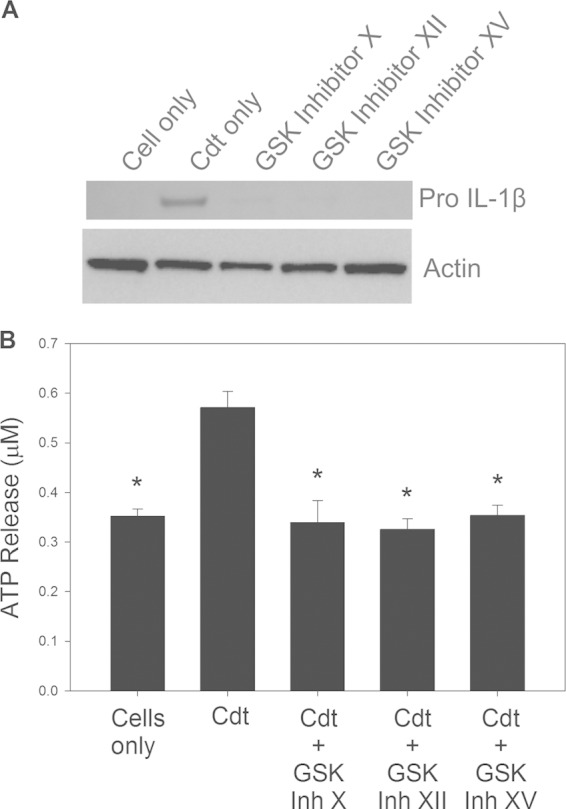
GSK3β activation contributes to Cdt-induced expression of pro-IL-1β and increased extracellular ATP levels. (A) THP-1-derived macrophages were pretreated with GSK inhibitors for 1 h, followed by the addition of 200 ng/ml Cdt. After 4 h, cells were fractionated by SDS-PAGE and analyzed by Western blotting for pro-IL-1β. Results are representative of three experiments. (B) Macrophages were pretreated with GSK inhibitors as described above and then treated with 200 ng/ml Cdt for 4 h. Supernatants were analyzed for ATP as described in Materials and Methods. Results represent the means ± standard errors of the means for three experiments; asterisks indicate statistical significance compared to control cells treated with Cdt alone (P ≤ 0.05).
DISCUSSION
In previous studies, we demonstrated that monocytes as well as macrophages derived from either human monocytes or the acute monocytic leukemia cell line THP-1 exhibit increases in cytokine gene expression and protein secretion following exposure to A. actinomycetemcomitans Cdt (19). Specifically, Cdt-treated macrophages were induced to produce IL-1β, TNF-α, and IL-6 within 5 h; we now show that IL-18 is also released by macrophages exposed to toxin for 48 h. Furthermore, previous studies demonstrated that Cdt-induced cytokine release was dependent upon the active Cdt subunit CdtB and its ability to function as a PIP3 phosphatase. This activity led to a perturbation of PI-3K signaling, resulting in decreased PIP3 levels and reduced phosphorylation of Akt and GSK3β. Decreased GSK3β phosphorylation resulted in increases in its kinase activity; moreover, the ability of Cdt to induce cytokine release was reduced when it was added to cells in the presence of GSK3β inhibitors. It should also be noted that CdtB mutants deficient in lipid phosphatase activity yet retaining DNase activity failed to induce cytokine release, as did mutants lacking both enzymatic activities (19).
IL-1β and IL-18 are synthesized as inactive precursors that require proteolytic cleavage for activation and secretion, resulting in the generation of mature forms with potent proinflammatory activities. In this study, we explored the events that govern the maturation of IL-1β and IL-18 following exposure of macrophages to Cdt. A key event in this process is the activation of caspase-1, which is also synthesized as an inactive proenzyme. Procaspase-1 requires further processing involving dimerization and autoproteolytic cleavage, which results in the generation of the subunits p20 and p10. Indeed, we demonstrate that Cdt induces time-dependent caspase-1 activation. Also, macrophage culture supernatants are typically utilized to detect the mature caspase-1 subunits, as they are not only found in the cytoplasm but also secreted along with active cytokines (30, 41). Thus, it was not surprising that Cdt-treated macrophages released caspase-1 at elevated levels, which were dependent upon the Cdt concentration. Furthermore, pretreatment of cells with the caspase-1 inhibitor Z-WEHD-FMK blocked the Cdt-induced release of both IL-1β and IL-18 but did not affect Cdt-induced expression of pro-IL-1β. In addition to using the caspase-1 inhibitor, we also confirmed that Cdt-induced IL-1β and IL-18 release was caspase-1 dependent by silencing its expression in THP-1 cells using RNA interference. Cdt-treated caspase-1 knockdown cells exhibited significantly reduced levels of caspase-1 release; additionally, a concomitant reduction in IL-1β and IL-18 release was also observed in these cells. It should be noted that the caspase-1-deficient cells were not altered in their ability to synthesize pro-IL-1β in response to Cdt.
Caspase-1 activation is dependent upon the formation of multiprotein complexes known as inflammasomes (35, 36). Typically, the inflammasome is composed of a receptor or sensor, such as the nucleotide-binding domain leucine-rich repeat (NLR) protein, and an adaptor molecule, such as apoptosis-associated speck-like protein containing a CARD (caspase recruitment domain) (ASC), that tethers the complex to procaspase-1. The group of human NLR proteins contains several members, 14 of which belong to the NLR protein family; one member of this family, NLRP3, is a component of one of the best-studied inflammasome complexes and has also been reported to be activated by microbial-derived ligands, including toxins (reviewed in references 36 and 42). Furthermore, it has been shown that exposure of human monocytes to A. actinomycetemcomitans results in the upregulation of NLRP3, regardless of whether the bacteria were able to express toxins such as Cdt and the leukotoxin; however, that report did not indicate whether the inflammasome was activated or if mature cytokines were secreted (43). Therefore, we chose to initially focus our studies on the role of the NLRP3 inflammasome and determine if, in addition to caspase-1, NLRP3 as well as the adaptor molecule ASC are required for Cdt-induced caspase-1 activation and cytokine release. Indeed, silencing of the expression of either NLRP3 or ASC confirmed the requirement for these components in the Cdt-induced proinflammatory response. We observed a significant reduction in the release of caspase-1 following treatment of macrophages with Cdt; moreover, the release of both IL-1β and IL-18 was also reduced. It should be noted that the ASC- and NLRP3-deficient cells were not impaired in their ability to upregulate the synthesis of pro-IL-1β in response to Cdt.
Interestingly, the Cdt-induced release of TNF-α was partially reduced (∼50%) by pretreatment with the caspase-1 inhibitor. Moreover, cells transfected with shRNA for caspase-1, ASC, or NLRP3 also exhibited a reduction in TNF-α release (∼80%) in response to the toxin. The reduced release of TNF-α was unexpected, as this cytokine typically does not require further processing by caspase-1. These results suggest that a portion of Cdt-induced TNF-α release may be the result of toxin-induced IL-1β production and its action on autocrine networks. In this context, it should be noted that several investigators have demonstrated cross-regulation between IL-1β/IL-18 processing and the processing of other inflammatory cytokines (44). For example, it has been shown that ASC is responsible for the induced expression of several cytokines, including TNF-α. It is not clear at this time if any of these cross-regulatory events are involved in the proinflammatory cytokine response induced by Cdt.
Inflammasome activation and the subsequent release of caspase-1, IL-1β, and IL-18 require additional signals, often referred to as danger signals, from stressed or infected cells. Of particular interest is the role of extracellular ATP, which is involved in intercellular communication that involves both lytic and nonlytic stimuli; in this capacity, ATP serves as an agonist for P2 nucleotide receptors such as P2X7 (reviewed in references 45 and 46). The P2X7 receptor is highly expressed on monocytes, macrophages, and lymphocytes, and its activation following an association with ATP contributes to the assembly of the NLRP3 inflammasome (37, 47). Thus, our observations are consistent with a mechanism involving Cdt-induced ATP-dependent activation of the purinergic receptor, as not only do toxin-treated cells exhibit increased levels of extracellular ATP, but the addition of apyrase also reduces these ATP levels as well as the release of caspase-1 and mature IL-1β. Moreover, pretreatment of cells with the P2X7 receptor inhibitor AZ11645373 reduced both caspase-1 release and the secretion of IL-1β and IL-18; no effect on the toxin-induced expression of pro-IL-1β was observed. Similar results were also observed with oxidized ATP, another P2X7 inhibitor (data not shown).
It is noteworthy that the NLRs were originally proposed to serve as sensors of activating signals such as microbial ligands; however, their role has been expanded to include serving as switches for cellular signals in response to stress resulting from infection (reviewed in references 35 and 36). Furthermore, it has been proposed that NLRP3 is capable of sensing imbalances in the concentrations of ROS and intracellular ions, in particular K+. Thus, Cdt-mediated increases in extracellular ATP levels and ATP-induced P2X7 activation likely contribute to NLRP3 assembly and activation through ROS generation and/or reduced intracellular concentrations of K+. To address these possibilities, we employed high levels of extracellular K+ as well as the K+ channel blocker glibenclamide to impede ion efflux. Our results clearly demonstrate that potassium efflux is critical to the Cdt-induced proinflammatory response, as inhibition by both agents prevented the activation of caspase-1 and the release of IL-1β and IL-18. These inhibitors did not interfere with Cdt-induced expression of pro-IL-1β. As ROS are typically generated from the mitochondrial electron transport chain and/or plasma membrane-associated NADPH oxidase, two inhibitors were employed to demonstrate that ROS generation was involved in the Cdt-induced activation of caspase-1 and the release of cytokines: DPI, an inhibitor of NADPH oxidase, and NAC, an antioxidant. Both agents reduced the release of caspase-1 as well as IL-1β and IL-18; the Cdt-induced expression of pro-IL-1β was not affected by these agents. Since reduced intracellular levels of K+ have been linked to triggering of ROS generation (31), we also assessed the effect of Cdt on macrophage ROS generation. While THP-1-derived macrophages constitutively generate low levels of ROS, Cdt treatment did not alter these levels (data not shown). Thus, it appears that endogenous levels of ROS along with ATP-dependent reductions in the intracellular levels of K+ contribute to NLRP3 inflammasome activation in the presence of Cdt.
We propose that Cdt induces a proinflammatory cytokine response via a two-step process, as summarized in Fig. 6. First, exposure to the toxin results in upregulation of both cytokine gene expression and protein synthesis, which results in elevated intracellular levels of both pro-IL-1β and IL-18 as well as TNF-α and IL-6. As previously demonstrated and as corroborated by this study, this response is dependent upon the active toxin subunit CdtB functioning as a PIP3 phosphatase. Depletion of PIP3 results in a blockade of the PI-3K signaling cascade and, in particular, a decrease in the phosphorylation of GSK3β and a concomitant increase in its kinase activity (19). Activation of GSK3β is critical for NF-κB activation and expression of proinflammatory cytokines. Although these events are dependent upon GSK3β activation, they alone are not sufficient to induce the maturation and release of IL-1β and IL-18. Thus, we further propose that a second signal is required, which involves the activation of the NLRP3 inflammasome. Our studies support a model in which Cdt-mediated activation of the inflammasome is dependent upon toxin-induced increases in extracellular ATP levels; moreover, these observations suggest that the increases in ATP levels are also dependent upon Cdt-induced GSK3β activation. Furthermore, we propose that the increase in the extracellular ATP level contributes to the activation of the purinergic receptor P2X7 and K+ efflux. These events, together with the endogenous generation of ROS, lead to the activation of the NLRP3 inflammasome and the release of mature IL-1β and IL-18.
FIG 6.

Summary diagram depicting the mechanism by which Cdt induces a proinflammatory cytokine response in macrophages. Two signals are proposed. The first signal (pink arrows) involves the upregulation of inflammatory cytokine gene and protein expression; this signal is dependent upon Cdt's abilities to function as a PIP3 phosphatase, block PI-3K signaling, and activate GSK3β, leading to the expression and synthesis of pro-IL-1β and IL-18. The second signal (blue arrows) involves the activation of the NLRP3 inflammasome, which requires the generation of extracellular ATP, an ATP-P2X7 interaction, K+ efflux, and utilization of endogenous ROS. Inhibitors of GSK3β also block ATP generation, suggesting that Cdt-induced activation of this kinase is also critical to ATP-dependent inflammasome activation.
It should be noted that in addition to lipid phosphatase activity, CdtB also exhibits DNase activity. While PIP3 phosphatase activity is almost comparable to those of other lipid phosphatases, its DNase activity is considerably less than that associated with DNase I (18). Nonetheless, DNase activity has been shown to be critical for toxicity to a number of cell types, such as CHO and HeLa cells, among others, which typically require high doses of toxin (1, 7). Lipid phosphatase activity is critical to lymphocyte toxicity, which requires low doses of toxin (18). Macrophages, which are not killed by Cdt, exhibit a proinflammatory cytokine response when exposed to toxin; this response to Cdt is dependent upon PIP3 phosphatase activity. We have previously demonstrated that Cdt induces a PI-3K blockade due to PIP3 depletion in macrophages, and furthermore, the proinflammatory response was eliminated with CdtB mutants lacking lipid phosphatase activity (19). Moreover, the Cdt-induced cytokine response from macrophages requires GSK3β activation, a result of a PI-3K signaling blockade (19). Consistent with this notion, we demonstrate that GSK3β inhibitors block the proinflammatory response, including synthesis of both cytokines and ATP.
Collectively, our results are consistent with the clinical picture associated with A. actinomycetemcomitans infection and periodontal disease, which are characterized by the presence of this organism in large numbers in biofilms associated with tooth surfaces, inflammation, and the destruction of periodontal tissue (48–51). We now propose that a significant contributing factor to A. actinomycetemcomitans-induced disease is derived from the proinflammatory actions of Cdt. However, it is not clear if these actions contribute to host defense and thereby serve to limit disease progression and/or severity or whether Cdt represents a virulence factor contributing to inflammation and tissue injury. It is also likely that a Cdt-mediated proinflammatory response could also promote and sustain infection by providing degraded tissue protein fragments and hemin that fuel the nutritional needs of bacteria (51). In conclusion, it is likely that these observations have implications not only for the pathogenesis of disease caused by A. actinomycetemcomitans but also for other infectious and chronic inflammatory disorders that occur in association with the wide range of Cdt-producing pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants DE06014, DE023071, and DE022465.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.03132-14.
REFERENCES
- 1.Comayras C, Tasca C, Peres SY, Ducommun B, Oswald E, De Rycke J. 1997. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun 65:5088–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okuda J, Fukumoto M, Takeda Y, Nishibuchi M. 1997. Examination of diarrheagenicity of cytolethal distending toxin: suckling mouse response to the products of the cdtABC genes of Shigella dysenteriae. Infect Immun 65:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okuda J, Kurazono H, Takeda Y. 1995. Distribution of the cytolethal distending toxin A gene (cdtA) among species of Shigella and Vibrio, and cloning and sequencing of the cdt gene from Shigella dysenteriae. Microb Pathog 18:167–172. [DOI] [PubMed] [Google Scholar]
- 4.Scott DA, Kaper JB. 1994. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun 62:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickett CL, Cottle DL, Pesci EC, Bikah G. 1994. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect Immun 62:1046–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer MP, Bueno LC, Hansen EJ, DiRienzo JM. 1999. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun 67:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickett CL, Whitehouse CA. 1999. The cytolethal distending toxin family. Trends Microbiol 7:292–297. doi: 10.1016/S0966-842X(99)01537-1. [DOI] [PubMed] [Google Scholar]
- 8.Shenker BJ, McKay TL, Datar S, Miller M, Chowhan R, Demuth DR. 1999. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J Immunol 162:4773–4780. [PubMed] [Google Scholar]
- 9.Shenker BJ, Hoffmaster RH, McKay TL, Demuth DR. 2000. Expression of the cytolethal distending toxin (Cdt) operon in Actinobacillus actinomycetemcomitans: evidence that the CdtB protein is responsible for G2 arrest of the cell cycle in human T-cells. J Immunol 165:2612–2618. doi: 10.4049/jimmunol.165.5.2612. [DOI] [PubMed] [Google Scholar]
- 10.Shenker BJ, Hoffmaster RH, Zekavat A, Yamguchi N, Lally ET, Demuth DR. 2001. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J Immunol 167:435–441. doi: 10.4049/jimmunol.167.1.435. [DOI] [PubMed] [Google Scholar]
- 11.De Rycke J, Oswald E. 2001. Cytolethal distending toxin (CDT): a bacterial weapon to control host cell proliferaton? FEMS Microbiol Lett 203:141–148. doi: 10.1111/j.1574-6968.2001.tb10832.x. [DOI] [PubMed] [Google Scholar]
- 12.Thelestam M, Frisan T. 2004. Cytolethal distending toxins. Rev Physiol Biochem Pharmacol 152:111–133. doi: 10.1007/s10254-004-0030-8. [DOI] [PubMed] [Google Scholar]
- 13.Nesic D, Hsu Y, Stebbins CE. 2004. Assembly and function of a bacterial genotoxin. Nature 429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- 14.Elwell CA, Chao K, Patel K, Dreyfus LA. 2001. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun 69:3418–3422. doi: 10.1128/IAI.69.5.3418-3422.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lara-Tejero M, Galan JE. 2001. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun 69:4358–4365. doi: 10.1128/IAI.69.7.4358-4365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boesze-Battaglia K, Besack D, McKay TL, Zekavat A, Otis L, Jordan-Sciutto K, Shenker BJ. 2006. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cell Microbiol 8:823–836. doi: 10.1111/j.1462-5822.2005.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boesze-Battaglia K, Brown A, Walker L, Besack D, Zekavat A, Wrenn S, Krummenacher C, Shenker BJ. 2009. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J Biol Chem 284:10650–10658. doi: 10.1074/jbc.M809094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shenker BJ, Dlakic M, Walker L, Besack D, Jaffe E, Labelle E, Boesze-Battaglia K. 2007. A novel mode of action for a microbial-derived immunotoxin: the cytolethal distending toxin subunit B exhibits phosphatidylinositol 3,4,5-triphosphate phosphatase activity. J Immunol 178:5099–5108. doi: 10.4049/jimmunol.178.8.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shenker B, Walker L, Zekavat A, Dlakic M, Boesze-Battaglia K. 2014. Blockade of the PI-3K signaling pathway by the Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces macrophages to synthesize and secrete pro-inflammatory cytokines. Cell Microbiol 16:1391–1404. doi: 10.1111/cmi.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antoniv T, Ivashkiv L. 2011. Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology 132:567–577. doi: 10.1111/j.1365-2567.2010.03402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arbibe L, Mira J-P, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Knaus U. 2000. Toll-like receptor 2-mediated NF-κB activation requires a Rac1-dependent pathway. Nat Immunol 1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 22.Brown J, Wang H, Hajishengallis H, Martin M. 2011. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 90:417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guha M, Mackman N. 2002. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 24.Hazeki K, Nigorikawa K, Hazeki O. 2007. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull 30:1617–1623. doi: 10.1248/bpb.30.1617. [DOI] [PubMed] [Google Scholar]
- 25.Molnarfi N, Gruaz L, Dayer J, Burger D. 2007. Opposite regulation of IL-1β and secreted IL-1 receptor antagonist production by phophatidylinositide-kinases in human monocytes activated by lipopolysaccharides or contact with T cells. J Immunol 178:446–454. doi: 10.4049/jimmunol.178.1.446. [DOI] [PubMed] [Google Scholar]
- 26.Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M, Takeuchi T, Matsuda S, Koyasu S. 2008. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood 112:635–643. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Garcia C, Rehani K, Cekic C, Alard P, Kinane D, Mitchell T, Martin M. 2008. IFN-β production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-β. J Immunol 181:6797–6802. doi: 10.4049/jimmunol.181.10.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier K, Kolbe T, Stulnig T, Horl W, Hengstschlager M, Muller M. 2008. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Said-Sadier N, Padilla E, Langsley G, Ojcius D. 2010. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 5:e10008. doi: 10.1371/journal.pone.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdul-Sater A, Said-Sadier N, Padilla E, Ojcius D. 2010. Chlamydial infection of monocytes stimulates IL-1β secretion through activation of the NLRP3 inflammasome. Microbes Infect 12:652–661. doi: 10.1016/j.micinf.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C, Tsai S, Hu S, Wu T, Huang T, Said-Sadier N, Ojcius D, Lai H. 2012. Activation of an NLRP3 inflammasome restricts Mycobacterium kansasii infection. PLoS One 7:e36292. doi: 10.1371/journal.pone.0036292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu Y, Jiang J, Said-Sadier N, Boxx G, Champion C, Tetlow A, Kickhoefer V, Rome L, Ojcius D, Kelly K. 2015. Activation of the NLRP3 inflammasome by vault nanoparticles expressing a chlamydial epitope. Vaccine 33:298–306. doi: 10.1016/j.vaccine.2014.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shenker BJ, Besack D, McKay TL, Pankoski L, Zekavat A, Demuth DR. 2004. Actinobacillus actinomycetemcomitans cytolethal distending toxin (Cdt). Evidence that the holotoxin is composed of three subunits: CdtA, CdtB, and CdtC. J Immunol 172:410–417. doi: 10.4049/jimmunol.172.1.410. [DOI] [PubMed] [Google Scholar]
- 34.Shenker B, Boesze-Battaglia K, Zekavat A, Walker L, Besack D, Ali H. 2010. Inhibition of mast cell degranulaton by a chimeric toxin containing a novel phsphatidylinositol-3,4,5-triphosphate phosphatase. Mol Immunol 48:203–210. doi: 10.1016/j.molimm.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariathasan S, Monack D. 2007. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 36.Pedra J, Cassel S, Sutterwala F. 2009. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol 21:10–16. doi: 10.1016/j.coi.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang T, Ojcius D, Young J, Wu Y, Ko Y, Wong T, Wu C, Lu C, Lai H. 2012. The anti-tumorigenic mushroom Agaricus blazei Murill enhances IL-1β production and activates the NLRP3 inflammasome in human macrophages. PLoS One 7:e41383. doi: 10.1371/journal.pone.0041383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JW, Dinarello CA. 2009. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood 113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yilmaz O, Sater A, Yao L, Koutouzis T, Pettengill M, Ojcius D. 2010. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol 12:188–198. doi: 10.1111/j.1462-5822.2009.01390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bratel J, Haraldson T, Meding B, Yontchev E, Ohman S-C, Ottosson J-O. 1997. Potential side effects of dental amalgam restorations. (I) An oral and medical investigation. Eur J Oral Sci 105:234–243. [DOI] [PubMed] [Google Scholar]
- 41.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. 2010. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franchi L, Munoz-Planillo R, Nunez G. 2012. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belibasakis G, Johansson A. 2012. Aggregatibacter actinomycetemcomitans targets NLRP3 and NLRP6 inflammasome expression in human mononuclear leukocytes. Cytokine 59:124–130. doi: 10.1016/j.cyto.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 44.Barker B, Taxman D, Ting J. 2011. Cross-regulation between the IL-1β/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol 23:591–597. doi: 10.1016/j.coi.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubyak G. 2012. P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell Microbiol 14:1697–1706. doi: 10.1111/cmi.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ciraci C, Janczy J, Sutterwala F, Cassel S. 2012. Control of innate and adaptive immunity by the inflammasome. Microbes Infect 14:1263–1270. doi: 10.1016/j.micinf.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 48.Andrukhov O, Ulm C, Reischl H, Nguyen P, Matejka M, Rausch-Fan X. 2011. Serum cytokine levels in periodontitis patients in relation to the bacterial load. J Periodontol 82:885–892. doi: 10.1902/jop.2010.100425. [DOI] [PubMed] [Google Scholar]
- 49.de Brito Bezerra B, Andriankaja O, Kang J, Pacios S, Bae HJ, Li Y, Tslagbe V, Schreiner H, Fine DH, Graves DT. 2012. A. actinomycetemcomitans-induced periodontal disease promotes systemic and local responses in rat periodontium. J Clin Periodontol 39:333–341. doi: 10.1111/j.1600-051X.2011.01847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. 2011. Impaired phagocytosis in localized aggressive periodontitis: rescue by resolvin E1. PLoS One 6:e24422. doi: 10.1371/journal.pone.0024422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hajishengallis G, Darveau R, Curtis M. 2012. The keystone-pathogen hypothesis. Nat Rev Microbiol 10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.