

Abstract
Nicotinamide (NAM) has been shown to suppress reactive oxygen species (ROS) production in primary human fibroblasts, thereby extending their replicative lifespan when added to the medium during long-term cultivation. Based on this finding, NAM is hypothesized to affect cellular senescence progression by keeping ROS accumulation low. In the current study, we asked whether NAM is indeed able to reduce ROS levels and senescence phenotypes in cells undergoing senescence progression and those already in senescence. We employed two different cellular models: MCF-7 cells undergoing senescence progression and human fibroblasts in a state of replicative senescence. In both models, NAM treatment substantially decreased ROS levels. In addition, NAM attenuated the expression of the assessed senescence phenotypes, excluding irreversible growth arrest. N-acetyl cysteine, a potent ROS scavenger, did not have comparable effects in the tested cell types. These data show that NAM has potent antioxidative as well as anti-senescent effects. Moreover, these findings suggest that NAM can reduce cellular deterioration caused by oxidative damage in postmitotic cells in vivo.
Keywords: aging, antioxidant, nicotinamide, ROS, senescence
INTRODUCTION
Reactive oxygen species (ROS) accumulate at high levels as cells approach the end of their replicative potential-i.e., replicative senescence (Hwang et al., 2009). Conversely, senescence is accelerated by high ROS levels that cause oxidative damage to cell constituents. ROS-induced telomeric DNA strand breaks lead to accelerated telomere shortening, which results in an early onset of senescence (Serra et al., 2003; von Zglinicki, 2002). Mitochondrial damage is another important factor affecting cellular senescence and age-related diseases, because mitochondrial dysfunctions are the most prominent events leading to ROS generation (Finkel, 2000). Electron transport chain complexes are prone to electron leakage (Genova et al., 2004; Loschen and Azzi, 1975), and free electrons bind to nearby oxygen molecules producing superoxide radicals that cause damage to nearby mitochondria. This leads to the production of even higher levels of superoxide radicals. This vicious cycle of ROS production and mitochondrial damage is believed to be a major cause of senescence and aging (Dai et al., 2014; Passos, 2007). This hypothesis is well supported by the increased replicative lifespan of fibroblasts and mesenchymal stem cells cultivated under low-oxygen conditions (Packer and Fuehr, 1977; Parrinello, 2003). Therefore, suppression of mitochondrial ROS generation could be an effective strategy for delaying the onset of replicative senescence and preventing certain aging-associated diseases.
Nicotinamide (NAM), an amide derivative of vitamin B3, has been shown to positively effect the survival of variety of cell types (Maiese and Chong, 2003), and high-dose NAM treatments are currently being tested for their applicability to several conditions such as acute lung injury following ischemia/ reperfusion (Zhao et al., 2014). Ischemia/reperfusion-induced injury is caused by acute activation of poly (ADP-ribose) polymerase (PARP), which is triggered by ROS-induced DNA strand breaks (Liaudet et al., 2000). Activated PARP depletes cellular pools of NAD+ and ATP, causing cytotoxicity. Once taken up by cells, NAM is readily converted to NAD+ through the salvage pathway (Jackson et al., 1995; Liu et al., 1982), and has therefore been proposed to attenuate the effects of ischemia/reperfusion injury by providing cells with a pool of NAD+ or by inhibiting PARP activity (Burkart, 1999). However, whether NAM has antioxidative effects has not been investigated in these studies. NAM has been shown to inhibit protein oxidation and lipid peroxidation (Kamat and Devasagayam, 1999); however, the underlying mechanisms are not well understood.
Previously, we showed that treatment with 5 mM NAM substantially decreased cellular ROS levels (Kang et al., 2006), and prolonged NAM treatment delayed ROS accumulation and substantially increased the replicative lifespan of human fibroblasts (Kang et al., 2006). However, NAM treatment eventually stopped suppressing the increase in ROS levels, and ROS levels spiked in the NAM-treated fibroblasts toward the end of their extended lifespan. Therefore, whether NAM is able to suppress the high levels of ROS generated when cells begin or enter senescence is not clear. In the current study, we tested the effect of NAM on ROS levels by using two different senescence models: cancer cells in which senescence was induced by an Adriamycin pulse (induced senescence model) (Cho, 2011; Song, 2005) and senescent fibroblasts induced by long-term cultivation (replicative senescence model). In both sensecence models, NAM treatment substantially reduced ROS levels and the majority of the assessed senescence phenotypes. These data suggests that NAM, although incapable of reversing the state of senescence, can keep ROS level low, thereby preventing cellular deterioration caused by oxidative damage during the latter stage of the cellular lifespan as well as in cells in a postmitotic state.
MATERIALS AND METHODS
Cell cultivation and senescence induction
Normal human fibroblasts from a newborn foreskin were provided by Dr. Jinho Chung (Seoul National University, Korea) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Lonza, USA) in the presence or absence of 5 mM NAM in an ambient atmosphere. Fibroblasts were passaged (1:4 dilution), and the cultures had stopped growth at p36.5. Most cells at passage displayed senescence phenotypes. To assess the effects of NAM through flow cytometry, an equal number of cells were seeded on culture dishes. The next day, one dish was added NAM and another dish was mock treated, and both were incubated for 3 more days, and serve “3 days” and “0” sample, respectively. One additional dish was treated with NAM at the 3rd day, and incubated for one additional day, and the serves “1 day” sample. MCF-7 cells were cultured in DMEM supplemented with 10% FBS. To induce senescence, MCF-7 cells were pulsed with 0.25 μM Adriamycin (Sigma-Aldrich Co., USA) for 4 h and chased in fresh medium, which was replaced every 2 days.
Western blot analysis
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS). Typically, 30–40 μg of protein was separated by SDS-PAGE, transferred to nitrocellulose membrane (Hybond ECL; Amersham, USA), and immunoblotted using one of the following primary antibodies: human PARP1, p21WAF1, or ERK 1/2 (Santa Cruz Biotechnology, USA). Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies and SuperSignal WestFemto substrate (Pierce, USA).
ROS, lipofuscin, cell volume, and granule content measurements
To quantify cellular ROS levels, cells were incubated with 5 μM DHE, 0.1 μM MitoSox (Invitrogen), or 15 μM DHR123 (Anaspec) for 30 min. Cells were then washed with phosphate-buffered saline (PBS), trypsinized, collected in PBS containing 1 mM EDTA, and analyzed using a FACS Canto II cell analyzer (BD Biosciences, USA). To measure the lipofuscin levels, the collected cells were washed in PBS and directly analyzed by flow cytometry (488 nm excitation and 530 nm emission). Forward and side scattering values in the flow cytometry scatter plots were used to determine cell volume and granule content, respectively.
Cell cycle distribution analysis
At each time point, 1 × 106 cells were collected and stored in 70% ethanol. Cells were then stained with 10 μg/ml of propidium iodide in PBS containing 1 mM of EDTA and 0.2 mg/ml RNaseA. The raw flow cytometry data were analyzed using CellQuest 3.2 software (BD Biosciences).
In situ staining of senescence-associated β-galactosidase activity and β-galactosidase assay in solution
A senescence-associated β-galactosidase (SA β-Gal) assay was performed as described by Dimri et al. (1995). Briefly, cells that had been fixed with 3% formaldehyde were washed in PBS (pH 6.0) containing 2 mM MgCl2. After incubation overnight at 37°C in β-galactosidase staining solution (1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, 5 mM K3Fe[CN]6, 5 mM K4Fe[CN]6, and 2 mM MgCl2), cells were examined using bright-field microscopy. β-Gal activity in the solution was quantified as described previously (Lee et al., 2006). Briefly, cells were lysed in PBS by freeze-thawing. The supernatant was incubated at 37°C for 12 h in reaction buffer (10 mM MgCl2, 1.1 mg/ml chlorophenol red-β-d-galactopyranoside in 0.1 M phosphate buffer pH 6.0). The reaction was stopped by adding 1 M Na2CO3, and the absorbance was measured at 562 nm.
RESULTS
NAM treatment suppresses the increase in ROS levels and expression of senescence phenotypes in senescing MCF-7 cells
NAM has been shown to have an antioxidative effect in rapidly proliferating cells including human fibroblasts (Kang et al., 2006) and cancer cells (Supplementary Fig. 1). In this study, the effect of NAM was examined in cells in which proliferation had slowed and eventually stopped because of senescence. In this model of induced senescence, MCF-7 cells were chased after pulsing them with 0.25 μM Adriamycin (Song et al., 2005). During the chase period, cell proliferation slowed and senescence phenotypes progressively developed (Figs. 1 and 2), as had been reported previously (Cho et al., 2011; Song et al., 2005). As shown in Fig. 1, senescence progression was evidenced by the increase in SA β-Gal activity and cell volume. The level of lipofuscins-aggregates of oxidatively damaged proteins and lipids-increased substantially, indicating senescence was occurring (Fig. 2B). Similarly, the level of mitochondrial superoxide steadily increased. NAM treatment attenuated this increase; mitochondrial superoxide levels were increased during the first day, but were maintained at low levels during the chase period (Fig. 2A). Furthermore, NAM treatment suppressed the expression of senescence phenotypes: increased lipofuscin levels (Fig. 2B), cell size (Fig. 2C), and cellular granule content (Fig. 2D). SA β-Gal activity, as quantified by measuring β-Gal activity in solution, was reduced by approximately 13% in NAM-treated cells at all the tested time points (Fig. 2E). However, this suppressive effect was not robust enough to be detected in an SA β-Gal activity in situ assay (data not shown). Treatment of cells with 5 mM NAM after day 4 of the chase period was also sufficient to lower ROS levels and reduced the development of senescence phenotypes, which had been apparent at that stage (Supplementary Fig. 2). Together, these data show that the high levels mitochondrial ROS and the phenotypes in senescing MCF-7 cells were suppressed by NAM treatment.
Fig. 1.
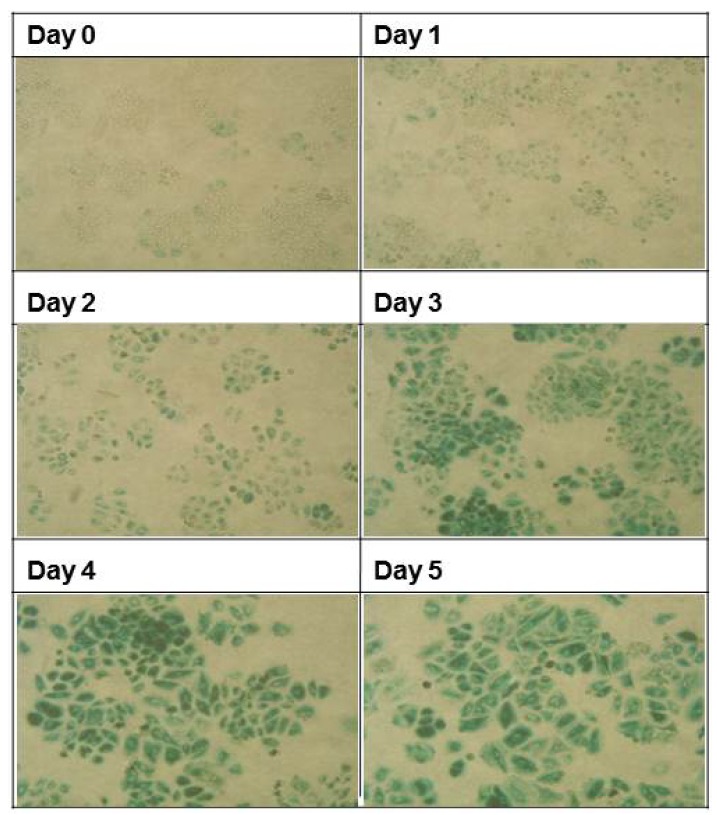
Changes in SA β-Gal activity in cells undergoing senescence progression. MCF-7 cells were pulsed with 0.25 μM Adriamycin for 4 h and further incubated in the absence of Adriamycin for the indicated times (chase phase). Cells were then stained for SA β-Gal activity in situ. Representative fields were chosen for imaging. The proportion of cells positive for SA β-Gal activity as well as cell size progressively increased over the course of the chase period.
Fig. 2.
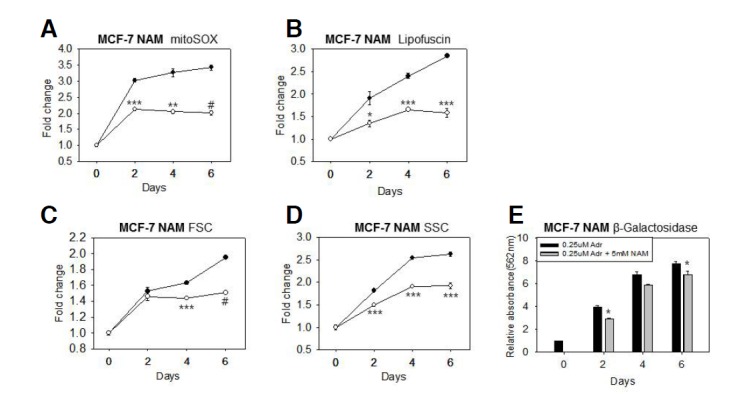
Effects of NAM on ROS levels and cellular changes associated with senescence progression. Adriamycin-treated MCF-7 cells were chased in the absence (─•─) or presence (─○─) of 5 mM NAM for 2, 4, or 6 days. Cells were then stained with MitoSox to determine the levels of mitochondrial superoxides (A) and/or analyzed directly by using flow cytometry to measure autofluorescence, which was used to determine lipofuscin levels (B). The forward (FSC) and side scattering (SSC) values from the flow cytometry data were plotted to show the change in cell volume (C) and granule content (D). In (E), the lysates of 1.8 × 105 cells were analyzed by performing a β- galactosidase assay in solution by using CPRG. The relative light absorbance values at 562 nm were plotted. In all the experiments, three independent biological samples were analyzed, and the most representative figures were presented. Statistical significance was assessed by two-tailed paired Student’s t-test. n = 3, *p < 0.05; **p < 0.01; ***p < 0.005; #p < 0.001.
NAM treatment reduces ROS levels and the appearance of senescence phenotypes in senescent fibroblasts
In senescent cells, high ROS levels and senescence phenotypes are already present and may not be readily suppressed by antioxidants (McFarland and Holliday, 1994; Rattan and Saretzki, 1994). Whether the high level of ROS in senescent cells can be reduced by NAM treatment was tested using fibroblasts that had already entered replicative senescence. A population of senescent fibroblasts (p36.5), along with two earlier passage populations (p19 and p26), was treated with 5 mM NAM for 3 days, and the ROS levels were then measured. NAM treatment of p19 and p26 fibroblasts reduced mitochondrial superoxide levels by approximately 10% and 15%, respectively, on day 1 of the treatment. Under our experimental conditions, the reduction in mitochondrial superoxide levels in response to NAM treatment was slightly smaller than what had been reported previously (Kang et al., 2006) (Fig. 3A). In senescent cells, mitochondrial superoxide levels were maintained at much higher levels, as we had predicted. Importantly, NAM treatment caused an even greater reduction in mitochondrial superoxide levels in these cells (∼20% on day 1) (Fig. 3A). NAM is effective in controlling levels of both mitochondrial and cytosolic superoxides, but had stronger effects on cytosolic superoxide (32%, 33%, and 45%, in p19, p26, and senescent cultures, respectively). Therefore, these results suggest that NAM treatment is able to reduce oxidative damage in senescent cells. Indeed, NAM treatment was effective in reducing lipofuscin levels in senescent cells by approximately 22% as well as in cells from the earlier passages (5–9% and 7% in p19 and p26 cells, respectively) (Fig. 3C). The increases in cell volume and granule content were also attenuated in senescent cell population (Figs. 3D and 3E). Importantly, there was a substantial reduction in SA β-Gal activity in the senescent cells in response to NAM treatment. Both the number of cells positive for β-Gal activity and the degree of activity were substantially reduced by NAM treatment (Fig. 4A). In addition, the overall β-galactosidase activity in the cells, measured in cell lysates, was reduced by 17% and 23% on days 1 and 3 of NAM treatment, respectively (Fig. 4B). Together, these data indicate that NAM treatment is able to suppress at least some of the senescence phenotypes that are already present in senescent cells. Moreover, these findings indicate that some of the phenotypes in senescent cells are reversible.
Fig. 3.

Effects of NAM on ROS levels and phenotypes expressed in senescent fibroblasts. Fibroblasts at an early passage (p19, ─•─), a late passage (p28, ─○─), or in senescence (p36.5, ─▾─) were incubated in the presence of 5 mM NAM for 1 or 3 days. The cells were then stained with MitoSox (A) or DHR123 (B) to determine the changes in the levels of mitochondria and cytosolic superoxide, respectively. Autofluorescence as well as the forward and side scattering values measured using flow cytometry were plotted to show the change in the lipofuscin levels (C), cell volume (D), and granule content (E). Statistical significance was assessed by two-tailed paired Student’s t-test. n = 3, *p < 0.05; **p < 0.01; ***p < 0.005; #p < 0.001.
Fig. 4.
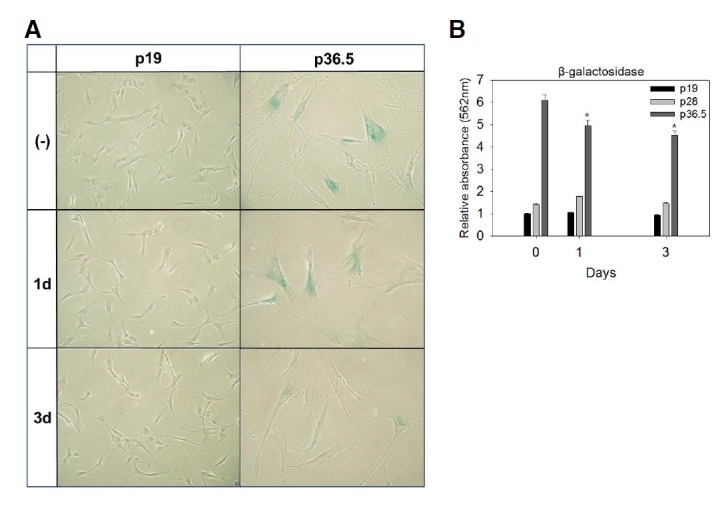
Effect of NAM on SA β-Gal activity in senescent fibroblasts. Early passage (p19) and senescent (p36.5) fibroblasts were incubated with 5 mM NAM for 1 or 3 days, and were then stained for SA β-Gal activity in situ. A group of well-stained cells were imaged in bright field. Cells at p36.5 were bigger and highly positive for SA β-Gal activity. In contrast, the NAM-treated cells were decreased in size and had a lower level of SA β-Gal activity (A). (B) Quantitative measurements by using CPRG also indicated that there was a reduction in the β-galactosidase activity in the NAM-treated senescent cells (p36.5). NAM treatment had least effect on the late-passage (p28) and early-passage cells (p19). The mean values from triplicate experiments were used to make the plot. The [*P < 0.05 (compared to the mock-treated cells (without NAM) by ANOVA test (Dunnett’s test)].
N-acetyl cysteine fails to suppress senescence-associated elevated ROS levels and phenotypes
The mechanism by which NAM reduces ROS levels is not yet known. We next tested if a ROS scavenger could induce similar changes in ROS levels and senescence phenotypes. To achieve this, senescence-induced MCF-7 cells and senescent fibroblasts were treated with N-acetyl cysteine (NAC), a potent ROS scavenger (Verhasselt et al., 1999) that has been shown to lower the basal levels of mitochondrial and cytosolic superoxides in various cell types at doses ranging from 1–5 mM (Spagnuolo et al., 2006; So-Young Jang, data not shown). Treatment of MCF-7 cells with 5 mM NAC from the start of the chase period after the Adriamycin pulse, however, did not affect the levels of superoxide or lipofuscin. Lipofuscin levels did decrease on day 6 of the chase (Figs. 5A–5C). Similarly, in fibroblasts, NAC treatment at doses of 1 and 5 mM failed to reduce mitochondrial and cytosolic superoxide levels. Instead, NAC treatment increased the levels of both mitochondrial and cytosolic superoxides as well as that of lipofuscins (Figs. 5D–5F). The cell volume and granule content were also not marked affected by NAC treatment (data not shown). The failure of NAC treatment to attenuate the increase in ROS during senescence and its progression indicates that this effect is not mediated generally by ROS-scavenging chemicals, but instead by certain activities of NAM. We speculate that the effects of NAM are mediated by the suppression of ROS generation, rather than through ROS scavenging.
Fig. 5.
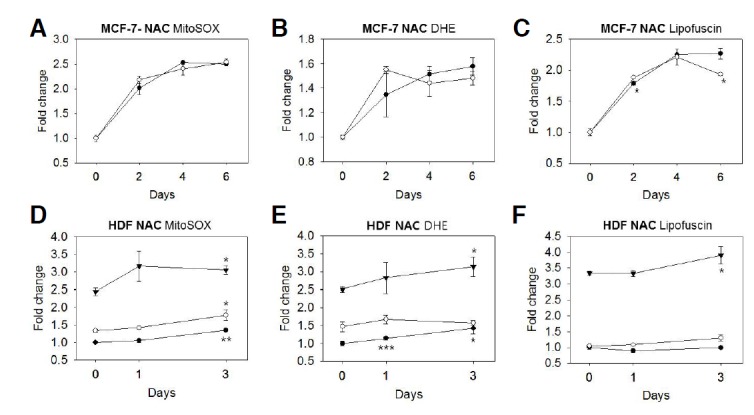
Effect of NAC on ROS levels and senescence-associated phenotype expression. (A–C) To determine the effect of NAC on the levels of ROS and lipofuscins during senescence progression, MCF-7 cells were cultivated in medium containing 5 mM NAM (─○─) or not containing NAM (─•─) from the start of the chase period and after the Adriamycin pulse. On days 2, 4, and 6 of the chase, cells were stained with MitoSox, DHR123, and/or were analyzed directly by using flow cytometry. (D–F) Fibroblasts at p19 (─•─) and p28 (─○─), and senescent cells (─▾─) were incubated in the presence of 5 mM NAM for 1 or 3 days, and then stained with Mito- and/or analyzed directly by using flow cytometry. In both MCF-7 cells and fibroblasts, the means of the measured values from one representative result among three biological repeats were presented as the relative levels of mitochondrial superoxide (A, D), cytosolic superoxide (B, E), or lipofuscins (C, F). Similarly, NAC treatment at a dose of 1 mM failed to effect these phenotypes (data not shown). Statistical significance was assessed by two-tailed paired Student’s t-test. n = 3, *p < 0.05; **p < 0.01; ***p < 0.005.
NAM alters cell cycle profiles but does not reinitiate proliferation of senescent cells
Senescent cells have higher levels of p53/p21WAF1 growth-arrest signaling and, therefore, become arrested at either the G1 or G2/M phase of the cell cycle (Hwang et al., 2009). Adriamycin-induced senescence is an example of G2/M arrest (Ling et al., 1996). We tested whether NAM-induced suppression of ROS and certain senescence phenotypes affected the state of the cell cycle. No increase in cell numbers or cell death were observed in senescence-induced MCF-7 cells or non-senescent fibroblasts during the 2-week period of cultivation in the presence of 5 mM NAM (data not shown), indicating that NAM was unable to reinitiate cell proliferation. Previously, NAM treatment was shown to attenuate increased p21WAF1 levels in a p53-independent manner (Lee et al., 2008). In this study, NAM treatment indeed attenuated the Adriamycin-induced increase in p21WAF1 levels in MCF-7 cells during the chase period (Fig. 6A, lanes 3 and 4 vs. 6 and 7), suggesting that the cell cycle profile was changed. To determine whether the cell cycle in MCF-7 cells is affected by NAM treatment during senescence progression, ploidy analysis was performed (Fig. 6B). Adriamycin-induced changes in cell cycle profile became apparent after 12 h of the chase; the S phase became shorter in duration, while the G2/M phase increased in duration (Fig. 6B). Importantly, the duration of the S phase was not markedly changed by NAM treatment (12 h + NAM), and became shorter at later time points (24 h + NAM), indicating that cells did not resume proliferation upon NAM treatment. Unexpectedly, however, NAM treatment caused a substantial decrease in the duration of the G2/M phase (from 44.9% to 28.0%) and an increase in the G1 phase (from 39.5% to 61.35%) at 24 h. We hypothesize that cells that were initially arrested at the G2/M phase in response to Adriamycin treatment somehow escaped the effects of NAM treatment and progressed through the cell cycle until they became arrested in the G1 phase. The decrease in p21WAF1 levels potentially contribute to this.
Fig. 6.
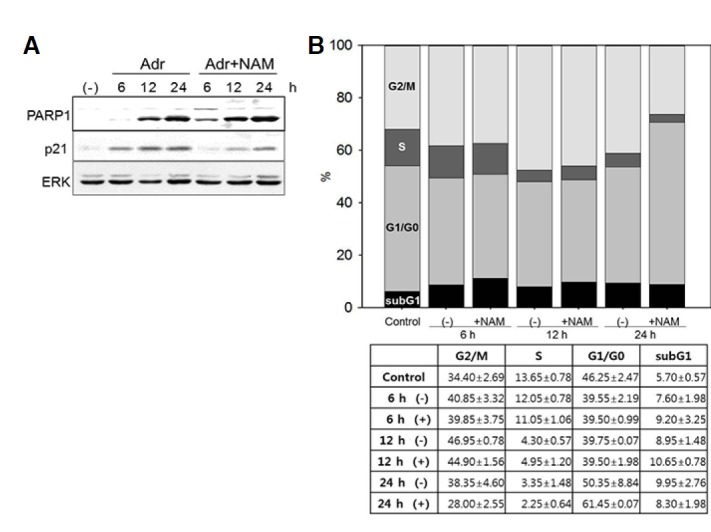
Effect of NAM on the cell cycle profiles of Adriamycin-pulsed MCF-7 cells. The Adriamycin-pulsed MCF-7 cells were chased in the absence or presence of 5 mM NAM. (A) Equal amounts of cell extracts were by performing Western blotting for PARP1, p21WAF1, and Erk proteins. The levels of Parp1 and p21WAF1 increased during the chase period. The increase of p21 levels were attenuated by the presence of NAM, while Parp1 levels were not. (B) At 6, 12, and 24 h of the chase period, cells were collected, stained with propidium iodide, and analyzed by flow cytometry. The proportions of cells in the G2/M (top section), S (second from the top) G1 (second from the bottom), and subG1 (bottom section) phases were plotted as bars. An increase in G2/M phase as well as a dramatic decrease in the S phase was apparent at the 12-h time point regardless of the presence of NAM. However, at the 24-h time point, G1 phase dramatically increased while G2/M decreased substantially in the NAM-treated population (+NAM, 24 h). Western blotting and cell cycle analyses were carried out with multiple repeats, and representative figures are presented. In (B), the averages of two measurements of the proportions of cells in the four different cell cycle phases are listed with their standard deviations.
DISCUSSION
In this study, we showed that NAM suppresses increasing ROS levels in senescent cells and cells undergoing senescence progression-two contexts in which ROS accumulate at high levels. The strong antioxidative effect of NAM in senescent cells suggests it has potential use in the prevention of aging-associated degenerative diseases that are attributed to accumulation of oxidative damage. For example, Alzheimer’s disease (AD) and Parkinson’s disease (PD) are known to be caused in part by ROS-induced oxidative stress in neurons. Certain nutritional antioxidants such as vitamin E have been shown to block neuronal death in vitro, and possibly exert protective effects against neurodegenerative diseases (Yatin et al., 2000). The protective effect of NAM in ischemia/reperfusion-induced tissue damage caused by acute oxidative stress (Zhao et al., 2014) suggests that NAM also functions in protecting neurons in AD, PD, and other oxidative stress-related neuronal diseases. The reduction of lipofuscin levels in senescent cells strongly suggests that NAM treatment can help reduce oxidative damage in postmitotic cells such as neurons.
NAM also suppressed certain senescence phenotypes. Accumulation of lipofuscins as well as the increases in cell volume, granule content, and SA β-Gal activity are reported here. Other senescence-associated changes that are caused by ROS or ROS-induced damage were also expected to be affected. Notably, the growth arrest state was not affected by NAM treatment. The cell cycle arrest caused by DNA damage signaling during cellular senescence cannot be reversed even if ROS levels are lowered. This implies that NAM cannot rejuvenate senescent tissues. However, NAM may still be useful in reducing the rate of deterioration in non-senescent postmitotic tissues.
As shown in our previous studies, an important result of long-term NAM treatment in human cells is a substantial delay in the expression of senescence phenotypes as well as an increase in replicative potential of primary human fibroblasts (over a 1.5-fold increase) (Kang et al., 2006). This is apparently different from the effects of other antioxidants. For example, the two potent antioxidants kinetin (Rattan and Clark, 1994) and carnosine (McFarland and Chong, 1994) only slightly delayed senescence progression in fibroblasts. In addition, N-t-butyl hydroxylamine, a spin-trapping agent, reportedly extends the lifespan of fibroblasts greatly by acting as a hydroxyl radical scavenger when it is applied to cells nearing senescence (Atamna et al., 2000). However, the magnitude of the effect of NAM is dependent on treatment duration and is cumulative. Even after NAM is removed, approximately 20 additional population doublings (PDs) occurred (Kang et al., 2006), suggesting that certain cellular pathways involved in controlling cell proliferation and ROS generation might be affected. In this regard, it is important to elucidate how NAM treatment reduces ROS levels. ROS levels in cells can be reduced by a decrease in proliferation or through cell removal. To date, the former possibility has not been thoroughly explored. However, in our previous work on the mechanism of NAM action, we proposed the involvement of a decrease in mitochondria via activation of autophagy (Jang et al., 2012). Mitochondria, especially in the cells passaged many times, produce high levels of superoxide radicals through a vicious cycle of ROS–mitochondrial damage (Melov, 2000). Therefore, rapid mitochondria turnover via autophagy would maintain ROS at low levels. However, it has yet to be shown that NAM-induced autophagy activation is indeed responsible for the reduction in ROS generation. Meanwhile, it cannot be ruled out that NAM directly reduces ROS levels by acting as a scavenger. NAM treatment reduces ROS levels before the mitochondria content is reduced (SY Jang, unpublished data). Therefore, an in-depth investigation into the mechanism underlying NAM-mediated ROS suppression is warranted.
The experimental paradigm used in this study to examine the effects of NAM on ROS levels and senescence phenotypes can be utilized as cellular model systems for pre-screening chemicals with anti-senescence or anti-aging potential. Cell must be cultivated for a couple of weeks before late-passage fibroblasts are available for testing the anti-senescence effects of chemicals.
Acknowledgments
This study was supported by a grant from University of Seoul, 2012 for Eun Seong Hwang and also by the R&D program of MOTIE/KIAT. (N0000697, Establishment of Infra Structure for Anti-aging Industry Support) for Cheol Min Kim.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Atamna H., Paler-Martinez A., Ames B.N. N-t-butyl hydroxylamine, a hydrolysis product of alpha-phenyl-N-t-butyl nitrone, is more potent in delaying senescence in human lung fibroblasts. J. Biol. Chem. 2000;275:6741–6748. doi: 10.1074/jbc.275.10.6741. [DOI] [PubMed] [Google Scholar]
- Burkart V., Blaeser K., Kolb H. Potent beta-cell protection in vitro by an isoquinolinone-derived PARP inhibitor. Horm. Metab. Res. 1999;12:641–644. doi: 10.1055/s-2007-978813. [DOI] [PubMed] [Google Scholar]
- Cho S., Park J., Hwang E.S. Kinetics of the cell biological changes occurring in the progression of DNA damage-induced senescence. Mol. Cells. 2011;6:539–546. doi: 10.1007/s10059-011-1032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai D.F., Chiao Y.A., Marcinek D.J., Szeto H.H., Rabinovitch P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan. 2014;3:6. doi: 10.1186/2046-2395-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O., et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- Genova M.L., Pich M.M., Bernacchia A., Bianchi C., Biondi A., Bovina C., Falasca A.I., Formiggini G., Castelli G.P., Lenaz G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann. N Y Acad. Sci. 2004;1011:86–100. doi: 10.1007/978-3-662-41088-2_10. [DOI] [PubMed] [Google Scholar]
- Hwang E.S., Yoon G., Kang H.T. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 2009;66:2503–2524. doi: 10.1007/s00018-009-0034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson T.M., Rawling J.M., Roebuck B.D., Kirkland J.B. Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP-ribose) levels but do not affect diethylnitrosamine-induced altered hepatic foci in Fischer-344 rats. J. Nutr. 1995;125:1455–14561. doi: 10.1093/jn/125.6.1455. [DOI] [PubMed] [Google Scholar]
- Jang S.Y., Kang H.T., Hwang. E.S. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012;287:19304–19314. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat J.P., Devasagayam T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox. Rep. 1999;4:179–184. doi: 10.1179/135100099101534882. [DOI] [PubMed] [Google Scholar]
- Kang H.T., Lee H.I., Hwang E.S. NAM extends replicative lifespan of human cells. Aging Cell. 2006;5:423–436. doi: 10.1111/j.1474-9726.2006.00234.x. [DOI] [PubMed] [Google Scholar]
- Lee B.Y., Han J.A, Im J.S., Morrone A., Johung K., Goodwin E.C., Kleijer W.J., DiMaio D., Hwang E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- Lee H.I., Jang S.Y., Kang H.T., Hwang E.S. p53-, SIRT1-, and PARP-1-independent downregulation of p21WAF1 expression in nicotinamide-treated cells. Biochem. Biophys. Res. Commun. 2008;368:298–304. doi: 10.1016/j.bbrc.2008.01.082. [DOI] [PubMed] [Google Scholar]
- Liaudet L., Soriano F.G., Szabó E., Virág L., Mabley J.G., Salzman A.L., Szabo C. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc. Natl. Acad. Sci. USA. 2000;97:10203–10208. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y.H., el-Naggar A.K., Priebe W., Perez-Soler R. Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol. Pharmacol. 1996;49:832–841. [PubMed] [Google Scholar]
- Liu G., Foster J., Manlapaz-Ramos P., Olivera B.M. Nucleoside salvage pathway for NAD biosynthesis in Salmo nella typhimurium. J. Bacteriol. 1982;152:1111–1116. doi: 10.1128/jb.152.3.1111-1116.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschen G., Azzi A. On the formation of hydrogen peroxide and oxygen radicals in heart mitochondria. Recent Adv. Stud. Cardiac. Struct. Metab. 1975;7:3–12. [PubMed] [Google Scholar]
- Maiese K., Chong Z.Z. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol. Sci. 2003;24:228–232. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- McFarland G.A., Holliday R. Retardation of the senescence of cultured human diploid fibroblasts by carnosine. Exp. Cell Res. 1994;212:167–175. doi: 10.1006/excr.1994.1132. [DOI] [PubMed] [Google Scholar]
- Melov S. Mitochondrial oxidative stress. Physiologic consequences and potential for a role in aging. Ann. N Y Acad. Sci. 2000;908:219–225. doi: 10.1111/j.1749-6632.2000.tb06649.x. [DOI] [PubMed] [Google Scholar]
- Packer L., Fuehr K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature. 1997;267:423–425. doi: 10.1038/267423a0. [DOI] [PubMed] [Google Scholar]
- Parrinello S., Samper E., Krtolica A., Goldstein J., Melov S., Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos J.F., Saretzki G., von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattan S.I., Clark B.F. Kinetin delays the onset of ageing characteristics in human fibroblasts. Biochem. Biophys. Res. Commun. 1994;201:665–672. doi: 10.1006/bbrc.1994.1752. [DOI] [PubMed] [Google Scholar]
- Serra V., von Zglinicki T., Lorenz M., Saretzki G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J. Biol. Chem. 2003;278:6824–6830. doi: 10.1074/jbc.M207939200. [DOI] [PubMed] [Google Scholar]
- Song Y.S., Lee B.Y., Hwang E.S. Distinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech. Ageing Dev. 2005;126:580–590. doi: 10.1016/j.mad.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Spagnuolo G., D’Anto V., Cosentino C., Schmalz G., Schweikl H., Rengo S. Effect of N-acetyl-L-cysteine on ROS production and cell death caused by HEMA in human primary gingival fibroblasts. Biomaterials. 2006;27:1803–1809. doi: 10.1016/j.biomaterials.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Verhasselt V., Vanden Berghe W., Vanderheyde N., Willems F., Haegeman G., Goldman M. N-acetyl-L-cysteine inhibits primary human T cell responses at the dendritic cell level: association with NF-kappaB inhibition. J. Immunol. 1999;162:2569–2574. [PubMed] [Google Scholar]
- Von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Yatin S.M., Varadarajan S., Butterfield D.A. Vitamin E prevents Alzheimer's amyloid beta-peptide (1–42)-induced neuronal protein oxidation and reactive oxygen species production. J. Alzheimers Dis. 2000;2:123–131. doi: 10.3233/jad-2000-2212. [DOI] [PubMed] [Google Scholar]
- Zhao W., Gan X., Su G., Wanling G., Li S., Hei Z., Yang C., Wang H. The interaction between oxidative stress and mast cell activation plays a role in acute lung injuries induced by intestinal ischemia-reperfusion. J. Surg. Res. 2014;187:542–552. doi: 10.1016/j.jss.2013.10.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.