

Abstract
Germline mutations in the breast cancer type 2 susceptibility gene (BRCA2) are linked to familial breast cancer and the progressive bone marrow failure syndrome Fanconi anaemia. Established Brca2 mouse knockout models show embryonic lethality, but those with a truncating mutation at the C-terminus survive to birth and develop thymic lymphoma at an early age. To overcome early lethality and investigate the function of BRCA2, we used T cell-specific conditional Brca2 knockout mice, which were previously shown to develop thymic lymphoma at a low penetrance. In the current study we showed that the number of peripheral T cells, particularly naïve pools, drastically declined with age. This decline was primarily ascribed to improper peripheral maintenance. Furthermore, heterozygous mice with one wild-type Brca2 allele manifested reduced T cell numbers, suggesting that Brca2 haploinsufficiency might also result in T cell loss. Our study reveals molecular events occurring in Brca2-deficient T cells and suggests that both heterozygous and homozygous Brca2 mutation may lead to dysfunction in T cell populations.
Keywords: breast cancer type 2 susceptibility gene (BRCA2), knockout mouse, T cell
INTRODUCTION
Germline mutations in the breast cancer type 2 susceptibility gene (BRCA2) are linked to familial breast cancer and the progressive bone marrow failure syndrome Fanconi anaemia (FA) (Ford et al., 1998; Howlett et al., 2002). Deleterious mutation in one BRCA2 allele predisposes carriers to breast and ovarian cancer with a 30–60% and 2–19% cumulative risk respectively (King et al., 2003; Risch et al., 2006). The incidence of other cancers is also increased, but at a much lower rate (Moran et al., 2012). BRCA2 is also known as FANCD1, one of 16 genes shown to be responsible for FA (Kupfer, 2013). Fanconi anaemia is a recessive disorder and both alleles of BRCA2 are mutated or inactivated in FA type D1 (Howlett et al., 2002). Bone marrow failure is the most common pathology in FA and approximately 30% of patients develop haematologic and solid tumours (Alter et al., 2003).
Since the identification of BRCA2 mutations in patients with hereditary breast cancer, various Brca2 mutant mice have been generated (Evers and Jonkers, 2006). None of the heterozygous mice display strong tumour predisposition whereas homozygous mice with a truncating mutation exhibit embryonic lethality (Bennett et al., 2000; Ludwig et al., 1997; Sharan et al., 1997; Suzuki et al., 1997; Yan et al., 2004). In some knockout models, 10–20% of the mice survive to birth and develop thymic lymphoma (Connor et al., 1997; Friedman et al., 1998). Despite the discrepancy in tumour susceptibility and tumour spectrum, mouse models have enhanced our understanding of the biology associated with human BRCA2 mutation (Lee et al., 1999; Patel et al., 1998). However, detailed analysis of Brca2 function has been hampered by this lethality, therefore conditional knockout mice have been generated (Cheung et al., 2004; Jonkers et al., 2001; Ludwig et al., 2001; McAllister et al., 2002). These mice have been vital tools for delineating the tumour suppressor activity and molecular function of BRCA2.
We aimed to use the conditional knockout system to study the role of Brca2 in T cells because these are the primary cell type affected by Brca2 deficiency in mice. We bred mice with a floxed Brca2 allele (Jonkers et al., 2001) to Lck-Cre transgenic mice, and previously reported that the [Brca2F11/F11; Lck-Cre] mice developed thymic lymphoma at a low penetrance (Park and Lee, 2008). Except for the small number of mice that developed thymic lymphoma, the mice remained normal and healthy. In this study, we closely examined the Brca2-deficient T cells and found a gradual loss of T cell populations, particularly the naïve pools. Brca2-deficient T cells showed activation of the p53 pathway, supporting the notion that activation of p53 induces apoptosis of Brca2-deficient cells (Cheung et al., 2002; Jonkers et al., 2001). Importantly, heterozygous mice also exhibited a gradual loss of T cells. Taken together, our findings suggest that a heterozygous germline BRCA2 mutation may confer immune dysfunction and that mature naïve T cell populations are highly susceptible to death triggered by Brca2 deficiency.
MATERIALS AND METHODS
Mice and preparation of tissues
Brca2F11/F11 and lck-Cre mice were kind gifts from Dr. Anton Berns (The Netherland Cancer Institute, The Netherlands). These mice were backcrossed to the FVB/N background for more than 10 generations to generate Brca2 conditional knockout mice. All experiments were approved by the Institutional Animal Care and Use Committees of Seoul National University, and followed the guidelines of Policy and Regulation for the Care and Use of Laboratory Animals.
The thymus and spleen of mice were placed in ice-cold PBS and ground with frosted slides to give a single cell suspension. The suspension was centrifuged at 400 × g for 10 min and red blood cells were lysed with ACK lysis buffer (155 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA). Cells were washed with PBS and resuspended in RPMI-1640 medium (Hyclone, USA) supplemented with 10% FCS (Hyclone), penicillin/streptomycin, L-glutamine, HEPES, sodium pyruvate, NEAA, and β-mercaptoethanol. Supplements and chemicals were obtained from Sigma (USA)
Flow cytometry analysis
The lymphocyte suspensions were washed in PBS containing 1% BSA and 0.01% sodium azide and incubated with various antibodies for 45 min at 4°C. Stained cells were analysed using the FACS Canto (BD Biosciences, USA). The following antibodies were used for staining: FITC-anti-B220, PE-anti-CD3, FITC-anti-CD44, PE-anti-CD62L from Biolegend (USA); and PerCP-anti-CD4 and APC-anti-CD8β.2 (Ly-3.2) from BD Pharmingen (USA).
Western blot analysis
Mouse tissues or cell pellets were homogenised in NETN buffer (150 mM NaCl, 20 mM Tris-Cl pH8.0, 0.5% v/v Nonidet P-40, 1 mM EDTA, 1 mM phenylmethanesulphonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml pepstatinA, 2 μg/ml Na3VO4, and 1 μg/ml leupeptin). Lysates (100–200 μg) were heated at 55°C for 15 min and separated by SDS-PAGE for Western blotting. The following antibodies were used: sheep-anti-BRCA2 antibody made in our laboratory (Choi et al., 2012), anti-p53 (rabbit polyclonal) and anti-p21 antibodies from Santa Cruz Biotechnology (USA), anti-phospho-p53 (human pSer15/mouse pSer18) antibody from Cell Signaling Technologies (USA), anti-PUMA antibody from AbCam (UK), and anti-actin antibody from Sigma-Aldrich.
Purification and culture of CD4+ T cells
CD4+ T cells were purified from spleen using anti-CD4 (L3T4) antibody-magnetic beads (Miltenyi Biotech, Germany), according to the manufacturer’s instructions. Purified CD4+ T cells (2 × 106 cells/ml) were stimulated by plate-bound anti-CD3 (2C11, Biolegend) and anti-CD28 (PV-1, Southern Biotech, USA) antibodies.
T cell receptor spectratyping
Total RNA was extracted from CD4+ splenic T cells using Trizol (Invitrogen, USA) and reverse transcribed with oligo-dT primer using the SuperScript II RT kit (Invitrogen). cDNA was amplified using various primers flanking the CDR3 regions to determine the CDR3 size distribution according to the manufacturer’s instructions (BioMed Immunotech, USA).
Cytokine ELISA
Interleukin-2, IFN-γ, and IL-4 levels were measured by capture ELISA using a standard protocol. Briefly, 96-well plates (Nunc, Denmark) were coated with 2 μg/ml anti-IL-2 (JES6-1A12), anti-IFN-γ (XMG-1.2), or anti-IL-4 (11B11) antibodies in PBS. After blocking in PBS/2% BSA, culture supernatants were incubated for 2 h Captured cytokines were labelled with biotinylated antibodies (clone JES6-5H4, R4-6A2, or BVD6-24G2 for each cytokine) and streptavidin-HRP, and developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrates. All reagents for ELISA were from Biolegend except for the recombinant cytokines used as standards, which were from R&D Systems (USA).
Measurement of anti-ovalbumin antibody titres
Mice were immunized with chicken ovalbumin (Sigma) in complete Freund’s adjuvant twice at 2-week intervals, and bled 4 weeks later. Sera were collected and subjected to an anti-ovalbumin specific ELISA. Briefly, 96-well plates (Greiner, Germany) were coated with 2 mg/ml ovalbumin and blocked with PBS/2% BSA. Sera were diluted in PBS/1% BSA and incubated in the ovalbumin-coated plates for 2 h. Captured ovalbumin-specific antibodies were labelled with biotinylated anti-IgM, IgG1, or IgG2a mouse antibodies (Calbiochem, USA) and streptavidin-HRP, and then developed with a TMB substrate.
RESULTS
Mutant protein expression in [Brca2F11/F11; Lck-Cre] mice
We used Brca2F11 (Brca2tm1Brn) mice harbouring loxp sites flanking exon 11 (Jonkers et al., 2001) to monitor the molecular events occurring in Brca2-deficient T cells. In previous studies, Brca2 knockout mice targeting different regions of the gene showed variable degrees of lethality and tumour susceptibility, suggesting a partial function of truncated or mutant Brca2 proteins (Evers and Jonkers, 2006). To clarify the status of Brca2 expression in [Brca2F11/F11; Lck-Cre] mice, we generated a sheep polyclonal antibody using the Brca2 C-terminal fragment (Choi et al., 2012). This antibody detected the Brca2 protein at highest levels in the thymus, spleen, and testis of young mice (Fig. 1A). This expression profile recapitulated the Brca2 expression pattern previously assessed at the mRNA level (Callens et al., 2002; Flores et al., 2002; Rajan et al., 1997). Interestingly, we detected a band similar in size to the exon 11-deleted Brca2 protein in the [Brca2F11/F11; Lck-Cre] thymus (Fig. 1B). The truncated Brca2 is likely to span all Brca2 exons except for the floxed-out exon 11, as indicated by the RT-PCR results (Fig. 1C). These findings demonstrate that [Brca2F11/F11; Lck-Cre] mice express a mutant Brca2 protein. Notably, many BRCA2 mutations found in patients with breast cancer generate truncated or mutated proteins (Couch et al., 1996; Goggins et al., 1996; Spain et al., 1999).
Fig. 1.
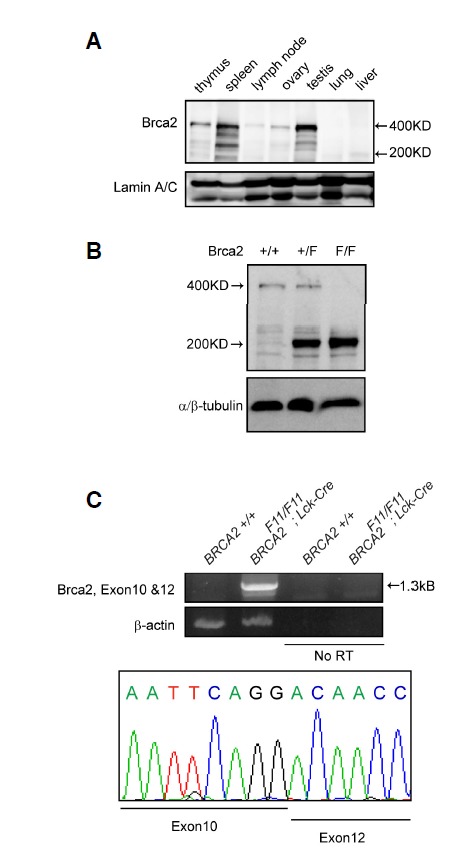
BRCA2 protein lacking exon11 is expressed in [Brca2F11/F11; Lck-Cre] mice. (A) Brca2 protein expression was detected in various mouse tissues. Each tissue was freshly isolated from 3-week-old mice, and 120 μg of protein was separated by SDS-PAGE and subjected to Western blot analysis. Lamin A/C was used as the loading control. (B) Western blot analysis of Brca2 was performed with thymic lysates from WT, [Brca2F11/+; Lck-Cre], and [Brca2F11/F11; Lck-Cre] mice. WT mice express the 400-kDa full-length Brca2 protein and [Brca2F11/F11; Lck-Cre] mice express the ∼220-kDa Brca2 fragment. (C) To analyse the Brca2 fragment from the [Brca2F11/F11; Lck-Cre] mice, RT-PCR was performed on thymic T cells. Sequence analysis of the 1.3-kb PCR product demonstrated joining of exons 10 and 12.
Loss of splenic T cells in [Brca2F11/F11; Lck-Cre] mice
In our mouse model Brca2 deletion should occur from the thymocytes because [Brca2F11/F11;Lck-Cre] mice express Cre recombinase under the proximal lck (p56) promoter (Gu et al., 1994). Therefore, to define the effects of Brca2 deletion, we first examined thymocytes and found that the number of thymocytes was slightly diminished in [Brca2F11/F11; Lck-Cre] mice (Fig. 2A). The diminution was more apparent in the double positive T cells compared to the single CD4+ or CD8+ T cells. Nevertheless, the reduction was not statistically significant and thymic T cell profiles remained unaltered (Fig. 2B). It was previously reported that Brca2 is not required for T cell development (Cheung et al., 2002; Patel et al., 1998). The non-involvement of Brca2 in the thymic developmental process was also evident from the identical T cell receptor (TCR) repertoire in splenic T cells from [Brca2F11/F11; Lck-Cre] mice compared with that in wild type (WT) mice (Fig. 2D). With age, the thymus degenerated and the number of thymocytes declined drastically in both WT and [Brca2F11/F11; Lck-Cre] mice, resulting in the same low cell numbers by week 12.
Fig. 2.
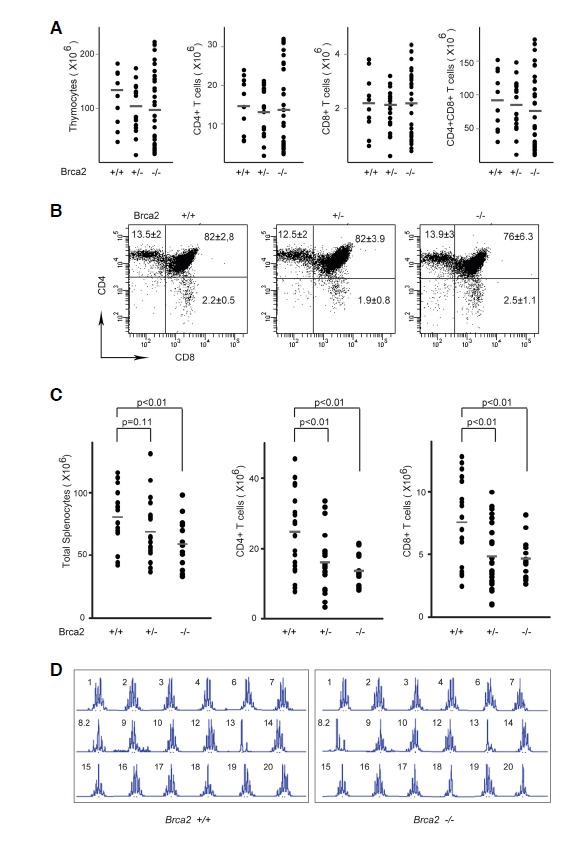
Brca2 deficiency leads to T cell loss. (A) Thymic T cells were isolated from 3- to 5-week-old WT, [Brca2F11/+; Lck-Cre], or [Brca2F11/F11; Lck-Cre] mice and trypan blue-negative cells were counted. Numbers of CD4+, CD8+, or CD4+CD8+ cells were calculated from flow cytometric analysis. Data were generated from 10–34 mice for each group and each dot represents an individual mouse. (B) The thymic T cell profile showed comparable composition of double negative (DN), double positive (DP), and single positive (SP) cells. The percent of cells in each quadrant is shown as the average+/−SD from 10–34 mice per group. (C) Splenocytes were isolated from 24- to 42-week-old WT, [Brca2F11/+; Lck-Cre], or [Brca2F11/F11; Lck-Cre] mice and trypan blue-negative cells were counted. Numbers of CD4+ or CD8+ cells were calculated from flow cytometric analysis. CD4:CD8 ratios for each group were 3.3 ± 0.8, 3.5 ± 0.8, 2.9 ± 0.3 for the WT, heterozygous, and homozygous mutant mice respectively. Each dot represents an individual mouse (14–25 mice per group). (D) Splenic CD4+ T cells showed a similar level of CDR3 diversity in WT and [Brca2F11/F11;Lck-Cre] mice. Peaks represent the intensity of the PCR product at a region from Vβ1 to Vβ24. Experiments were repeated twice.
Next, we examined mature T cells in the spleen. Although thymic T cells were not much affected by Brca2 deficiency, splenic T cell numbers were significantly reduced in the [Brca2F11/F11; Lck-Cre] mice (Fig. 2C). This decrease was observed for both CD4+ and CD8+ T cell populations, but not for B cells (Fig. 3A). Interestingly, the reduction in splenic T cell number was also evident in the heterozygous [Brca2F11/+; Lck-Cre] mice, suggesting haploinsufficiency of Brca2.
Fig. 3.
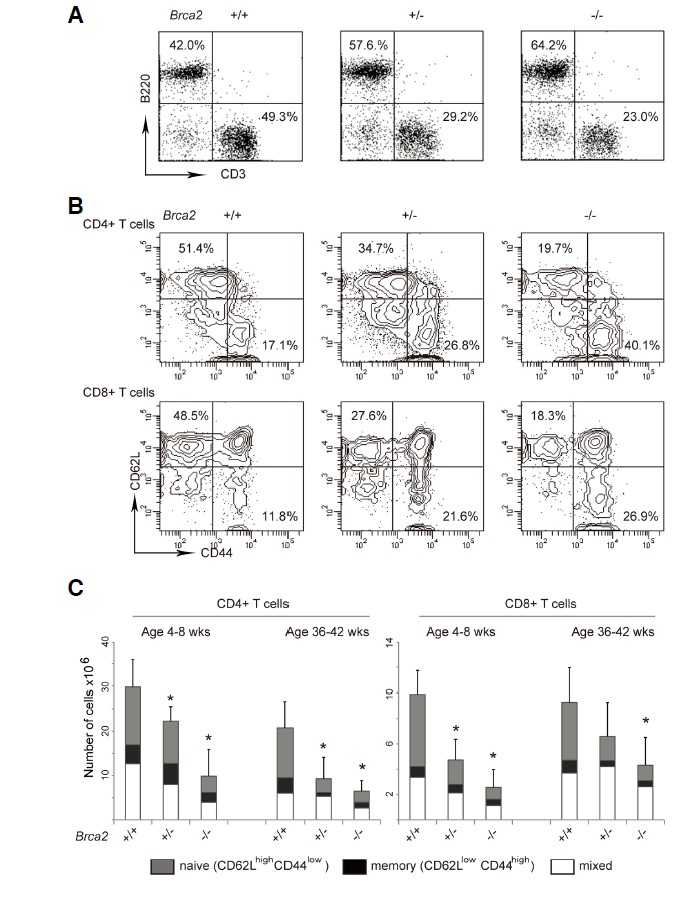
Loss of naïve T cells in [Brca2F11/F11; Lck-Cre] mice. Splenic T cells were isolated from 4–8-week-old or 36–42-week-old WT, [Brca2F11/+; Lck-Cre], or [Brca2F11/F11; Lck-Cre] mice. (A) Cells were stained with FITC-ati-B220 and PE-anti-CD3 antibodies. Decreased T cell and increased B cell proportions are shown in the representative plots. Overall number of B cells remained unaltered. (25.9 × 106 ± 8.2; 20 × 106 ± 6.5; 21.7 ± 8 for WT; heterozygous; and homozygous mice respectively) (B) Cells were stained with [PerCP-anti-CD4, APC-anti-CD8, FITC-anti-CD44, and PE-anti-CD62L] antibodies. CD4+ or CD8+ T cells were gated and percentages of cells with CD44low CD62Lhigh expression were calculated from the flow cytometry data. Representative contour plots for activation marker expression (CD44 vs.CD62L) on the gated CD4+ or CD8+ cells are shown. (C) Data were shown as average +/− SD from 3–8 mice for each genotype. P values < 0.05 from t-tests with the WT are marked as asterisks in the figure.
Loss of naïve T cells in [Brca2F11/F11; Lck-Cre] mice
To further characterize the T cell populations susceptible to Brca2 deficiency, we examined surface markers that distinguish naïve or activated/memory T cells, i.e. CD44 and CD62L (Dutt et al., 2007; Dutton et al., 1998). As shown in Figs. 3B and 3C, we found that the decrease in cell number was most dramatic for T cell populations with CD44lowCD62Lhigh naïve surface marker expression. The reduction in total T cell numbers and disappearance of naïve T cell populations suggest that Brca2 is required for the proliferation/survival of slowly proliferating peripheral T cells. Disappearance of the naïve T cell pool also suggests that immune function might be compromised in the [Brca2F11/F11; Lck-Cre] mice.
Reduced immune response in [Brca2F11/F11; Lck-Cre] mice
In the next experiment, we examined whether Brca2 deficient cells manifest functional impairment. First, we stimulated CD4+ T cells with plate-bound anti-CD3 and anti-CD28 antibodies to examine cytokine production. Because Brca2-deficient mice have a lower percentage of T cells in the spleen, we purified CD4+ T cells and used a constant number of cells in the assay. As shown in Fig. 4A, Brca2-deficient T cells produced similar levels of IL-2 to wild-type cells, indicating that Brca2-deficient T cells were competent for IL-2 production. However, these cells produced less IFN-γ and IL-4, suggesting a functional defect of Brca2-deficient T cells. Notably, there was more profound reduction in the IL-4 production.
Fig. 4.

T cells from [Brca2F11/F11;Lck-Cre] mice show reduced immune responses. (A) CD4+ T cells were isolated from WT or [Brca2F11/F11; Lck-Cre] mice and stimulated with plate-bound anti-CD3 and anti-CD28 antibodies. Culture supernatants were collected after 48 h and subjected to cytokine ELISA. Experiments were repeated twice. The p values < 0.05 are marked as asterisks. (B) Sera were collected from WT and [Brca2F11/F11; Lck-Cre] mice 4 weeks after 2nd immunization with chicken ovalbumin, and the level of ovalbumin-specific antibodies were determined. Experiments were repeated twice and data points show average +/− SEM from 6 mice. Statistical significance was determined at dilution points giving ∼50% maximum O.D. readings, and the p-values < 0.05 are marked with asterisks for IgM and IgG1.
Second, we determined the effect of Brca2 deficiency on T cell function in vivo by assessing T cell-dependent antibody responses. We immunized Brca2 WT [Brca2+/+; Lck-Cre] or Brca2-deficient [Brca2F11/F11; Lck-Cre] mice with chicken ovalbumin in complete Freund’s adjuvant twice at a two-week interval. After 4 weeks, we collected the immune serum and measured ovalbumin-specific IgM, IgG1, and IgG2a isotype antibody levels. As shown in Fig. 4B, Brca2-deficient mice produced lower levels of ovalbumin-specific antibodies than WT mice. Notably, Brca2 deficiency had a more profound effect on production of IgM (9-fold reduction) compared with IgG1 (3-fold reduction) or IgG2a (less than 3-fold reduction) isotypes. Overall, these data demonstrate that Brca2 deficiency compromised T cell function.
Activation of the p53 pathway in Brca2-deficient T cells
Brca2 is an essential regulator of homologous recombination, a vital pathway for error-free repair of DNA double strand breaks (Thorslund and West, 2007). Therefore, in the absence of functional Brca2 proliferating cells accumulate DNA double strand breaks, which activate the p53 checkpoint (Connor et al., 1997; Sharan et al., 1997). The activation of p53 might trigger cell cycle arrest or cell death and could explain the T cell deficit in Brca2-deficient mice. To investigate whether p53 was activated in the Brca2-deficient T cells, we assessed the expression and phosphorylation of p53. In response to DNA damages, p53 is known to undergo extensive post-translational modifications and to become stabilized and activated (Dai and Gu, 2010). Phosphorylation at Ser15 in human p53 has been shown to relieve the inhibition or degradation of p53 by MDM2 (Shieh et al., 1997), whereas the mouse equivalent pSer18 has been implicated in the pro-apoptotic function of p53 (Sluss et al., 2004). As shown in Fig. 5, p53 protein expression was increased to a moderate extent in thymic and splenic T cells of the [Brca2F11/F11; Lck-Cre] mice (Figs. 5A and 5B). Upon T cell activation, up-regulation and phosphorylation (pSer18) of p53 was apparent in the Brca2-deficient T cells (Fig. 5C). We then examined several p53 downstream targets, such as p21 and Puma. The induction of p21 and Puma has been linked to cell cycle arrest and apoptosis respectively (Jung et al., 2010; Yu et al., 2003). Moderate induction of both p21 and Puma was observed in thymic and splenic T cells in the [Brca2F11/F11; Lck-Cre] mice (Figs. 5A and 5B). Upon T cell activation, up-regulation of p21 and Puma was clearly observed in Brca2-deficient T cells (Fig. 5C). Together, these results demonstrate the activation of p53 pathway in Brca2-deficient T cells and suggest that the p53 checkpoint might be responsible for the loss of Brca2-deficient T cells.
Fig. 5.
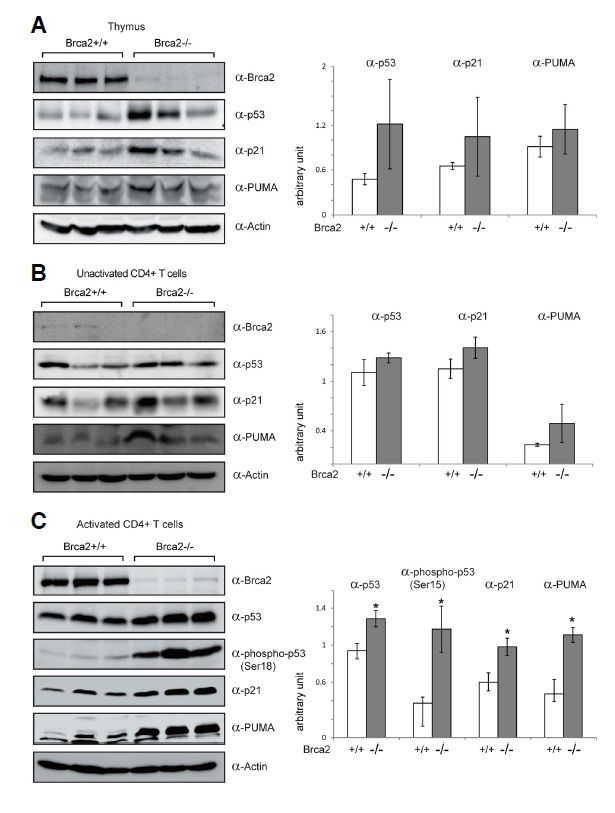
The p53 pathway is activated in T cells of [Brca2F11/F11; Lck-Cre] mice. Western blot analysis was performed on the thymic (A) and CD4+ splenic T cells as fresh (B) or 48 h after anti-CD3 and anti-CD28 antibody stimulation (C), from the WT and [Brca2F11/F11; Lck-Cre] mice. Actin was used as a loading control. Signals for p53 phosphorylation (mouse pSer18) were detectable only in activated T cells. Western blot data were quantified by densitometry and normalized by Actin signals (bar graphs on the right). On the bar graphs, Y axis is an arbitrary unit and each bar represents average +/− SD from 3 mice. The p values < 0.05 are marked as asterisks.
DISCUSSION
In this study we used conditional Brca2 knockout mice to investigate the role of BRCA2 in T cells. The [Brca2F11/F11; Lck-Cre] mice exhibited gradual loss of splenic T cells and impaired T cell-dependent immune function. These findings suggest that BRCA2 is required to maintain adequate T cell numbers as well as functional capacity. As heterozygous [Brca2F11/+; Lck-Cre] mice also manifested T cell loss, albeit to a lower extent, our findings suggest that individuals with a single allelic BRCA2 mutation may suffer from T cell deficiency.
Previous reports that mice with a Brca2 deletion had no overt T cell phenotype suggested that Brca2 might be dispensable for T cells (Cheung et al., 2002; Patel et al., 1998). We demonstrated that this is not the case, as our mice developed T cell loss and dysfunction. One difference between our mice and others is that the conditional allele targeted a different region. The targeted allele for our mice was selected based on the frequent mutations found in BRCA2 exon 11 (Jonkers et al., 2001; Tavtigian et al., 1996). The 6174delT mutation found in the Ashkenazi Jewish population (Neuhausen et al., 1996) is also present within exon 11 and generates a truncated BRCA2 protein (Goggins et al., 1996; Spain et al., 1999). Our mice expressed a mutant form of Brca2 that spanned all exons except for the floxed-out exon 11. Because exon 11 contains most of the BRC repeats, a series of unique BRCA2 domains that associate with Rad51 (Pellegrini et al., 2002; Wong et al., 1997), the mutant Brca2 would not be fully functional. The variable phenotypes among mouse models suggest that each truncated or mutant Brca2 protein retains BRCA2 function to a different degree.
The best characterized molecular function of BRCA2 is the control of homologous recombination through recruitment of the Rad51 recombinase to damaged DNA sites (Holloman, 2011). BRCA2 deficiency therefore results in the failure of Rad51-mediated DNA double strand break repair, leading to accumulation of DNA damage (Marx, 1997). In [Brca2F11/F11; Lck-Cre] mice, the T cell loss initiated in the thymus but only became apparent in the periphery. Our interpretation of this observation is that T cell loss is a cumulative phenotype associated with accumulation of DNA damage during cell division. Alternatively, but not exclusively, mature naïve T cells could be more susceptible to cell death or cell cycle arrest inflicted by Brca2 deficiency. The differential effect of Brca2 deficiency in cytokines or antibody isotypes may also indicate differential susceptibility of the responsible cell types. Further studies are required to address whether T cell loss has been caused by the intrinsic property of particular cell types.
The Brca2-deficient T cells of our mouse model showed prominent activation of the p53 pathway. Activation of p53 has been suggested in many other Brca2-deficient mouse models and is thought to determine the cell fate between survival and death (Lee et al., 1999; Patel et al., 1998). Brca2-deficient cells may be lost in the presence of fully functional p53, as in our study. In other situations, where inactivation or mutation of p53 is achieved, Brca2-deficient cells may survive and become tumorigenic. This dual hypothesis is well corroborated by the pathology related to BRCA2 deficiency (bone marrow failure or cancer) and supported by two major observations. First, breast cancer patients with BRCA2 mutation have more p53 mutations than those with sporadic tumours (Gretarsdottir et al., 1998; Ramus et al., 1999). Second, heterozygous Brca2-deficient mice rarely develop spontaneous tumours whereas crosses on the p53-deficient background show a greatly increased tumour incidence (Cheung et al., 2002; Jonkers et al., 2001). Although we have not directly addressed whether the activation of p53 caused T cell loss, concomitant up-regulation of p53 and its downstream targets strongly suggests that the dual hypothesis applies to Brca2-deficient T cells. Taken together, these studies uncover molecular events occurring in Brca2-deficient T cells and suggest that T cells may be a major cell type affected by both heterozygous and homozygous mutations of human BRCA2.
Acknowledgments
Brca2f11and Lck-Cre mice were a gift from A. Berns (NCI, The Netherlands). This study was supported by the Basic Science Research Program Grant 2012-R1A1A3010579 awarded to Hae-Ock Lee and 2011-0018630 awarded to Hyunsook Lee, through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology.
REFERENCES
- Alter B.P., Greene M.H., Velazquez I., Rosenberg P.S. Cancer in Fanconi anemia. Blood. 2003;101:2072. doi: 10.1182/blood-2002-11-3597. [DOI] [PubMed] [Google Scholar]
- Bennett L.M., McAllister K.A., Blackshear P.E., Malphurs J., Goulding G., Collins N.K., Ward T., Bunch D.O., Eddy E.M., Davis B.J., et al. BRCA2-null embryonic survival is prolonged on the BALB/c genetic background. Mol. Carcinog. 2000;28:174–183. doi: 10.1002/1098-2744(200007)28:3<174::aid-mc6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Callens N., Dumont M., Begue A., Lint C., Baert J.L., Simard J., de Launoit Y. Genomic organization and expression of the mouse Brca2 gene. Mamm. Genome. 2002;13:352–358. doi: 10.1007/s00335-002-2300-6. [DOI] [PubMed] [Google Scholar]
- Cheung A.M., Hande M.P., Jalali F., Tsao M.S., Skinnider B., Hirao A., McPherson J.P., Karaskova J., Suzuki A., Wakeham A., et al. Loss of Brca2 and p53 synergistically promotes genomic instability and deregulation of T-cell apoptosis. Cancer Res. 2002;62:6194–6204. [PubMed] [Google Scholar]
- Cheung A.M., Elia A., Tsao M.S., Done S., Wagner K.U., Hennighausen L., Hakem R., Mak T.W. Brca2 deficiency does not impair mammary epithelium development but promotes mammary adenocarcinoma formation in p53(+/−) mutant mice. Cancer Res. 2004;64:1959–1965. doi: 10.1158/0008-5472.can-03-2270. [DOI] [PubMed] [Google Scholar]
- Choi E., Park P.G., Lee H.O., Lee Y.K., Kang G.H., Lee J.W., Han W., Lee H.C., Noh D.Y., Lekomtsev S., et al. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Dev. Cell. 2012;22:295–308. doi: 10.1016/j.devcel.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Connor F., Bertwistle D., Mee P.J., Ross G.M., Swift S., Grigorieva E., Tybulewicz V.L., Ashworth A. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat. Genet. 1997;17:423–430. doi: 10.1038/ng1297-423. [DOI] [PubMed] [Google Scholar]
- Couch F.J., Farid L.M., DeShano M.L., Tavtigian S.V., Calzone K., Campeau L., Peng Y., Bogden B., Chen Q., Neuhausen S., et al. BRCA2 germline mutations in male breast cancer cases and breast cancer families. Nat. Genet. 1996;13:123–125. doi: 10.1038/ng0596-123. [DOI] [PubMed] [Google Scholar]
- Dai C., Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt S., Tseng D., Ermann J., George T.I., Liu Y.P., Davis C.R., Fathman C.G., Strober S. Naive and memory T cells induce different types of graft-versus-host disease. J. Immunol. 2007;179:6547–6554. doi: 10.4049/jimmunol.179.10.6547. [DOI] [PubMed] [Google Scholar]
- Dutton R.W., Bradley L.M., Swain S.L. T cell memory. Ann. Rev. Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- Evers B., Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–5897. doi: 10.1038/sj.onc.1209871. [DOI] [PubMed] [Google Scholar]
- Flores K.G., McAllister K.A., Greer P.K., Wiseman R.W., Hale L.P. Thymic model for examining BRCA2 expression and function. Mol. Carcinog. 2002;35:103–109. doi: 10.1002/mc.10081. [DOI] [PubMed] [Google Scholar]
- Ford D., Easton D.F., Stratton M., Narod S., Goldgar D., Devilee P., Bishop D.T., Weber B., Lenoir G., Chang-Claude J., et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman L.S., Thistlethwaite F.C., Patel K.J., Yu V.P., Lee H., Venkitaraman A.R., Abel K.J., Carlton M.B., Hunter S.M., Colledge W.H., et al. Thymic lymphomas in mice with a truncating mutation in Brca2. Cancer Res. 1998;58:1338–1343. [PubMed] [Google Scholar]
- Goggins M., Schutte M., Lu J., Moskaluk C.A., Weinstein C.L., Petersen G.M., Yeo C.J., Jackson C.E., Lynch H.T., Hruban R.H., et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- Gretarsdottir S., Thorlacius S., Valgardsdottir R., Gudlaugsdottir S., Sigurdsson S., Steinarsdottir M., Jonasson J.G., Anamthawat-Jonsson K., Eyfjord J.E. BRCA2 and p53 mutations in primary breast cancer in relation to genetic instability. Cancer Res. 1998;58:859–862. [PubMed] [Google Scholar]
- Gu H., Marth J.D., Orban P.C., Mossmann H., Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Holloman W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011;18:748–754. doi: 10.1038/nsmb.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett N.G., Taniguchi T., Olson S., Cox B., Waisfisz Q., De Die-Smulders C., Persky N., Grompe M., Joenje H., Pals G., et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Jonkers J., Meuwissen R., van der Gulden H., Peterse H., van der Valk M., Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Jung Y.S., Qian Y., Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010;22:1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M.C., Marks J.H., Mandell J.B., New York Breast Cancer Study. G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- Kupfer G.M. Fanconi anemia: a signal transduction and DNA repair pathway. Yale J. Biol. Med. 2013;86:491–497. [PMC free article] [PubMed] [Google Scholar]
- Lee H., Trainer A.H., Friedman L.S., Thistlethwaite F.C., Evans M.J., Ponder B.A., Venkitaraman A.R. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol. Cell. 1999;4:1–10. doi: 10.1016/s1097-2765(00)80182-3. [DOI] [PubMed] [Google Scholar]
- Ludwig T., Chapman D.L., Papaioannou V.E., Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- Ludwig T., Fisher P., Murty V., Efstratiadis A. Development of mammary adenocarcinomas by tissue-specific knockout of Brca2 in mice. Oncogene. 2001;20:3937–3948. doi: 10.1038/sj.onc.1204512. [DOI] [PubMed] [Google Scholar]
- Marx G. Possible function found for breast cancer genes. Science. 1997;276:531–532. doi: 10.1126/science.276.5312.531. [DOI] [PubMed] [Google Scholar]
- McAllister K.A., Bennett L.M., Houle C.D., Ward T., Malphurs J., Collins N.K., Cachafeiro C., Haseman J., Goulding E.H., Bunch D., et al. Cancer susceptibility of mice with a homozygous deletion in the COOH-terminal domain of the Brca2 gene. Cancer Res. 2002;62:990–994. [PubMed] [Google Scholar]
- Moran A., O’Hara C., Khan S., Shack L., Woodward E., Maher E.R., Lalloo F., Evans D.G. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam. Cancer. 2012;11:235–242. doi: 10.1007/s10689-011-9506-2. [DOI] [PubMed] [Google Scholar]
- Neuhausen S., Gilewski T., Norton L., Tran T., McGuire P., Swensen J., Hampel H., Borgen P., Brown K., Skolnick M., et al. Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer. Nat. Genet. 1996;13:126–128. doi: 10.1038/ng0596-126. [DOI] [PubMed] [Google Scholar]
- Park P.G., Lee H. Development of thymic lymphomas in mice disrupted of Brca2 allele in the thymus. Exp. Mol. Med. 2008;40:339–344. doi: 10.3858/emm.2008.40.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K.J., Yu V.P., Lee H., Corcoran A., Thistlethwaite F.C., Evans M.J., Colledge W.H., Friedman L.S., Ponder B.A., Venkitaraman A.R. Involvement of Brca2 in DNA repair. Mol. Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- Pellegrini L., Yu D.S., Lo T., Anand S., Lee M., Blundell T.L., Venkitaraman A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- Rajan J.V., Marquis S.T., Gardner H.P., Chodosh L.A. Developmental expression of Brca2 colocalizes with Brca1 and is associated with proliferation and differentiation in multiple tissues. Dev. Biol. 1997;184:385–401. doi: 10.1006/dbio.1997.8526. [DOI] [PubMed] [Google Scholar]
- Ramus S.J., Bobrow L.G., Pharoah P.D., Finnigan D.S., Fishman A., Altaras M., Harrington P.A., Gayther S.A., Ponder B.A., Friedman L.S. Increased frequency of TP53 mutations in BRCA1 and BRCA2 ovarian tumours. Genes Chromosomes Cancer. 1999;25:91–96. doi: 10.1002/(sici)1098-2264(199906)25:2<91::aid-gcc3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Risch H.A., McLaughlin J.R., Cole D.E., Rosen B., Bradley L., Fan I., Tang J., Li S., Zhang S., Shaw P.A., et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006;98:1694–1706. doi: 10.1093/jnci/djj465. [DOI] [PubMed] [Google Scholar]
- Sharan S.K., Morimatsu M., Albrecht U., Lim D.S., Regel E., Dinh C., Sands A., Eichele G., Hasty P., Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ikeda M., Taya Y., Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Sluss H.K., Armata H., Gallant J., Jones S.N. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 2004;24:976–984. doi: 10.1128/MCB.24.3.976-984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain B.H., Larson C.J., Shihabuddin L.S., Gage F.H., Verma I.M. Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc. Natl. Acad. Sci. USA. 1999;96:13920–13925. doi: 10.1073/pnas.96.24.13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., de la Pompa J.L., Hakem R., Elia A., Yoshida R., Mo R., Nishina H., Chuang T., Wakeham A., Itie A., et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev. 1997;11:1242–1252. doi: 10.1101/gad.11.10.1242. [DOI] [PubMed] [Google Scholar]
- Tavtigian S.V., Simard J., Rommens J., Couch F., Shattuck-Eidens D., Neuhausen S., Merajver S., Thorlacius S., Offit K., Stoppa-Lyonnet D., et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat. Genet. 1996;12:333–337. doi: 10.1038/ng0396-333. [DOI] [PubMed] [Google Scholar]
- Thorslund T., West S.C. BRCA2: a universal recombinase regulator. Oncogene. 2007;26:7720–7730. doi: 10.1038/sj.onc.1210870. [DOI] [PubMed] [Google Scholar]
- Wong A.K., Pero R., Ormonde P.A., Tavtigian S.V., Bartel P.L. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- Yan D.H., Wen Y., Su L.K., Xia W., Wang S.C., Zhang S., Gan L., Lee D.F., Spohn B., Frey J.A., et al. A delayed chemically induced tumorigenesis in Brca2 mutant mice. Oncogene. 2004;23:1896–1901. doi: 10.1038/sj.onc.1207314. [DOI] [PubMed] [Google Scholar]
- Yu J., Wang Z., Kinzler K.W., Vogelstein B., Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA. 2003;100:1931–1936. doi: 10.1073/pnas.2627984100. [DOI] [PMC free article] [PubMed] [Google Scholar]