

Abstract
Athletic performance is an important criteria used for the selection of superior horses. However, little is known about exercise-related epigenetic processes in the horse. DNA methylation is a key mechanism for regulating gene expression in response to environmental changes. We carried out comparative genomic analysis of genome-wide DNA methylation profiles in the blood samples of two different thoroughbred horses before and after exercise by methylated-DNA immunoprecipitation sequencing (MeDIP-Seq). Differentially methylated regions (DMRs) in the pre-and post-exercise blood samples of superior and inferior horses were identified. Exercise altered the methylation patterns. After 30 min of exercise, 596 genes were hypomethylated and 715 genes were hypermethylated in the superior horse, whereas in the inferior horse, 868 genes were hypomethylated and 794 genes were hypermethylated. These genes were analyzed based on gene ontology (GO) annotations and the exercise-related pathway patterns in the two horses were compared. After exercise, gene regions related to cell division and adhesion were hypermethylated in the superior horse, whereas regions related to cell signaling and transport were hypermethylated in the inferior horse. Analysis of the distribution of methylated CpG islands confirmed the hypomethylation in the gene-body methylation regions after exercise. The methylation patterns of transposable elements also changed after exercise. Long interspersed nuclear elements (LINEs) showed abundance of DMRs. Collectively, our results serve as a basis to study exercise-based reprogramming of epigenetic traits.
Keywords: DNA methylation, exercise, MeDIP-Seq, thoroughbred horse, transposable elements
INTRODUCTION
In England, thoroughbred horses have been bred to improve their speed, stamina, and agility for the past 400 years. Thoroughbred horses possess several specific genetic features associated with their racing ability, including single nucleotide polymorphisms (SNPs) and gene expression of variant transcripts as adaptive responses to exercise (Kim et al., 2013a; Petersen et al., 2013). Transcript expression of these gene variants in thoroughbred horses results in distinguished phenotypes such as sprint, endurance, and hardiness (Hill et al., 2010a; 2010b; Petersen et al., 2013; Suontama et al., 2013). Thoroughbred horses have high oxygen and energy supply demands during exercise (Dempsey and Wagner, 1999; Eivers et al., 2010; Park et al., 2012). These findings provide a biological model to study the exercise-induced epigenetic processes that occur in the blood. To date, many traits have been identified and are used to select superior racehorse breeds (Dempsey and Wagner, 1999; Eivers et al., 2010; Park et al., 2012). However, the roles of DNA methylation in the athletic performance of horses remain unknown.
The regulation and variation of gene expression play important roles in the athletic performance of horses (Eivers et al., 2010; 2012; McGivney et al., 2009; 2010). Appropriate transcriptional response of exercise-related genes and exercise-induced adaptive physiological changes are important determinants of racing ability. Comparison of gene expression profiles in pre- and post-exercise samples across species identified candidate genes that are likely to be important for improving athletic performance. These genes include those related to exercise metabolism (TNNC2, ACTN3, TPM1; upregulated after exercise in the equine skeletal muscle), glucose metabolism (PDK4, LDHA), angiogenesis (VEGFA; upregulated after exercise in the equine skeletal muscle), and glucose transport (SLC2A3, SLC2A4) (Barres et al., 2012; Eivers et al., 2010; Gim et al., 2014; Handschin et al., 2007a; Jemiolo and Trappe, 2004; McGivney et al., 2010). Previous reports were focused on the oxidative stress response (Eivers et al., 2012; Vider et al., 2001), fatigue recovery pathways (Norton and Layman, 2006), metabolic pathways (Burgomaster et al., 2008; Gim et al., 2014; Russell et al., 2005), and transcriptional regulation (Barres et al., 2012; Louis et al., 2007; Russell et al., 2005; Seip et al., 1995) associated with running ability or muscle-related diseases. Genome-wide analysis of gene expression profiles related to athletic performance identified many exercise-induced differentially expressed genes (Kim et al., 2013a; Park et al., 2012). However, epigenetic regulation of gene expression due to exercise in thoroughbred horses remains unknown. Although the genetic makeup of all cells in an organism is identical, various stimuli, including exercise, can induce epigenetic changes in certain cells. A recent study analyzed the exercise-driven epigenetic changes in humans and concluded that exercise induces acute gene expression together with a dynamic change in DNA hypomethylation on the promoters involved (Barres et al., 2012). However, these studies did not analyze the differences in methylation between athletically superior and inferior individuals. We studied whole-genome methylation patterns in preand post-exercise blood samples collected from superior and inferior horses.
Analysis of DNA methylation patterns provides insight into the regulation of gene expression. Epigenetic control of gene transcription plays important roles in X chromosome inactivation (Chang et al., 2006), embryonic development (Liang et al., 2011), genomic imprinting (Sha, 2008), and oncogenesis (Dawson and Kouzarides, 2012; Goeppert et al., 2014). A particular DNA methylation state is typically stable in the absence of environmental stimuli, but factors such as aging, development, and change of diet have the capacity to alter DNA methylation patterns in organisms (Jaenisch and Bird, 2003). Exercise can modify DNA methylation profiles to control glucose metabolism, recovery, and dissimilation of lactate (Barres et al., 2012). In each of these processes, DNA methyltransferases play important roles in the de novo methylation of genomic DNA by adding a methyl group to the fifth carbon of cytosine (Jair et al., 2006; Smith et al., 1992). Thus, methylation state is sustained in resting state, and environmental changes alter the methylation pattern to maintain homeostasis.
A number of techniques have been developed to analyze genome-wide DNA methylation profiles (Laird, 2010; Pomraning et al., 2009). Because blood samples are easy to obtain, methylation state of blood can be readily analyzed. In this respect, whole-genome methylation analysis of human blood has been used to identify cancer biomarkers (Bosviel et al., 2012; Dauksa et al., 2012; Hsiung et al., 2007). Different patterns of whole-genome DNA methylation have been detected in control and cancer cells, as well as in various sample conditions such as gender, race, and ages. Not only the promoter regions of oncogenes or tumor suppressor genes have different DNA methylation patterns, but also long interspersed elements (LINEs) and short interspersed elements (SINEs) show distinctive DNA methylation patterns in the blood of cancer patients (Dauksa et al., 2012). The methylation patterns of LINEs are a distinctive factor in human genomic DNA derived from blood. LINEs are highly methylated in the males compared to females (El-Maarri et al., 2007; 2011; Zhang et al., 2011), whereas these are differentially methylated among races (Zhang et al., 2011). After exercise, differences in the patterns of DNA methylation could be detected according to gender, racing ability, and other conditions. Intravascular hemolysis occurs in the blood of female thoroughbred horses after exercise, whereas this does not occur in males (Cywinska et al., 2011). However, studies analyzing whole-genome DNA methylation in blood in various cases are limited. Therefore, we chose blood samples to study differentially methylated regions (DMRs) in the genomes of thoroughbred horses between the two genders before and after exercise.
In the study reported here, using the methylated-DNA immunoprecipitation sequencing (MeDIP-Seq) method, we analyzed the genome-wide DNA methylation patterns in the blood samples from superior and inferior horses before and after exercise. We screened exercise-related genes and compared their methylation patterns. Our study reveals, for the first time, exercise-induced transient alterations in promoter methylation in thoroughbred horses, and identifies differences in methylation patterns between superior and male, and inferior and female horses.
MATERIALS AND METHODS
Ethics statement
All animal experiments were approved by the Institutional Animal Care and Use Committee of Pusan National University (approval number PNU-2013-0417). Sampling was carried out in the presence of a veterinarian and care was taken to minimize the suffering of horses.
DNA and RNA samples
Blood samples from two healthy retired racing horses were used for experiments. Information regarding the horses is listed in Supplementary Table 1. One horse (GEUMBIT SESANG) was superior and male, and the other (JIGUSANG SERYEOK) was inferior and female. Total blood samples were drawn from the jugular vein. A veterinarian collected blood immediately before and after exercise to avoid changes in DNA methylation caused by external factors. The before exercise samples were collected at pre-exercise rest in two horses. The horses were trotted for 30 min, and then two samples were immediately collected. A total of four samples were collected in vacutainers and stored at −80°C until analysis. We utilized the blood sampling procedure of a previous study, wherein the time interval was not considered as an external factor (Nostell et al., 2012). Genomic DNA was extracted from four different blood samples of the two horses using DNeasy Blood & Tissue Kit (QIAGEN; Germany) according to the manufacturer’s guidelines. The quality and concentration of DNA were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop; USA). The integrity and quality of all DNA samples were analyzed. DNA samples with an A260/A280 ratio of 1.6–2.2 and an A260/A230 ratio of >1.6 were chosen, and their main band was confirmed by agarose gel electrophoresis. Samples that satisfied all criteria were used for MeDIP-Seq and bisulfite-treatment experiments.
Total RNA was isolated from four blood samples from each horse using TRIzol (Invitrogen, USA) for gene expression validation experiments. Each sample was treated with Turbo DNA-free DNase (Ambion, USA) and quantified using NanoDrop ND-1000. Moloney murine leukemia virus reverse transcriptase with RNase inhibitor (Promega) was used to synthesize cDNA from 500 ng total RNA at an annealing temperature of 42°C.
Methylated DNA immunoprecipitation and sequencing
Genomic DNA was fragmented to 100–500 bp fragments by sonication. The end of each DNA fragment was ligated to a 3′-dA overhang. Illumina sequencing adapters were then ligated to the ends using the Paired-End DNA Sample Prep Kit (Illumina; USA). Double-stranded DNA was denatured and DNA fragments were immunoprecipitated using 5-methylcytosine antibody beads (Diagenode; USA). Quantitative real-time polymerase chain reaction (qPCR) analysis was performed to validate the quality of immunoprecipitated fragments. DNA fragments of 200–300 bp were excised from the gel and purified using a gel extraction kit (Qiagen). The extracted fragments were quantified using Quant-iT™ dsDNA High Sensitivity Assay Kit (Invitrogen; USA) on an Agilent 2100 Analyzer (Agilent Technologies; USA). After qPCR analysis, DNA libraries were subjected to paired-end sequencing with a 50-bp read length using the Illumina HiSeq 2000 platform (Illumina). After the completion of a sequencing run, raw image files were processed using Illumina Real-Time Analysis (RTA) for image analysis and base calling. Sequencing reads were submitted to the NCBI Short Read Archive (SRA) under an SRA accession.
Bisulfite sequencing
To validate the methylation state of the methylation peak based on MeDIP-Seq data, bisulfite genomic sequencing PCR (BSP) was performed (Sigma, USA). We designed a bisulfite-modified PCR primer pair from horse genomic sequences for the promoter regions of three genes using the MethPrimer program (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi). The primer information is listed in Supplementary Table 2. PCR amplification was performed in each DNA sample using the following conditions: 94°C for 4 min, followed by 35 cycles of 94°C for 40 s, annealing at 55°C for 40 s, elongation at 72°C for 1 min, and final elongation at 72°C for 7 min using a Mastercycler® pro S thermal cycler (Eppendorf, Germany). The PCR products were separated on a 1.5% agarose gel, purified with a Labopass gel extraction kit (Cosmogenetech, Korea), and cloned into the pGEM-T Easy vector (Promega). Ten individual clones were sequenced by an automated 3730 DNA analyzer (Applied Biosystems, USA).
Real-time RT-PCR amplification
The total reaction mixture (15 μl) consisted of 7 μl H2O, 5 μl QuantiTech SYBR Green PCR master Mix (Qiagen), 1 μl each of forward and reverse primers, and 1 μl cDNA template. In addition, to confirm non-specific background amplification, we amplified a no template control (NTC). Real-time RT-PCR amplifications for target genes and housekeeping genes (GAPDH) were conducted as follows: 50 cycles of 94°C for 10 s, 58°C for 15 s, and 72°C for 15 s. Melting curve analysis was performed for 30 s at 55–99°C. Information on primers used for assessing expression level is given in Supplementary Table 2. To guarantee reproducibility, we amplified all samples in triplicate. We determined the level of significance (paired t-test) of expression for all samples.
Bioinformatics analysis
Raw sequence data were first processed to filter out adapters and low-quality reads, and then aligned to the horse reference genome (equCab2) using the SOAPaligner software (version 2.21) with mismatches of no more than 2 bp (Li et al., 2009). Uniquely mapped reads were retained for further analyses. To identify genomic regions that were enriched in a pool of specifically immunoprecipitated DNA fragments, genome-wide peak scanning was carried out by Model-based Analysis of ChIP-Seq (MACS, version 1.4.2) with a P-value threshold of 1 × 10−4 to exclude false positive peaks or noises (Zhang et al., 2008). In addition, an option of ‘--mfold’ to select the regions with MFOLD range of high-confidence enrichment ration against the background to build a model was used, with a lower limit of 10 and an upper limit of 30. The distribution of methylation peaks in different regions of the horse genome in each sample, including the promoter, 5′-untranslated region (UTR), 3′-UTR, exons, introns, intergenic regions, CpG islands (CGIs), and repeats, were analyzed. Methylated peaks corresponding to different genomic regions were selected by mapping at least 50% of the peaks on a particular genomic region. Methylated peaks overlapping at two different genomic regions (i.e., exon/intron) were preferentially selected by mapping at least 50% of the peaks to a particular genomic region. A CGI was defined using three criteria: greater than 200 bp in length, GC content ≥ 50 %, and CpG observed/expected ratio ≥ 0.6. The methylation densities in different regions of the genome were also compared.
To identify DMRs in the samples, their peaks were merged and the difference in the number of reads within those peaks between the two groups were analyzed using the negative binominal (NB) test (P < 0.01), applying DESeq coupled with DESeq normalization implemented in the R package TCC (Sun et al., 2013). In addition, DMRs with a greater than two-fold difference in read numbers were selected and classified as hyper- or hypo-methylated regions. The average DMR density was calculated by dividing each gene into 50 non-overlapping bins. All genes containing DMRs were used for subsequent gene ontology (GO) enrichment analyses using the DAVID Functional Annotation Tool with P < 0.05 (Huang da et al., 2009).
RESULTS
Genome-wide DNA methylation analyses of thoroughbred horse blood DNA
We performed MeDIP-Seq analysis of genomic DNA isolated from blood samples collected from two different horses, a superior male (GEUMBIT SESANG) and an inferior female (JIGU-SANG SERYEOK) (Supplementary Table 1). Up to 1.20 Gb of MeDIP-Seq data were generated. After the removal of adapter sequences, contamination, and low-quality reads from raw reads, the number of sequenced reads was 24 million on average (Supplementary Table 3). Total reads were mapped to the reference genome (equCab2) (Supplementary Table 4), and mapping rates of 84.68–86.99 % were obtained. The uniquely mapped rate was also detected at 71.05–75.71% of the total reads. Reads were distributed across g-nomic regions, showing good genome-wide coverage (Supplementary Fig. 1) (Chavez et al., 2010).
The regions enriched in methyl-cytosine are shown as highly methylated peaks. Methylation peaks are crucial parameters for analyzing global DNA methylation profiles (Hu et al., 2013; Yan et al., 2010). The peak coverage represented 3.05–3.83% of the reference genome (Supplementary Table 3), and the number of total predicted peaks was 81,140–105,303 (Table 1). We analyzed the distribution of peaks across different genomic regions in each sample. Intergenic regions showed more peaks than gene-body regions (Table 1). We confirmed the frequency of peaks in each region, including the promoter at the transcription start site (upstream 2 kb), 5′-UTR, exon, intron, 3′-UTR, downstream 2 kb at the transcription termination site, repeat, and other regions (Supplementary Table 5). Introns showed the most abundant peaks in each genomic region. Promoters showed relatively higher peak frequencies than other genomic regions (Supplementary Table 5), suggesting that promoter methylation plays a crucial role in the regulation of gene expression. The frequency of downstream DMRs was similar to that of the promoter (Supplementary Table 5). In addition, methylation peaks showed positive correlation with chromosomal length and gene number (Supplementary Fig. 2). We also predicted DMRs in two horses before and after exercise based on a two-fold change and P-value (Supplementary Fig. 3), and classified those DMRs into different genomic regions, including upstream (2 kb at transcription start site), 5′-UTR, exon, intron, 3′-UTR, and downstream (2 kb at transcription termination site) regions (Supplementary Table 6). To validate DNA methylation changes before and after exercise, DMRs that were found in the upstream region in ZNF80, POU1F1, MFGE8, and ANAPC1 were subjected to bisulfite sequencing for (Supplementary Figs. 4A–4D), and the findings were consistent with those of MeDIP-Seq. ZNF80 is a member of the zinc finger family; POU1F1 is a growth hormone transcription factor; and MFGE8 has a role in cell adhesion for connecting smooth muscle to elastic fibers in arteries (Larsson et al., 2006). In addition, changes in the expression of ANAPC1 in GEUMBIT SESANG, which was hypermethylated in an upstream region, was identified by real-time RT-PCR before and after exercise, suggesting a likely relationship between DNA methylation and the expression of these genes (Supplementary Fig. 4D).
Table 1.
Distribution of methylation peaks in different genomic regions
| Promoter1 | 5′ UTR | Exon | Intron | 3′ UTR | Downstrem1 | Repeat | Others | Total | ||
|---|---|---|---|---|---|---|---|---|---|---|
| JIGUSANG | Before exercise | 2,070 | 61 | 4,632 | 29,998 | 155 | 2,016 | 29,208 | 37,163 | 105,303 |
| SERYEOK | After exercise | 1,854 | 69 | 4,259 | 26,014 | 128 | 1,818 | 25,411 | 33,416 | 92,969 |
| GEUMBIT | Before exercise | 1,556 | 54 | 3,892 | 22,830 | 109 | 1,532 | 22,190 | 28,977 | 81,140 |
| SESANG | After exercise | 1,897 | 57 | 4,155 | 27,902 | 151 | 1,942 | 26,920 | 35,186 | 98,210 |
Promoter and downstream is defined as upstream 2 kb at TSS and downstream 2 kb at TTS, respectively.
Different methylation patterns were found in different genomic regions. The frequency of methylation was lower at TSSs. By contrast, gene-body regions showed higher frequency of methylation (Fig. 1). A moderate change in methylation pattern of promoter regions was seen in both horses before and after trotting. In the genomic region, TSS shores are essential for the regulation of gene expression, and accordingly, these regions were less methylated than other regions. This pattern was consistent with the results of previous studies (Feber et al., 2011; Hu et al., 2013; Sati et al., 2012).
Fig. 1.
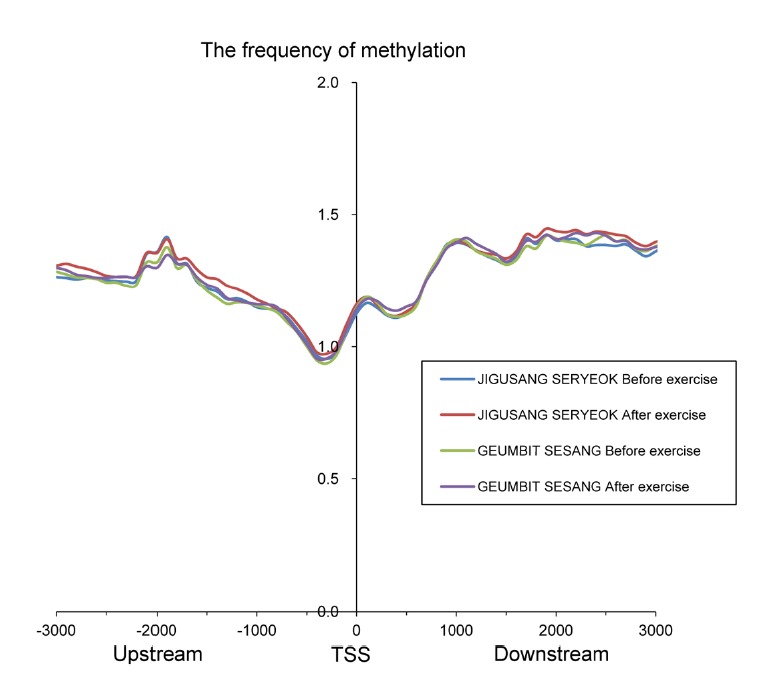
Distribution of methylation peaks adjacent to the transcription start site (approximately 3 kb).
DNA methylation patterns between the two thoroughbred horses before and after exercise
We analyzed DMRs in blood samples collected from the horses before and after exercise (Fig. 2). We found 1954 DMRs in JIGUSANG SERYEOK (female inferior horse) and 1602 DMRs in GEUMBIT SESANG (male superior horse) with even distribution in the genomes (Circos plot, Fig. 3). Overall, exon regions showed a higher level of DNA methylation than other regions of the genes. These data indicated differences in methylation between samples collected before and after exercise, and between superior and inferior horses. As shown in Fig. 2, of the total DMRs the frequency of hypermethylated regions after exercise was 51.8% and 44.7% in in JIGUSANG SERYEOK and GEUMBIT SESANG, respectively, after exercise. Thus, there was a tendency for hypomethylation in the superior horse, and hypermethylation in the inferior horse. We plotted the mean density of DMRs relative to that of genes after exercise (Fig. 4) and found similar patterns in both horses. In both horses, hyper- and hypomethylated DMRs were enhanced in gene-body regions. The gene-body regions showed relatively greater change in methylation patterns after exercise, whereas the magnitude of change was lower in the upstream and downstream gene regions. The gene-body regions in JIGUSANG SERYEOK were more hypermethylated than those in GEUMBIT SESANG. Hypomethylation patterns were similar in both horses after exercise (Fig. 4).
Fig. 2.
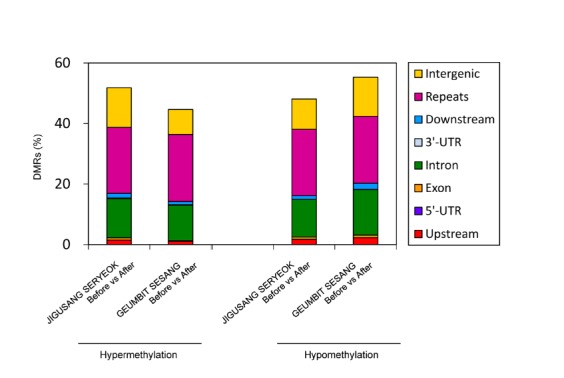
Differentially methylated regions (DMRs) were analyzed by comparing genetic features in the two horses before and after exercise. The percentage of hyper- and hypomethylation in different genetic regions such as intergenic, gene-body, and promoter regions, and repeats.
Fig. 3.
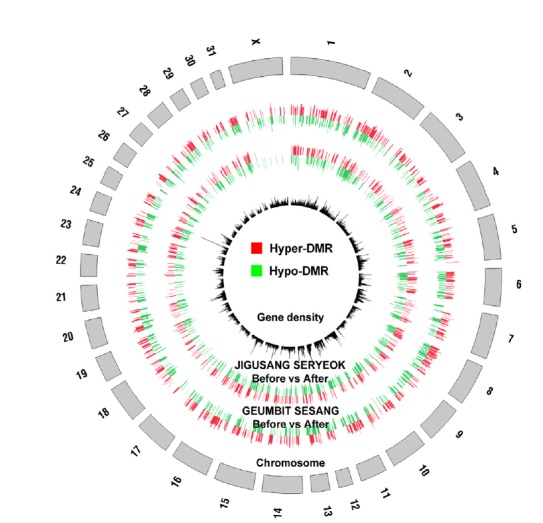
Comprehensive maps of the whole-genome methylome in two horse blood before and after exercise. Inner black circle represents gene density corresponding to each chromosome. Outer two circles are apparent two horse distribution of DMRs after exercises.
Fig. 4.
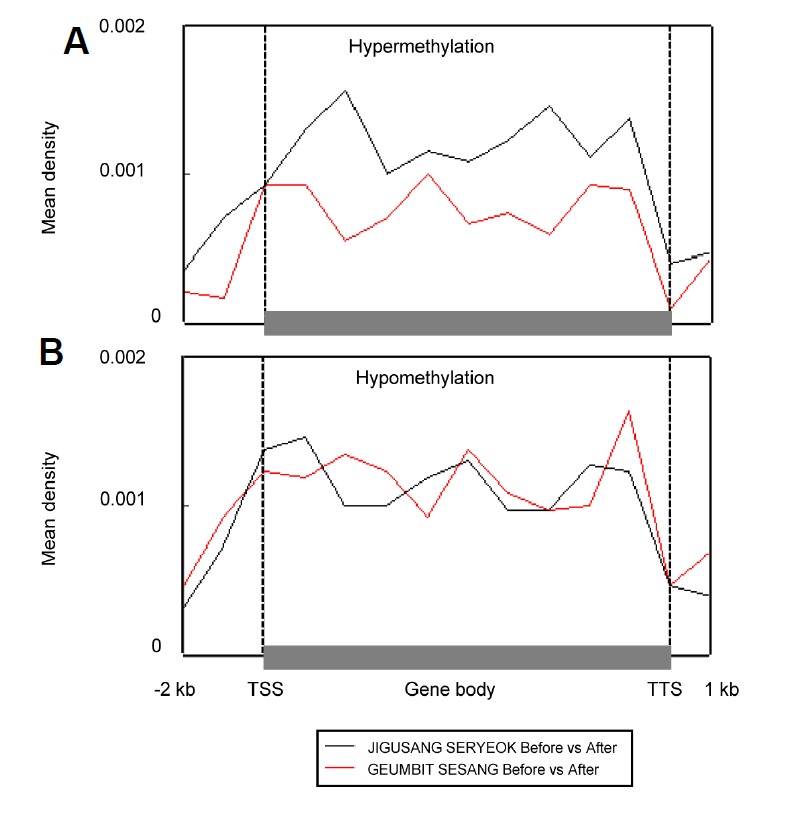
Changes in average methylation density associated with position relative to the gene body and both ends in the genome of the two horses after exercise. Methylation density at 2 kb upstream and 1 kb downstream of the gene body was calculated by dividing the peak summit count of each genomic region. Average methylation density in hypermethylated (A) and hypomethylated (B) genes after exercise.
To understand the relationship between DNA methylation and gene expression changes related to exercise, RNA-Seq data of the corresponding samples were analyzed (Park et al., 2012). Differentially expressed genes (DEGs) were selected on the basis of ≥1.5-fold change in gene expression and P < 0.05 and were linked to DMRs that might be correlated with gene expression. For RNA-Seq and MeDIP-Seq, 11 and 13 genes, respectively, showed a negative correlation between gene expression level and DNA methylation level (Supplementary Table 7). DNA methylation and the expression of these genes have been found to be strongly correlated in earlier studies (Barres et al., 2012; Jaenisch and Bird, 2003); therefore, exercise-derived DMRs of these genes could alter gene expression levels in the two horses.
CGIs play important roles in the epigenetic regulation of gene expression. With the exception of genomic imprinting loci and inactivated X chromosomes, 60–90% of the CGIs exist in methylated form in the mammalian genome (Bird, 2002; Tucker, 2001). Promoter DNA methylation caused by DNA methyltransferases is considered as a signal for downregulating gene expression. A total of 109,505 CGIs were detected in the horse genome. Of these CGIs, approximately 6.27% (n = 6861) were hypomethylated and 3.26% (n = 3566) were hypermethylated after exercise in JIGUSANG SERYEOK. In GEUMBIT SESANG, approximately 8.47 % (n = 9275) of the CGIs were hypomethylated and 2.90% (n = 3179) were hypermethylated after exercise (Table 2). In these horses, there was a higher abundance of hypomethylated regions than hypermethylated regions after exercise. These results suggested that exercise contributed to hypermethylation of CGIs in the whole genome. Analysis of the frequencies of nucleotides around DMRs revealed that the percentage of cytosine and guanine was increased and that of adenine and thymine decreased in the centers of DMRs (Supplementary Fig. 5). Thus, CGIs appear to play an important role in altering the methylation patterns in the horse genome.
Table 2.
Distribution of methylated CpG islands in the genomes of blood samples from the two thoroughbred horses after trotting
| Upstream 2 kb | Gene body | Others | Total | Total CGIs | Methylation frequency (%) | ||
|---|---|---|---|---|---|---|---|
| JIGUSANG SERYEOK after exercise | Hypomethylation | 588 | 1,559 | 4,714 | 6,861 | 109,505 | 6.27 |
| Hypermethylation | 450 | 185 | 2,931 | 3,566 | 109,505 | 3.26 | |
| GEUMBIT SESANG after exercise | Hypomethylation | 1,342 | 4,277 | 3,656 | 9,275 | 109,505 | 8.47 |
| Hypermethylation | 245 | 577 | 2,357 | 3,179 | 109,505 | 2.90 |
Transposable elements (TEs) occupy approximately 30–50% of the mammalian genome. Since the expression of TEs introduces genetic instability, DNA methylation patterns in TE regions are maintained in organisms to inhibit their expression (Carnell and Goodman, 2003; Girardot et al., 2006; Walsh et al., 1998). Changes in DNA methylation patterns of TE regions could be related to the genome defense system. We examined DMRs within the TEs and found that prominent changes in the methylation of LINEs and SINEs occurred after exercise. The DMRs within TEs were more abundant in JIGUSANG SERYEOK than in GEUMBIT SESANG. However, the TE to DMR ratio among different TE families was similar in both horses (Fig. 5). After exercise, the LINEs were more hypomethylated in JIGUSANG SERYEOK than in GEUMBIT SESANG. Similarly, LTR elements were more hypermethylated in JIGUSANG SERYEOK after exercise. This experiment showed that the TE regions of JIGUSANG SERYEOK underwent more changes in methylation, particularly LINEs and LTR elements.
Fig. 5.
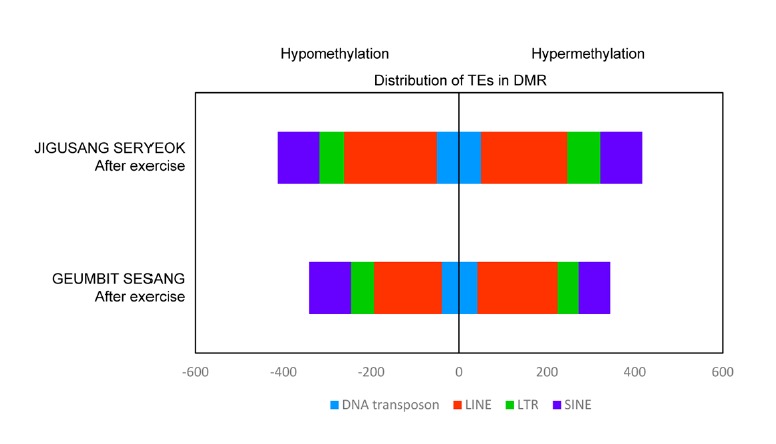
Genomic distribution of transposable elements among DMRs. Hypo- and hypermethylation density in transposable elements was classified according to the TE family. DNA here refers to DNA transposon, a class of repetitive sequences.
To characterize the changes in the methylation profiles of TEs, we analyzed the peak distribution of TEs in the horse genomes (Supplementary Table 8). LINEs and SINEs were found most frequently. LTR elements and DNA transposons were also abundantly present in the horse genomes. We analyzed the frequency of peaks by calculating the number of peaks per repeat in the genome. The highest frequency of peaks was observed in small recognition particle RNA (srpRNA) and this frequency decreased after exercise. Satellite DNA showed a high frequency of peaks and did not show significant changes after exercise. A higher frequency of peaks was observed in LINEs and SINEs than in LTR elements and DNA transposons (Supplementary Table 8). Thus, after exercise, the total number of methylation peaks in TE regions decreased in JIGUSANG SERYEOK, whereas it increased in GEUMBIT SESANG. No significant change in the methylation pattern of LTR elements was observed in JIGUSANG SERYEOK, whereas in GEUMBIT SESANG, there was an increase in the frequency of LTR peaks.
GO analysis of DMR genes as functional categories
We classified the genes differentially methylated in pre- and post-exercise samples according to their putative functions. A previous study found that the genes with methylation in both promoter and gene-body regions may be considered as methylated genes (Hu et al., 2013). Therefore, we identified genes containing DMRs among methylated genes in pre- and post-exercise samples from the two thoroughbred horses. GO analysis of these genes was performed using the DAVID program (Huang da et al., 2009). Then, we depicted GO analysis as a heatmap to easily understand the significance of GO pathway distribution after exercise (Supplementary Fig. 6). Among these genes, 328 and 388 genes differently methylated after exercise in JIGUSANG SERYEOK and GEUMBIT SESANG, respectively, were confirmed. In both the horses, nine genes were hypomethylated and eight genes were hypermethylated after exercise (Table 2). The ZNF80 gene of the two horses was hypomethylated after exercise, and their methylation level was confirmed through bisulfite sequencing (Supplementary Fig. 4). These genes were related to one or more of the following categories: biological process (BP), cellular component (CC), and molecular function (MF). In JIGUSANG SERYEOK 868 and 794 DMRs were identified as hypomethylated and hypermethylated, respectively, after exercise. Approximately 596 DMRs were identified as hypomethylated and 715 as hypermethylated in GEUMBIT SESANG after exercise. In the two horses, channel activity-related regions were also hypomethylated after exercise (Supplementary Table 9). In GEUMBIT SESANG, regions related to cell division and adhesion were hypermethylated after exercise, whereas cell signaling- and transport-related regions were hypermethylated in JIGUSANG SERYEOK (Supplementary Fig. 6). These genes likely play important roles in exercise-related physiological processes in the horse.
We used the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database to predict the putative functions of the identified genes containing DMRs. The output revealed that genes related to the tight junction pathway were hypermethylated after exercise in both horses. Genes related to the cell signaling pathway were also hypomethylated after exercise in both horses. Our results suggest that exercise activated cell signaling pathways in the horses (Supplementary Fig. 7).
Among the genes contained in the GO and KEGG list in the present study, we chose a set of genes that are related to an exercise-related function. We selected a total of 23 genes in two horses before and after exercise. We have submitted the methylation peaks of these genomic regions between before and after exercise as custom tracks in the UCSC Genome Browser for visualization (Supplementary Fig. 7).
DISCUSSION
DNA methylation plays essential roles in the regulation of gene expression (Gao et al., 2010; Jaenisch and Bird, 2003) and inactivation of TEs (Carnell and Goodman, 2003; Girardot et al., 2006; Walsh et al., 1998). The majority of reports on genome-wide DNA methylation studies of blood samples are restricted to cancer (Al-Moundhri et al., 2010; Hsiung et al., 2007), smokers (Zeilinger et al., 2013), and asthma patients (Kim et al., 2013b). This is the first study that compared the genome-wide DNA methylation profiles of pre- and post-exercise blood samples from thoroughbred horses. Our results reveal that the DNA methylation profiles in the whole blood of thoroughbred horses are similar to those of various tissues of mammalian and plant origin, and that exercise produces different methylation patterns in male superior and female inferior horses.
The objective of this study was to screen for candidate genes and metabolic pathways regulated by exercise. We analyzed the effect of exercise on whole-genome DNA methylation in the blood of thoroughbred horses and showed that exercise changes methylation profiles of exercise-related genes. Our results show differences in the methylation pattern between the genomes of male superior, and female inferior horses. Genomes of these two horses showed subtle differences in the patterns of methylated peaks after exercise. Several studies compared gene expression profiles of pre- and post-exercise blood samples and identified significant changes in expression patterns of exercise-related genes (Gano et al., 2011; Garcia-Lopez et al., 2007; Radom-Aizik et al., 2009). We also demonstrated the effect of exercise on DNA methylation patterns, as well as the gene expression level of exercise-related genes. After exercise, gene expression patterns might be changed by circulating factors such as hormones or neurotransmitters (Roberts et al., 2009). In our study, gene expression was found to be related to methylation remodeling after exercise. As further studies, analysis of the correlation between our DNA methylation data and gene expression will reveal the mechanisms responsible for adaptive responses to exercise and exercise-induced changes in physiological processes.
Methylation peaks can be used to confirm the whole-genome methylation patterns. In this study, methylation peaks were compared before and after exercise. Consistent with previous reports, our results showed minor variation in methylation patterns between the two horses before and after exercise (Ball et al., 2009; Hu et al., 2013; Sati et al., 2012). However, MeDIP-Seq analysis showed more methylation in the intronic regions than exon regions (Supplementary Table 5), a pattern similar to that found in the chicken genome (Hu et al., 2013) and in the genome of malignant peripheral nerve sheath tumors (Feber et al., 2011). By contrast, previous studies have shown higher methylation level of exons than of introns in the human and rat genomes (Laurent et al., 2010; Sati et al., 2012). We also provided the patterns of methylation peaks in the shore of gene body, and repeat sequences. In intron, the frequency of methylation peaks were higher than the shore of gene body (promoter and downstream), which shows similar patterns. When the gene body is methylated, it is possible to be influenced together with the shore of gene body. The correlation between gene expression and the shore of gene body methylation is needed to elucidate as a further study. Our results serve species-specific methylation patterns.
We observed differences and changes in methylation patterns after exercise in female and male individuals. In the human genome, the methylation level of LINEs is lower in women than in men (El-Maarri et al., 2007; 2011; Zhang et al., 2011). Interestingly, the methylation level of LINEs was lower in the male horse than in the female horse (Supplementary Table 8). Moreover, the female horse showed a higher level of hypomethylation in its LINEs and hypermethylation of LTR elements (Fig. 5). The expression of LINEs induces genomic instability, with the host cell maintaining the methylation state of the LINEs (Carnell and Goodman, 2003). In the horse transcriptome, LINE-containing transcripts blood cells were upregulated after exercise, and this study demonstrated that exercise-induced stress leads to change in LINEs in the host (Capomaccio et al., 2010). The present study examined whole-genome methylation patterns as well as changes in LINEs in two horses before and after exercise. The racing activity, gender, age, and other factors in relation to methylation patterns should be validated using a larger horse population. The findings of the present study differ from that of the human genome, and differences in methylation patterns based on gender should be considered when assessing the ability of horses.
We also identified individual-specific DMRs and DEGs after exercise at the whole-genome level. We compared two sets of NGS data, including the present data and previous RNA-Seq data of the corresponding samples (Park et al., 2012). The methylation state of 24 genes was negatively correlated with gene expression levels (Supplementary Table 7). In GEUMBIT SESANG, DEDD2 is hypomethylated in the promoter region and then highly expressed after exercise. DEDD2 contains the death effector domain, induces weak apoptosis, and affects cell death (Stegh et al., 1998). TGM2, a gene encoding a transglutaminase, also has the same pattern in JIGUSANG SERYEOK. This gene induces an isopeptide bond between a free amine group and causes high resistance to the proteolytic degradation of the peptide (Griffin et al., 2002). These gene lists provide clues to the exercise-induced pathway in the host.
We compared nucleotide frequencies of DMRs in the whole genome of four samples: JIGUSANG SERYEOK and GEUMBIT SESANG before and after exercise, respectively. The percentage of cytosine and guanine increased at the centers of DMRs, whereas the percentage of adenine and thymine increased in regions adjacent to the centers of the DMRs (Supplementary Fig. 5). These results suggested that CGIs play key role in altering the methylation patterns in the horse genome. Methylation of promoter and gene-body regions play important roles in epigenetic regulation of gene expression. Therefore, we sorted genes methylated in promoter regions and intragenic regions. Gene body methylation could influence chromatin structure and efficiency of transcription elongation (Klose and Bird, 2006; Lorincz et al., 2004). Our results showed that 1497 genes were hypomethylated and 1438 genes hypermethylated in their genic region after exercise. Since these genes are likely to be involved in exercise metabolism, we performed GO and KEGG pathway analyses.
Exercise has been shown to induce genetic or epigenetic changes in the genome (Barres et al., 2012). In our study, we selected genes containing DMRs and performed GO and KEGG pathway analyses to characterize their roles in exercise. We presented the top three GO terms among three GO categories, and the top two KEGG pathways. Based on the GO terms and pathways identified, these DMRs may play exercise-related roles by altering metabolic processes (Supplementary Fig. 6). Genes related to the cell junction pathway were hypermethylated in the horses. Tight junction- and adherens junction-related genes showed enriched methylation patterns. Tight junctions are involved in the regulation of cellular growth and differentiation (Matter and Balda, 2003), whereas the adherens junctions play key roles in cellular growth (Dejana et al., 2008). The pattern of GO terms showed subtle differences between the two horses, which could be useful to discriminate traits of superior and inferior horses following exercise. Methylated genes in superior and inferior horses were classified into different GO molecular function categories. Genes related to “intercellular pathway” were methylated in JIGUSANG SERYEOK, and those related to “cell activity” were methylated in GEUMBIT SESANG. Notably, “glioma”-related genes were hypermethylated after exercise in JIGUSANG SERYEOK, suggesting that exercise induces epigenetic inhibition of the glioma pathway. “Cell signaling”-related genes were hypomethylated after exercise in both horses. Focal adhesions allow attachment to the substratum and transmit force through the sites of adhesion that regulate cell growth, survival, and gene expression (Sastry and Burridge, 2000).
The hypermethylated GO terms were primarily related to cellular physiology and ion transport. This finding suggested that the cells respond to exercise stimuli and regulate gene expression by altering the methylation state. Efficient ion transport in the skeletal muscles and motor neurons is important during exercise (Allen et al., 2008; Gardiner et al., 2006). Ion channel modulation-related terms were identified in DMRs, indicating a metabolic shift in blood.
The hypomethylated GO terms showed different patterns in superior and inferior horses. In JIGUSANG SERYEOK, plasma membrane, synapse, and receptor GO terms were identified. Cytoskeleton, nucleoplasm, and cell projection terms were identified in the cellular compartment domain. In the superior horse, genes related to cell signaling and neuronal functions tended to be hypomethylated, whereas cell structure-related genes were hypomethylated in the inferior horse. Exercise induces changes in synaptic plasticity (Farmer et al., 2004; Gomez-Pinilla et al., 2011). It has been shown that PGC-1α is up-regulated after exercise and promotes the transcription of synaptic genes in humans, mice, and rats (Goto et al., 2000; Handschin et al., 2007b; Russell et al., 2003). Consistent with these reports, our study identified changes in the methylation state of genes related to synaptic remodeling in the horse. Transmembrane transport and channel activity GO terms were consistently identified in both horses (Supplementary Fig. 6). Transmembrane transport is important for muscular and neuronal functions (Clausen, 2008). This result is also in agreement with previous studies, indicating that many genes undergo methylation to maintain homeostasis after exercise. After exercise in JIGUSANG SERYEOK, the GO term “cytoskeleton” was identified as both hypomethylated and hypermethylated. In order to resolve this contradiction, we analyzed the genes included in the GO term. In the “cytoskeleton” category, a total of 42 genes were hypermethylated and 38 genes were found to be hypomethylated. With the exception of four genes, these sets of genes were different, suggesting that genes involved in cytoskeletal remodeling and intercellular transport frequently undergo changes in their methylation patterns in response to exercise. These pathways are likely related to physiological changes occurring in the horse that are induced by exercise. Differences in gender in relation to horse racing is less noticeable than other abilities, although some studies have described sex-based differences in horses (Cywinska et al., 2011; Noble et al., 2007; Padalino et al., 2014). Our study has identified sex-based differences in the genomic response to exercise. Further studies are needed to identify the precise roles of these genes and pathways in athletic performance of horses and their adaptive responses to exercise.
We sorted out the DMR-containing genes categorized in the GO and KEGG analyses (Supplementary Table 9). Then, we presented the methylation peaks of genes contained in GO and KEGG (Supplementary Fig. 8). Akt activity is crucial in various cellular processes such as apoptosis, cell proliferation, cell migration, transcription, and glucose metabolism. After exercise, Akt phosphorylation and its pathway are induced in rats (Sakamoto et al., 2003). In the after exercise GO and KEGG data of the two horses, AKT1 is hypomethylated in GEUMBIT SESANG (Supplementary Fig. 8A), whereas AKT3 was hypermethylated in JIGUSANG SERYEOK (Supplementary Fig. 8B). Our data on DMR-containing genes in the Akt pathway indicate that specific epigenetic Akt pathway mechanisms are involved in exercise. The neurotransmitter glutamate plays crucial roles in cellular metabolism and in the mammalian central nervous system (CNS) by interacting with glutamate receptors. Various studies have demonstrated that exercise improves learning tasks or neurogenesis by influencing glutamate receptors (Dietrich et al., 2005; Farmer et al., 2004). Three glutamate receptor-related genes (GRID1, GRIK4, and GRIN2A) were visualized. As expected, two genes were hypomethylated (Supplementary Figs. 8D and 10E), and the GRID1 gene was hypermethylated after exercise (Supplementary Fig. 8C). The ABCC9 protein is predicted to form ATP-sensitive potassium channels in exercise-related tissues such as vascular and non-vascular smooth, skeletal, and cardiac muscle tissues (Olson and Terzic, 2010). The ABCC9 gene was hypermethylated in JIGUSANG SERYEOK (Supplementary Fig. 8G), whereas the ABCC6 gene was hypomethylated in the two horses (Supplementary Fig. 8F). Exercise induces changes in the ion transport-related pathway of suitable organs or situations; therefore, epigenetic should be conducted in future studies. Obscurin (OBSCN) organizes myofibrils and induces interactions between the sarcoplasmic reticulum and myofibrils. It also plays a crucial role in the formation and maintenance of A-bands (Carlsson et al., 2008). Therefore, it is essential to determine the roles and epigenetic pathways of obscurin in exercise as the OBSCN gene was shown to be hypermethylated in JIGUSANG SERYEOK (Supplementary Fig. 8K). As the largest protein, titin, which is derived from the TTN gene, influences elasticity and contraction of striated muscle tissues. Titin connects the Z line and M line in the sarcomere, and different expression patterns were identified in athletes and non-athletic individuals (McBride et al., 2003). The TTN gene is hypermethylated in JIGUSANG SERYEOK, and the patterns of epigenetic changes in TTN gene were strongly associated with racing ability (Supplementary Fig. 8M). These genes have undergone changes in their DNA methylation patterns after exercise, which could affect its role in regulating exercise in the horse. Our data on DMR-containing genes can provide further information on the pathways involved in exercise physiology.
In conclusion, for the first time, we analyzed genome-wide methylation profiles of athletically superior and inferior horses before and after exercise. Exercise leads to changes in DNA methylation patterns in the thoroughbred horse blood genome. Distribution of methylation peaks showed different patterns in the two horses. Thus, our data serve as a basis for understanding the exercise-induced epigenetic changes in racing horses and provide the basis for further studies on superior exercise-related traits, as well as differences in DMRs between the genomes of male and female horses.
Table 3.
Differentially methylated genes were consistent in the two horses both before and after exercise
| Methylation pattern | Ensembl_ID | Region | Gene | Description | Repeat_name | Repeat_family |
|---|---|---|---|---|---|---|
| Hypomethylation | ENSECAG00000005130 | Upstream | ZNF80 | Zinc finger protein 80 | HAL1 | LINE/L1 |
| ENSECAG00000022638 | Intron | PLD1 | Phospholipase D1 | MER2 | DNA/MER2_type | |
| ENSECAG00000020390 | Intron | MAMSTR | MEF2 activating motif and SAP domain containing transcriptional regulator | |||
| ENSECAG00000006609 | Intron | JAG2 | Jagged 2 | |||
| ENSECAG00000024035 | Intron | CSMD2 | CUB and sushi multiple domains 2 | |||
| ENSECAG00000008870 | Intron | ABCC6 | ATP-binding cassette sub-family C member 6 | ERE1 | SINE | |
| ENSECAG00000006703 | Upstream | |||||
| ENSECAG00000002853 | Upstream | Tigger5 | DNA/MER2_type | |||
| ENSECAG00000025495 | Downstream | |||||
| Hypermethylation | ENSECAG00000008844 | Downstream | SLC25A24 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 24 | ||
| ENSECAG00000014168 | Intron | PSMA7 | Proteasome (prosome, macropain) subunit, alpha type, 7 | L1MB1 | LINE/L1 | |
| ENSECAG00000021836 | Intron | OGFR | Opioid growth factor receptor | |||
| ENSECAG00000015042 | Intron | NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | |||
| ENSECAG00000016911 | Downstream | HEBP1 | Heme binding protein 1 | L2a | LINE/L2 | |
| ENSECAG00000012628 | Intron | EPB41L4A | Erythrocyte membrane protein band 4.1 like 4A | MIRb | SINE/MIR | |
| ENSECAG00000022933 | Intron | ELL2 | Elongation factor, RNA polymerase II, 2 | L2 | LINE/L2 | |
| ENSECAG00000003454 | Upstream |
Acknowledgments
This work was supported by a grant from the Next generation BioGreen 21 Program (No. PJ0081062011) of the Rural Development Administration, Republic of Korea.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Al-Moundhri M.S., Al-Nabhani M., Tarantini L., Baccarelli A., Rusiecki J.A. The prognostic significance of whole blood global and specific DNA methylation levels in gastric adenocarcinoma. PLoS One. 2010;5:e15585. doi: 10.1371/journal.pone.0015585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D.G., Lamb G.D., Westerblad H. Impaired calcium release during fatigue. J. Appl. Physiol. 2008;104:296–305. doi: 10.1152/japplphysiol.00908.2007. [DOI] [PubMed] [Google Scholar]
- Ball M.P., Li J.B., Gao Y., Lee J.H., LeProust E.M., Park I.H., Xie B., Daley G.Q., Church G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R., Yan J., Egan B., Treebak J.T., Rasmussen M., Fritz T., Caidahl K., Krook A., O’Gorman D.J., Zierath J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–411. doi: 10.1016/j.cmet.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bosviel R., Garcia S., Lavediaux G., Michard E., Dravers M., Kwiatkowski F., Bignon Y.J., Bernard-Gallon D.J. BRCA1 promoter methylation in peripheral blood DNA was identified in sporadic breast cancer and controls. Cancer Epidemiol. 2012;36:e177–182. doi: 10.1016/j.canep.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Burgomaster K.A., Howarth K.R., Phillips S.M., Rakobowchuk M., Macdonald M.J., McGee S.L., Gibala M.J. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J. Physiol. 2008;586:151–160. doi: 10.1113/jphysiol.2007.142109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capomaccio S., Verini-Supplizi A., Galla G., Vitulo N., Barcaccia G., Felicetti M., Silvestrelli M., Cappelli K. Transcription of LINE-derived sequences in exercise-induced stress in horses. Anim. Genet. 2010;41(Suppl 2):23–27. doi: 10.1111/j.1365-2052.2010.02094.x. [DOI] [PubMed] [Google Scholar]
- Carlsson L., Yu J.G., Thornell L.E. New aspects of obscurin in human striated muscles. Histochem. Cell Biol. 2008;130:91–103. doi: 10.1007/s00418-008-0413-z. [DOI] [PubMed] [Google Scholar]
- Carnell A.N., Goodman J.I. The long (LINEs) and the short (SINEs) of it: altered methylation as a precursor to toxicity. Toxicol. Sci. 2003;75:229–235. doi: 10.1093/toxsci/kfg138. [DOI] [PubMed] [Google Scholar]
- Chang S.C., Tucker T., Thorogood N.P., Brown C.J. Mechanisms of X-chromosome inactivation. Front. Biosci. 2006;11:852–866. doi: 10.2741/1842. [DOI] [PubMed] [Google Scholar]
- Chavez L., Jozefczuk J., Grimm C., Dietrich J., Timmermann B., Lehrach H., Herwig R., Adjaye J. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010;20:1441–1450. doi: 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T. Role of Na+, K+-pumps and transmembrane Na+, K+-distribution in muscle function. The FEPS lecture-Bratislava 2007. Acta Physiol. 2008;192:339–349. doi: 10.1111/j.1748-1716.2007.01798.x. [DOI] [PubMed] [Google Scholar]
- Cywinska A., Szarska E., Kowalska A., Ostaszewski P., Schollen-berger A. Gender differences in exercise--induced intra-vascular haemolysis during race training in thoroughbred horses. Res. Vet. Sci. 2011;90:133–137. doi: 10.1016/j.rvsc.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Dauksa A., Gulbinas A., Barauskas G., Pundzius J., Oldenburg J., El-Maarri O. Whole blood DNA aberrant methylation in pancreatic adenocarcinoma shows association with the course of the disease: a pilot study. PLoS One. 2012;7:e37509. doi: 10.1371/journal.pone.0037509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Dejana E., Orsenigo F., Lampugnani M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- Dempsey J.A., Wagner P.D. Exercise-induced arterial hypoxemia. J. Appl. Physiol. 1999;87:1997–2006. doi: 10.1152/jappl.1999.87.6.1997. [DOI] [PubMed] [Google Scholar]
- Dietrich M.O., Mantese C.E., Porciuncula L.O., Ghisleni G., Vinade L., Souza D.O., Portela L.V. Exercise affects glutamate receptors in postsynaptic densities from cortical mice brain. Brain Res. 2005;1065:20–25. doi: 10.1016/j.brainres.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Eivers S.S., McGivney B.A., Fonseca R.G., MacHugh D.E., Menson K., Park S.D., Rivero J.L., Taylor C.T., Katz L.M., Hill E.W. Alterations in oxidative gene expression in equine skeletal muscle following exercise and training. Physiol. Genomics. 2010;40:83–93. doi: 10.1152/physiolgenomics.00041.2009. [DOI] [PubMed] [Google Scholar]
- Eivers S.S., McGivney B.A., Gu J., MacHugh D.E., Katz L.M., Hill E.W. PGC-1alpha encoded by the PPARGC1A gene regulates oxidative energy metabolism in equine skeletal muscle during exercise. Anim. Genet. 2012;43:153–162. doi: 10.1111/j.1365-2052.2011.02238.x. [DOI] [PubMed] [Google Scholar]
- El-Maarri O., Becker T., Junen J., Manzoor S.S., Diaz-Lacava A., Schwaab R., Wienker T., Oldenburg J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum. Genet. 2007;122:505–514. doi: 10.1007/s00439-007-0430-3. [DOI] [PubMed] [Google Scholar]
- El-Maarri O., Walier M., Behne F., van Uum J., Singer H., Diaz-Lacava A., Nusgen N., Niemann B., Watzka M., Reinsberg J., et al. Methylation at global LINE-1 repeats in human blood are affected by gender but not by age or natural hormone cycles. PLoS One. 2011;6:e16252. doi: 10.1371/journal.pone.0016252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer J., Zhao X., van Praag H., Wodtke K., Gage F.H., Christie B.R. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience. 2004;124:71–79. doi: 10.1016/j.neuroscience.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Feber A., Wilson G.A., Zhang L., Presneau N., Idowu B., Down T.A., Rakyan V.K., Noon L.A., Lloyd A.C., Stupka E., et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res. 2011;21:515–524. doi: 10.1101/gr.109678.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gano L.B., Donato A.J., Pierce G.L., Pasha H.M., Magerko K.A., Roeca C., Seals D.R. Increased proinflammatory and oxidant gene expression in circulating mononuclear cells in older adults: amelioration by habitual exercise. Physiol. Genomics. 2011;43:895–902. doi: 10.1152/physiolgenomics.00204.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L., Geng Y., Li B., Chen J., Yang J. Genome-wide DNA methylation alterations of Alternanthera philoxeroides in natural and manipulated habitats: implications for epigenetic regulation of rapid responses to environmental fluctuation and phenotypic variation. Plant Cell Environ. 2010;33:1820–1827. doi: 10.1111/j.1365-3040.2010.02186.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Lopez D., Hakkinen K., Cuevas M.J., Lima E., Kauhanen A., Mattila M., Sillanpaa E., Ahtiainen J.P., Karavirta L., Almar M., et al. Effects of strength and endurance training on antioxidant enzyme gene expression and activity in middle-aged men. Scand. J. Med. Sci. Sports. 2007;17:595–604. doi: 10.1111/j.1600-0838.2006.00620.x. [DOI] [PubMed] [Google Scholar]
- Gardiner P., Dai Y., Heckman C.J. Effects of exercise training on alpha-motoneurons. J. Appl. Physiol. 2006;101:1228–1236. doi: 10.1152/japplphysiol.00482.2006. [DOI] [PubMed] [Google Scholar]
- Gim J.A., Ayarpadikannan S., Eo J., Kwon Y.J., Choi Y., Lee H.K., Park K.D., Yang Y.M., Cho B.W., Kim H.S. Transcriptional expression changes of glucose metabolism genes after exercise in thoroughbred horses. Gene. 2014;547:152–158. doi: 10.1016/j.gene.2014.06.051. [DOI] [PubMed] [Google Scholar]
- Girardot M., Guibert S., Laforet M.P., Gallard Y., Larroque H., Oulmouden A. The insertion of a full-length Bos taurus LINE element is responsible for a transcriptional deregulation of the Normande Agouti gene. Pigment Cell Res. 2006;19:346–355. doi: 10.1111/j.1600-0749.2006.00312.x. [DOI] [PubMed] [Google Scholar]
- Goeppert B., Konermann C., Schmidt C.R., Bogatyrova O., Geiselhart L., Ernst C., Gu L., Becker N., Zucknick M., Mehrabi A., et al. Global alterations of DNA methylation in cholangio-carcinoma target the Wnt signaling pathway. Hepatology. 2014;59:544–554. doi: 10.1002/hep.26721. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F., Zhuang Y., Feng J., Ying Z., Fan G. Exercise impacts brain-derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur. J. Neurosci. 2011;33:383–390. doi: 10.1111/j.1460-9568.2010.07508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M., Terada S., Kato M., Katoh M., Yokozeki T., Tabata I., Shimokawa T. cDNA Cloning and mRNA analysis of PGC-1 in epitrochlearis muscle in swimming-exercised rats. Biochem. Biophys. Res. Commun. 2000;274:350–354. doi: 10.1006/bbrc.2000.3134. [DOI] [PubMed] [Google Scholar]
- Griffin M., Casadio R., Bergamini C.M. Transglutaminases: nature’s biological glues. Biochem. J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C., Chin S., Li P., Liu F., Maratos-Flier E., Lebrasseur N.K., Yan Z., Spiegelman B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Biol. Chem. 2007a;282:30014–30021. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- Handschin C., Kobayashi Y.M., Chin S., Seale P., Campbell K.P., Spiegelman B.M. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007b;21:770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E.W., Gu J., Eivers S.S., Fonseca R.G., McGivney B.A., Govindarajan P., Orr N., Katz L.M., MacHugh D.E. A sequence polymorphism in MSTN predicts sprinting ability and racing stamina in thoroughbred horses. PLoS One. 2010a;5:e8645. doi: 10.1371/journal.pone.0008645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E.W., Gu J., McGivney B.A., MacHugh D.E. Targets of selection in the Thoroughbred genome contain exercise-relevant gene SNPs associated with elite racecourse perfor-mance. Anim. Genet. 2010b;41(Suppl 2):56–63. doi: 10.1111/j.1365-2052.2010.02104.x. [DOI] [PubMed] [Google Scholar]
- Hsiung D.T., Marsit C.J., Houseman E.A., Eddy K., Furniss C.S., McClean M.D., Kelsey K.T. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 2007;16:108–114. doi: 10.1158/1055-9965.EPI-06-0636. [DOI] [PubMed] [Google Scholar]
- Hu Y., Xu H., Li Z., Zheng X., Jia X., Nie Q., Zhang X. Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers. PLoS One. 2013;8:e56411. doi: 10.1371/journal.pone.0056411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Sys-tematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33 Suppl:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jair K.W., Bachman K.E., Suzuki H., Ting A.H., Rhee I., Yen R.W., Baylin S.B., Schuebel K.E. De novo CpG island methylation in human cancer cells. Cancer Res. 2006;66:682–692. doi: 10.1158/0008-5472.CAN-05-1980. [DOI] [PubMed] [Google Scholar]
- Jemiolo B., Trappe S. Single muscle fiber gene expression in human skeletal muscle: validation of internal control with exercise. Biochem. Biophys. Res. Commun. 2004;320:1043–1050. doi: 10.1016/j.bbrc.2004.05.223. [DOI] [PubMed] [Google Scholar]
- Kim H., Lee T., Park W., Lee J.W., Kim J., Lee B.Y., Ahn H., Moon S., Cho S., Do K.T., et al. Peeling back the evolutionary layers of molecular mechanisms responsive to exercise-stress in the skeletal muscle of the racing horse. DNA Res. 2013a;20:287–298. doi: 10.1093/dnares/dst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.J., Park S.W., Kim T.H., Park J.S., Cheong H.S., Shin H.D., Park C.S. Genome-wide methylation profiling of the bronchial mucosa of asthmatics: relationship to atopy. BMC Med. Genet. 2013b;14:39. doi: 10.1186/1471-2350-14-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R.J., Bird A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Laird P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Larsson A., Peng S., Persson H., Rosenbloom J., Abrams W.R., Wassberg E., Thelin S., Sletten K., Gerwins P., Westermark P. Lactadherin binds to elastin--a starting point for medin amyloid formation? Amyloid. 2006;13:78–85. doi: 10.1080/13506120600722530. [DOI] [PubMed] [Google Scholar]
- Laurent L., Wong E., Li G., Huynh T., Tsirigos A., Ong C.T., Low H.M., Kin Sung K.W., Rigoutsos I., Loring J., et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Yu C., Li Y., Lam T.W., Yiu S.M., Kristiansen K., Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Liang P., Song F., Ghosh S., Morien E., Qin M., Mahmood S., Fujiwara K., Igarashi J., Nagase H., Held W.A. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics. 2011;12:231. doi: 10.1186/1471-2164-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz M.C., Dickerson D.R., Schmitt M., Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- Louis E., Raue U., Yang Y., Jemiolo B., Trappe S. Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle. J. Appl. Physiol. 2007;103:1744–1751. doi: 10.1152/japplphysiol.00679.2007. [DOI] [PubMed] [Google Scholar]
- Matter K., Balda M.S. Functional analysis of tight junctions. Methods. 2003;30:228–234. doi: 10.1016/s1046-2023(03)00029-x. [DOI] [PubMed] [Google Scholar]
- McBride J.M., Triplett-McBride T., Davie A.J., Abernethy P.J., Newton R.U. Characteristics of titin in strength and power athletes. Eur. J. Appl. Physiol. 2003;88:553–557. doi: 10.1007/s00421-002-0733-y. [DOI] [PubMed] [Google Scholar]
- McGivney B.A., Eivers S.S., MacHugh D.E., MacLeod J.N., O’Gorman G.M., Park S.D., Katz L.M., Hill E.W. Transcriptional adaptations following exercise in thoroughbred horse skeletal muscle highlights molecular mechanisms that lead to muscle hypertrophy. BMC Genomics. 2009;10:638. doi: 10.1186/1471-2164-10-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGivney B.A., McGettigan P.A., Browne J.A., Evans A.C., Fonseca R.G., Loftus B.J., Lohan A., MacHugh D.E., Murphy B.A., Katz L.M., et al. Characterization of the equine skeletal muscle transcriptome identifies novel functional responses to exercise training. BMC Genomics. 2010;11:398. doi: 10.1186/1471-2164-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble G.K., Houghton E., Roberts C.J., Faustino-Kemp J., de Kock S.S., Swanepoel B.C., Sillence M.N. Effect of exercise, training, circadian rhythm, age, and sex on insulin-like growth factor-1 in the horse. J. Anim. Sci. 2007;85:163–171. doi: 10.2527/jas.2006-210. [DOI] [PubMed] [Google Scholar]
- Norton L.E., Layman D.K. Leucine regulates translation initiation of protein synthesis in skeletal muscle after exercise. J. Nutr. 2006;136:533S–537S. doi: 10.1093/jn/136.2.533S. [DOI] [PubMed] [Google Scholar]
- Nostell K.E., Essen-Gustavsson B., Brojer J.T. Repeated post-exercise administration with a mixture of leucine and glucose alters the plasma amino acid profile in Standard-bred trotters. Acta Vet. Scand. 2012;54:7. doi: 10.1186/1751-0147-54-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson T.M., Terzic A. Human K(ATP) channelopathies: diseases of metabolic homeostasis. Pflugers Arch. 2010;460:295–306. doi: 10.1007/s00424-009-0771-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padalino B., Rubino G., Lacinio R., Petazzi F. Observations on the hematology of standardbred horses in training and racing in Southern Italy. J. Equine Vet. Sci. 2014;34:398–402. [Google Scholar]
- Park K.D., Park J., Ko J., Kim B.C., Kim H.S., Ahn K., Do K.T., Choi H., Kim H.M., Song S., et al. Whole transcriptome analyses of six thoroughbred horses before and after exercise using RNA-Seq. BMC Genomics. 2012;13:473. doi: 10.1186/1471-2164-13-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J.L., Mickelson J.R., Rendahl A.K., Valberg S.J., Andersson L.S., Axelsson J., Bailey E., Bannasch D., Binns M.M., Borges A.S., et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9:e1003211. doi: 10.1371/journal.pgen.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomraning K.R., Smith K.M., Freitag M. Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods. 2009;47:142–150. doi: 10.1016/j.ymeth.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Radom-Aizik S., Zaldivar F., Jr., Leu S.Y., Cooper D.M. A brief bout of exercise alters gene expression and distinct gene pathways in peripheral blood mononuclear cells of early- and late-pubertal females. J. Appl. Physiol. 2009;107:168–175. doi: 10.1152/japplphysiol.00121.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts M.D., Dalbo V.J., Hassell S.E., Kerksick C.M. The expression of androgen-regulated genes before and after a resistance exercise bout in younger and older men. J. Strength Cond. Res. 2009;23:1060–1067. doi: 10.1519/JSC.0b013e3181a59bdd. [DOI] [PubMed] [Google Scholar]
- Russell A.P., Feilchenfeldt J., Schreiber S., Praz M., Crettenand A., Gobelet C., Meier C.A., Bell D.R., Kralli A., Giacobino J.P., et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes. 2003;52:2874–2881. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- Russell A.P., Hesselink M.K., Lo S.K., Schrauwen P. Regulation of metabolic transcriptional co-activators and transcription factors with acute exercise. FASEB J. 2005;19:986–988. doi: 10.1096/fj.04-3168fje. [DOI] [PubMed] [Google Scholar]
- Sakamoto K., Aschenbach W.G., Hirshman M.F., Goodyear L.J. Akt signaling in skeletal muscle: regulation by exercise and passive stretch. Am. J. Physiol. Endocrinol. Metab. 2003;285:E1081–1088. doi: 10.1152/ajpendo.00228.2003. [DOI] [PubMed] [Google Scholar]
- Sastry S.K., Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 2000;261:25–36. doi: 10.1006/excr.2000.5043. [DOI] [PubMed] [Google Scholar]
- Sati S., Tanwar V.S., Kumar K.A., Patowary A., Jain V., Ghosh S., Ahmad S., Singh M., Reddy S.U., Chandak G.R., et al. High resolution methylome map of rat indicates role of intragenic DNA methylation in identification of coding region. PLoS One. 2012;7:e31621. doi: 10.1371/journal.pone.0031621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seip R.L., Angelopoulos T.J., Semenkovich C.F. Exercise induces human lipoprotein lipase gene expression in skeletal muscle but not adipose tissue. Am. J. Physiol. 1995;268:E229–236. doi: 10.1152/ajpendo.1995.268.2.E229. [DOI] [PubMed] [Google Scholar]
- Sha K. A mechanistic view of genomic imprinting. Annu. Rev. Genomics Hum. Genet. 2008;9:197–216. doi: 10.1146/annurev.genom.122007.110031. [DOI] [PubMed] [Google Scholar]
- Smith S.S., Kaplan B.E., Sowers L.C., Newman E.M. Mechanism of human methyl-directed DNA methyltransferase and the fidelity of cytosine methylation. Proc. Natl. Acad. Sci. USA. 1992;89:4744–4748. doi: 10.1073/pnas.89.10.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegh A.H., Schickling O., Ehret A., Scaffidi C., Peterhansel C., Hofmann T.G., Grummt I., Krammer P.H., Peter M.E. DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. EMBO J. 1998;17:5974–5986. doi: 10.1093/emboj/17.20.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Nishiyama T., Shimizu K., Kadota K. TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics. 2013;14:219. doi: 10.1186/1471-2105-14-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suontama M., van der Werf J.H., Juga J., Ojala M. Genetic correlations for foal and studbook traits with racing traits and implications for selection strategies in the Finnhorse and Standardbred trotter. J. Anim. Breed Genet. 2013;130:178–189. doi: 10.1111/j.1439-0388.2012.01011.x. [DOI] [PubMed] [Google Scholar]
- Tucker K.L. Methylated cytosine and the brain: a new base for neuroscience. Neuron. 2001;30:649–652. doi: 10.1016/s0896-6273(01)00325-7. [DOI] [PubMed] [Google Scholar]
- Vider J., Lehtmaa J., Kullisaar T., Vihalemm T., Zilmer K., Kairane C., Landor A., Karu T., Zilmer M. Acute immune response in respect to exercise-induced oxidative stress. Pathophysiology. 2001;7:263–270. doi: 10.1016/s0928-4680(00)00057-2. [DOI] [PubMed] [Google Scholar]
- Walsh C.P., Chaillet J.R., Bestor T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- Yan H., Kikuchi S., Neumann P., Zhang W., Wu Y., Chen F., Jiang J. Genome-wide mapping of cytosine methylation revealed dynamic DNA methylation patterns associated with genes and centromeres in rice. Plant J. 2010;63:353–365. doi: 10.1111/j.1365-313X.2010.04246.x. [DOI] [PubMed] [Google Scholar]
- Zeilinger S., Kuhnel B., Klopp N., Baurecht H., Kleinschmidt A., Gieger C., Weidinger S., Lattka E., Adamski J., Peters A., et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8:e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.F., Cardarelli R., Carroll J., Fulda K.G., Kaur M., Gonzalez K., Vishwanatha J.K., Santella R.M., Morabia A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics. 2011;6:623–629. doi: 10.4161/epi.6.5.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.