

Abstract
Hyponatremia is frequently seen in patients with ascites secondary to advanced cirrhosis and portal hypertension. The development of ascites in patients with cirrhosis is multi-factorial. Portal hypertension and the associated systemic vasodilation lead to activation of the sodium-retaining neurohumoral mechanisms which include the renin-angiotensin-aldosterone system, sympathetic nervous system and antidiuretic hormone (ADH). The net effect is the avid retention of sodium and water to compensate for the low effective circulatory volume resulting in the development of ascites. Although not apparent in the early stages of cirrhosis, the progression of cirrhosis and ascites leads to impairment of the kidneys to eliminate solute- free water. This leads to additional compensatory mechanisms including non-osmotic secretion of ADH, also known as arginine vasopressin, further worsening excess water retention and thereby hyponatremia. Hyponatremia is associated with increased morbidity and mortality in patients with cirrhosis, and is an important prognostic marker both before and after liver transplant. The management of hyponatremia in this setting is a challenge as conventional therapy for hyponatremia including fluid restriction and loop diuretics are frequently inefficacious. In this review, we discuss the pathophysiology and various treatment modalities, including selective vasopressin receptor antagonists, for the management of hyponatremia in patients with cirrhosis.
Keywords: Hyponatremia in cirrhosis, Dilutional hyponatremia, Hypervolemic hyponatremia, Vasopressin receptor antagonists, Vaptans
Core tip: Hyponatremia is the most common electrolyte abnormality observed in hospitalized patients and is a common finding in patients with advanced cirrhosis. The management of hyponatremia in cirrhosis is challenging as conventional therapy for hyponatremia including fluid restriction and loop diuretics are frequently inefficacious. Vaptans, drugs that selectively antagonizes the effects of arginine vasopressin on the V2 receptors in the kidney tubules, represent a logical step in the treatment of hyponatremia. The currently available vaptans, however, are not approved for use in patients with cirrhosis due to the increased risk for hepatic failure and mortality.
INTRODUCTION
Hyponatremia is the most common electrolyte abnormality observed in hospitalized patients[1]. Hyponatremia in cirrhosis is currently defined as a serum sodium level of less than 130 meq/L[2]. It has been suggested that the prevalence of a serum sodium concentration less than 135, 130 and 120 meq/L in patients with cirrhosis and ascites is 49.4%, 21.6% and 1.2%, respectively[3]. Patients with cirrhosis may develop hyponatremia due to either hypovolemia (example: loss of extracellular fluid due to diuretics) or hypervolemia (expanded extracellular fluid volume due to the inability of the kidneys to excrete solute-free water proportionate to the amount of free water ingested). In this review, we will discuss the pathogenesis, prognostic value and management of dilutional hyponatremia (hypervolemic hyponatremia) in patients with cirrhosis and portal hypertension.
PATHOGENESIS
Systemic vasodilation
Systemic vasodilation and arterial underfilling play a major role in development of hyponatremia in patients with cirrhosis and portal hypertension (Figure 1). A hyperdynamic circulation, characterized by an increased cardiac output, markedly reduced systemic vascular resistance and reduced mean arterial pressure, is a common cardiovascular physiological manifestation of patients with cirrhosis and advanced portal hypertension[4,5]. The marked reduction in vascular resistance predominantly involves the splanchnic arterial circulation[6]. The opening of portasystemic collaterals[5] and the increased synthesis of circulating vasodilators, including nitric oxide (NO), glucagon, vasoactive intestinal peptide, substance P, platelet activating factor, prostaglandins and prostacyclins play a crucial role in the pathogenesis of splanchnic vasodilation[7] (Figure 2). The accumulating circumstantial evidence favors a key role for NO in the pathogenesis of splanchnic vasodilation in patients with advanced cirrhosis and portal hypertension[8-10]. The activation of nitric oxide synthase in the endothelial cells is multi-factorial, which include mechanical stimuli due to ‘shear stress’, vascular endothelial growth factors, tumor necrosis factor alpha, and more importantly endotoxins or bacterial DNA[11,12] that are less efficiently cleared from the gastrointestinal tract due to portal systemic shunting and defective reticuloendothelial cell function in cirrhosis.
Figure 1.
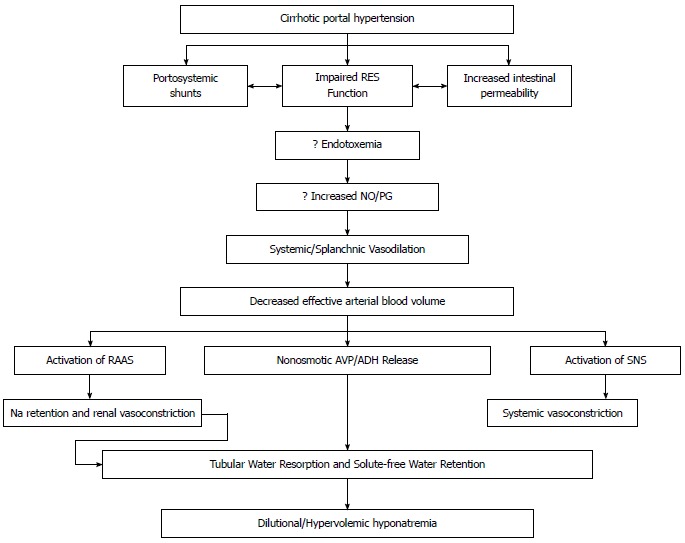
Proposed mechanisms for the development of hyponatremia. SNS: Sympathetic nervous system; RAAS: Renin-angiotensin-aldosterone system; NO: Nitric oxide; RES: Reticuloendothelial system; PG: Prostaglandin; AVP: Arginine vasopressin; ADH: Antidiuretic hormone.
Figure 2.
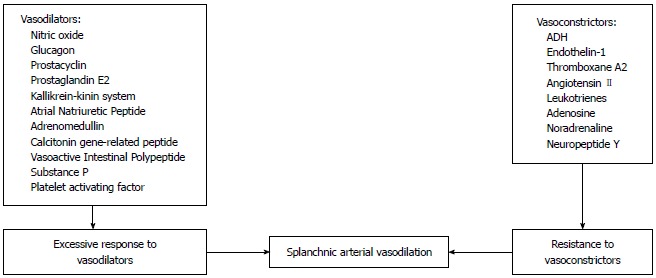
Mechanisms involved in the splanchnic vasodilation in cirrhosis. ADH: Antidiuretic hormone.
It has been suggested that endotoxemia may be a causative factor for increased systemic prostacyclin synthesis, and could be reversed to some extent by antibacterial agents[13]. It is possible that when one of the vasoactive mediators, such as NO or prostacyclin, is inhibited, other vasoactive pathways such as the angiotensin-II, norepinephrine, vasopressin and the augmented sympathetic tone, are up-regulated thereby preventing the correction of the splanchnic vasodilation. The complex relationship among these vasoactive systems implies that no one factor is likely to be solely responsible for the splanchnic vasodilation seen in patients with portal hypertension. This may explain the difficulty in developing pharmacological agents to counteract splanchnic vasodilation.
Water balance and role of antidiuretic hormone (also known arginine vasopressin)
The total body water and osmolality are maintained within normal limits in such a way that an increase in water intake (normally 1.5-3 L/d; may vary from 0.5-20 L/d under extreme conditions) is followed by an increase in renal solute-free water excretion and a decrease in water intake is ensued by a decrease in free water excretion. The serum osmolality (and hence serum sodium) is tightly regulated primarily at the level of hypothalamus via the release of antidiuretic hormone (ADH). A rise or fall in serum osmolality is accompanied by a corresponding increase or decrease of ADH secretion. Under normal physiologic conditions, the kidneys are in a state of antidiuresis with a 24-h urine osmolality higher than plasma osmolality[14].
The collecting duct has minimal water permeability under normal conditions, but permeability increases when ADH is released in response to hyperosmolality and hypovolemia. The enhanced binding of vasopressin to the V2 receptors on the basolateral membrane of the cells lining the renal collecting ducts leads to production of cyclic AMP and subsequent activation of protein kinase A. This in turn phosphorylates microtubular subunits that aggregate to form specific water channel, aquaporin-2 (AQP-2), that are translocated from the cytoplasmic vesicles to the apical plasma membrane. This process allows the reabsorption of large volumes of water from the collecting duct, which leads to an increase in body water content and hypervolemic hyponatremia[15-20] (Figure 3). Under physiologic conditions, when serum osmolality increases, ADH secretion increases, aquaporin channels in the renal collecting duct are activated, resulting in water reabsorption. A fall in serum osmolality leads to inactivation of the renal aquaporin channels and excretion of dilute urine to maintain the volume status and serum osmolality. The rapid adaptation of the free water excretion depends on the presence of intact osmoreceptors in the anterior hypothalamus, the release of ADH and the appropriate interaction between the ADH and AQP-2.
Figure 3.
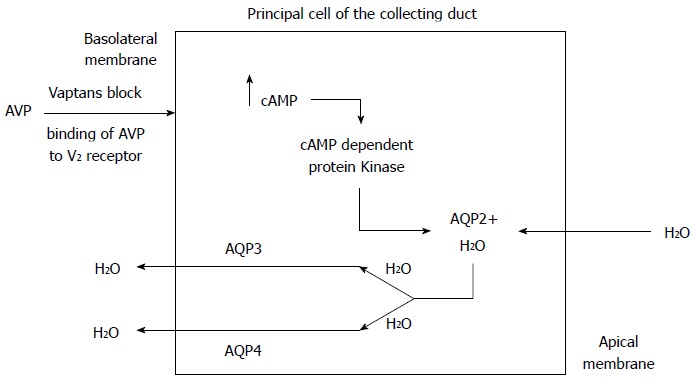
Mechanism of action of vaptans. AVP: Arginine vasopressin; AQP2: Aquaporin-2; AQP3: Aquaporin-3; AQP4: Aquaporin-4.
ADH is a polypeptide hormone that is synthesized in the supraoptic and paraventricular nuclei of the hypothalamus and stored in the posterior pituitary gland. Increased plasma osmolality and hypovolemia are the principal physiological stimuli for vasopressin secretion. Thus both osmotic and non-osmotic stimulations regulate ADH release. The osmotic pathway is mediated via osmoreceptors located in the anterior hypothalamus close to the supraoptic nuclei. These receptors sense the intracellular water content in the neurons (by their swelling and shrinking) and respond linearly to the changes in plasma osmolality[21]. The major non-osmotic pathway for ADH release involves the autonomic nervous system which is mediated via the baroreceptors located in the atria, ventricle, aortic arch, and carotid sinus. These baroreceptors communicate to the hypothalamus via parasympathetic pathways and cause a release of ADH in response to hypovolemia. Please refer to Table 1 for the details of vasopressin receptor subtypes.
Table 1.
Vasopressin receptors
| Receptor | Location | Function |
| V1a | Vascular smooth muscle | Vasoconstriction and myocardial hypertrophy |
| Platelets | Platelet aggregation | |
| Hepatocytes | Glycogenolysis | |
| Myometrium | Uterine contractions | |
| V1b | Anterior pituitary | ACTH release |
| V2 | Basolateral membrane in collecting tubule | Water reabsorption |
| Vascular endothelium | Release of vonWillebrand factor | |
| and factor VIII | ||
| Vascular smooth muscle | Vasodilation |
Non-osmotic stimulation of renin-angiotensin-aldosterone system, sympathetic nervous system and ADH
The systemic/splanchnic vasodilation and arterial underfilling in patients with cirrhosis and portal hypertension lead to a decrease in the effective circulatory volume and a reduction in stretch at the carotid and renal baroreceptors. In order to restore the effective circulatory volume, the sodium-retaining neurohumoral mechanisms, such as the renin-angiotensin-aldosterone system, sympathetic nervous system and ADH, are activated leading to maximal retention of sodium and water.
Water and sodium retention secondary to impaired renal elimination of solute-free water clearly has been shown to occur in decompensated cirrhotic patients with ascites and edema. This impairment could be subclinical and only detected by a water loading test in compensated cirrhosis[22-26]. The role that ADH plays in mediating this abnormal water excretion was studied by Bichet et al[27] who measured plasma ADH concentrations before and after the water load test in cirrhotic patients with and without ascites. There was a significant difference in the inability to suppress ADH after the water load test in decompensated cirrhotic patients with ascites as compared to compensated cirrhotic patients, despite the presence of a low serum osmolality in decompensated cirrhotic patients.
The relative role of osmotic and non-osmotic pathways for the hypersecretion of ADH in patients with cirrhosis has been debated. Most cirrhotic patients have low serum osmolality and sodium levels, and one would expect to see suppression of ADH release if the stimulation was primarily from the osmoreceptors[2]. The ADH, norepinephrine, and aldosterone levels as well as renin activity were significantly higher in cirrhotic patients with ascites after the water load test implying that there was activation of the sodium-retaining neurohumoral mechanisms. It appears that the decrease in systemic vascular resistance leads to effective arterial underfilling, which causes baroreceptor mediated nonosmotic stimulation of ADH and other vasoconstricting systems leading to the activation of sodium retaining neurohumoral mechanisms in order to restore perfusion pressure[6]. These findings may suggest that the hypo-osmotic stimuli to suppress ADH release are overridden by the nonosmotic stimuli secondary to arterial under filling[28]. Thus in order to prevent impending vascular collapse from effective circulatory volume depletion, the body sacrifices the osmolar homeostasis and releases ADH in response to the non-osmotic stimulus of the endogenous vasoconstrictor agents. The net result is enhanced sodium and water retention to correct the depletion of circulatory volume and this occurs despite the presence of increased total body extracellular sodium, plasma volume, and cardiac output. As suppression of ADH release is required to excrete a water load, the inability of kidneys to excrete water in the presence of the non-osmotically triggered ADH release leads to the development of a dilutional or hypervolemic hyponatremia. Thus the hyponatremia in this patient population is purely dilutional and does not reflect a sodium deficient state.
Many other factors including elevated atrial natriuretic peptide[29], decreased renal production of PGE-2[30-34] and decreased metabolism of ADH have been implicated in the development of hyponatremia in cirrhosis[35,36].
Prognostic value of hyponatremia in cirrhosis
In patients without cirrhosis, hyponatremia depending on its severity may lead to a range of symptoms including mild cognitive dysfunction, falls, seizures, coma and very rarely death[37]. Hyponatremia in cirrhosis is a chronic process and this allows the brain to adapt to the hypo-osmolality of the extracellular fluid. The most important factor in determining the severity of neurologic symptoms in patients with hyponatremia is the acuity of fall of serum sodium rather than the absolute reduction of serum sodium. Hence patients with cirrhosis and hyponatremia are less likely to have severe neurologic symptoms[38]. However, hyponatremia may pose a second osmotic hit to cerebral edema and astrocyte swelling, in addition to the astrocyte dysfunction caused by increased intracellular glutamine concentration from ammonia metabolism, thereby precipitating hepatic encephalopathy[38].
The quality of life is poor in patients with cirrhosis and hyponatremia due to the requirement for strict fluid restriction. Hyponatremia has been found to be an independent predictive factor of the impaired health related quality life[39] as well as hepatic encephalopathy[40]. Numerous studies have shown that the severity of hyponatremia and ascites is a major determinant of disease severity and prognosis in cirrhosis[26,41-48]. In one study, the serum sodium level before the onset of spontaneous bacterial peritonitis (SBP) was an independent predictor of renal failure triggered by SBP[49]. It has also been suggested that serum sodium is an earlier and more sensitive test than serum creatinine to detect circulatory dysfunction resulting in renal failure and/or death[47]. Although patients with hyponatremia are at a very high risk for developing hepatorenal syndrome, low serum sodium in hepatorenal syndrome is not only due to high ADH levels but also due to decreased GFR and proximal sodium reabsorption[38].
Patients with hyponatremia were found to have a higher risk of early death before transplantation independent of the severity of cirrhosis as assessed by the MELD scores[48]. Hence, some investigators have advocated an expedited liver transplantation under a ‘sickest first’ model in cirrhotic patients with MELD scores below 21, persistent ascites and hyponatremia[48]. It has been suggested that serum sodium could be incorporated into the MELD score[48], and this may provide a more accurate survival prediction than MELD alone[50]. Other studies have also identified hyponatremia to be a risk factor for increased morbidity and mortality after liver transplantation[51,52].
TREATMENT
Management of hyponatremia in the presence moderate to severe ascites is challenging for both physicians and patients. Hypovolemic hyponatremia should be treated with fluid resuscitation to restore the circulatory volume and withdrawal of the precipitating factor (usually diuretic therapy). On the other hand, hypervolemic/dilutional hyponatremia in cirrhosis is ideally managed with fluid restriction and measures to enhance the renal solute-free water excretion. The majority of patients find it difficult to adhere to fluid restriction, and discontinuation of diuretics may further worsen ascites and hydrothorax requiring repeated paracentesis or thoracocentesis.
Rapid correction (> 9 mEq/L in 24 h) of serum sodium may lead to serious neurological complications such as central pontine myelinolysis or seizures. Hyponatremia could pose significant risk to patients if they were to undergo liver transplantation where it is not always possible to maintain fluctuations in serum sodium levels to less than 10 mEq/L over 24 h. However, treatment for hyponatremia is indicated when the serum sodium is less than 120 meq/L or the patient has neurologic symptoms that might be due to hyponatremia. The general principles of the treatment of hyponatremia are broadly outlined below.
Water restriction
The mainstay of therapy of hyponatremia in patients with cirrhosis is fluid restriction (1-1.5 L/d) to a level sufficient to induce a negative water balance. Fluid restriction should be considered if the patient has neurologic symptoms that might be due to hyponatremia or when the serum sodium is less than 120 mEq/L, which occurs in about 1% of patients with cirrhosis[3]. There is no role for routine free water restriction in patients with mild, asymptomatic hyponatremia. To be effective, fluid intake should be less than urine output to account for the endogenous production of water by the body. In the authors’ experience, compliance with fluid restriction is poor in these patients even in the hospital setting and often difficult to achieve. Patients on strict fluid restriction may be encouraged to suck on ice chips or lollipops to quench the thirst. A good indicator of adequate water restriction is the change in plasma sodium concentration within the first 24-48 h. If there is no increase in the plasma sodium levels within the first 48-72 h, either the patient is not following the water restriction or a stricter water restriction is needed. Sodium restriction (2 g/d) should be continued in addition to fluid restriction as these patients also have ascites.
Hypertonic saline
Hypertonic saline is indicated only in symptomatic patients who are intolerant or unresponsive to free water restriction, those with profound hyponatremia (< 110 mEq/L), or within hours of liver transplantation to prevent the likelihood for an emergent rapid correction in the operating room when the serum sodium levels are somewhat higher (between 120-130 mEq/L). Extreme care should be exercised not to overcorrect the serum sodium levels above 9 mEq/L per 24 h to avoid the risks of central pontine myelinolysis, quadriplegia, coma or death. As hypertonic sodium chloride infusion leads to increasing ascites and edema, it is usually not recommended for the treatment of hypervolemic hyponatremia, except in cases of profound hyponatremia as discussed above.
Correction of hypokalemia
Correction of hypokalemia also appears to be important in patients with cirrhosis and hyponatremia for two reasons: hypokalemia promotes the development of hepatic encephalopathy; correction of hypokalemia tends to raise serum sodium concentration. Hypokalemia predisposes to hepatic encephalopathy by at least two mechanisms: hypokalemia increases renal ammonia synthesis; the concomitant alkalemia increases the fraction of unionized ammonia in the plasma. As potassium is as osmotically active as sodium, supplementation of potassium can raise serum sodium and osmolality in patients with hyponatremia[53].
Albumin infusion
Intravenous albumin infusion might be useful in the short term, although long term use has not been studied and this approach is expensive and impractical[54].
Pharmacological therapy
The objectives of pharmacological therapy are to increase solute-free water excretion. There have been many attempts to achieve this goal with varying success rates, and this is a field in evolution. The target of pharmacological therapy has focused on the release or action of ADH [arginine vasopressin (AVP)]. The potential options include use of κ-opioid agonists to block the central release of ADH, blockade of the V2 receptor of ADH with specific antagonists; and finally alteration of the effect of ADH at the level of the collecting duct in the kidney. Democycline could block the action of ADH in the collecting ducts, but could cause renal failure and hence is not recommended. Therefore only κ-opioid agonists and V2 receptor antagonists have been studied in both animals and humans.
κ-opioid agonists
κ-opioid agonists inhibit ADH release from the neurohypophysis, and have been shown to exert an aquaretic effect in animal models and in patients with cirrhosis. In the only human study, niravoline (0.5-2 mg, iv) was given to 18 patients with cirrhosis[55,56]. There was a marked aquaretic effect between 1 and 2 h after administration with a return to basal values at 24 h[57]. The aquaretic effect was not sustained, and moreover, it was associated with major neurological side effects including personality disorders and mild confusion. Hence, no large scale studies have been performed with κ-opioid agonists.
Vasopressin receptor antagonists or vaptans
The biologic effects of AVP (ADH) are mediated via specific receptors called V1a, V1b, and V2 receptors as discussed earlier in this review. Anti-diuretic properties of ADH are mediated primarily through the V2 receptors, which are found exclusively in the renal collecting ducts. Activation of V2 receptors is responsible for water reabsorption. The development of V2 receptor antagonists was therefore a logical step in the management of fluid overload and hyponatremia as effective V2 receptor antagonists could theoretically produce pure aquaresis (Figure 3).
The initial studies on vaptans in cirrhosis were done on patients with cirrhosis without hyponatremia[13,58]. These studies demonstrated the efficacy of oral vaptans in increasing the urine volume and solute-free water excretion resulting in a negative fluid balance. The subsequent studies with vaptans have consistently demonstrated their efficacy in improving serum sodium levels in the short term[59-64], but with increased risk for mortality in patients with cirrhosis, as explained below.
Tolvaptan, satavaptan and lixivaptan are all oral agents which selectively block the V2 receptor. The intravenous agent, conivaptan, which blocks both V2 and V1 receptors may lead to further reduction in blood pressure, increase the risk of variceal bleeding via the V1a receptor blockade[65]. Tolvaptan is a selective non-peptide V2 receptor antagonist and when this drug was added to standard diuretic therapy for periods ranging from 25 to 60 d in patients with heart failure[61,63], treated patients had significantly lower weight and improvement in edema as well as serum sodium levels compared to those who received placebo. This drug was initially approved by the United States Food and Drug Administration (FDA) for use in hyponatremia based on a randomized controlled trial involving a predominant patient population comprising of those with congestive heart failure. Sixty three patients with cirrhosis with a CTP score less than 10 and serum sodium less than 120 were also included in the above study[61]. However, based on the findings of multicenter trials evaluating the effect of tolvaptan on the progression of disease in polycystic kidney disease[66,67], the FDA determined that tolvaptan should not be used in patients with liver disease or cirrhosis due to the risks for liver failure and death. Similarly, a one year follow-up study on patients on satavaptan showed increased mortality compared to placebo, resulting in withdrawal of the drug by the pharmaceutical company[68].
Domeclocycline, another ADH antagonist, which increased free water excretion and thus corrects hyponatremia, should not be used in cirrhosis due to its nephrotoxic potential[69]. Since most patients with hyponatremia have advanced cirrhosis, the side-effect profile of single dose-finding studies must be interpreted with caution, as in the case of the preliminary studies on vaptans, because adverse events are more likely to occur after long term administration in an unselected population with greater co-morbidities.
Terlipressin, by virtue of its strong effect on vasopressin V1 receptor, has therapeutic potential in portal hypertensive bleeding as well as hepatorenal syndrome. Terlipressin is also a partial agonist of renal vasopressin V2 receptors and acute reduction in serum sodium level has been documented in patients who are initiated on terlipressin[70]. The resultant hyponatremia, although severe in some patients, is usually reversible after withdrawal of terlipressin therapy. Hence serum sodium levels should be monitored while patients are on therapy with terlipressin.
CONCLUSION
Hyponatremia is very common in patients with cirrhosis and the routine correction of asymptomatic hyponatremia is not recommended. The main indications for correction of hyponatremia are presence of neurologic symptoms that might be due to hyponatremia and serum sodium less than 120 mEq/L. The only exception is in patients who are likely to receive liver transplantation within hours when their serum sodium concentration is less than 130 mEq/L to avoid rapid correction in the operating room as it may be associated with serious neurological complications. Correction of hypokalemia and fluid restriction are the mainstays of treatment. Administration of hypertonic saline may be considered in a monitored setting to correct profound hyponatremia (serum sodium < 110 mEq/L) and in the immediate pre-liver transplant period to prevent the risk of osmotic demyelination syndrome. No vasopressin receptor antagonist is currently approved by the FDA for treatment of hyponatremia in patients with liver disease or cirrhosis. The availability of selective and efficacious oral V2 receptor antagonists, without major side effects, will be a major development for the management of hyponatremia.
Footnotes
Conflict-of-interest: None conflicting interest related to the manuscript submitted for publication.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 21, 2014
First decision: November 14, 2014
Article in press: January 30, 2015
P- Reviewer: Barreto R, Ruiz-Margain A, Stanciu C S- Editor: Yu J L- Editor: A E- Editor: Liu XM
References
- 1.Baran D, Hutchinson TA. The outcome of hyponatremia in a general hospital population. Clin Nephrol. 1984;22:72–76. [PubMed] [Google Scholar]
- 2.Ginés P, Berl T, Bernardi M, Bichet DG, Hamon G, Jiménez W, Liard JF, Martin PY, Schrier RW. Hyponatremia in cirrhosis: from pathogenesis to treatment. Hepatology. 1998;28:851–864. doi: 10.1002/hep.510280337. [DOI] [PubMed] [Google Scholar]
- 3.Angeli P, Wong F, Watson H, Ginès P. Hyponatremia in cirrhosis: Results of a patient population survey. Hepatology. 2006;44:1535–1542. doi: 10.1002/hep.21412. [DOI] [PubMed] [Google Scholar]
- 4.Abelmann WH. Hyperdynamic circulation in cirrhosis: a historical perspective. Hepatology. 1994;20:1356–1358. [PubMed] [Google Scholar]
- 5.Groszmann RJ. Hyperdynamic circulation of liver disease 40 years later: pathophysiology and clinical consequences. Hepatology. 1994;20:1359–1363. [PubMed] [Google Scholar]
- 6.Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodés J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151–1157. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 7.Ginès P, Fernández-Esparrach G, Arroyo V, Rodés J. Pathogenesis of ascites in cirrhosis. Semin Liver Dis. 1997;17:175–189. doi: 10.1055/s-2007-1007196. [DOI] [PubMed] [Google Scholar]
- 8.Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337:776–778. doi: 10.1016/0140-6736(91)91384-7. [DOI] [PubMed] [Google Scholar]
- 9.Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology. 2006;43:S121–S131. doi: 10.1002/hep.20993. [DOI] [PubMed] [Google Scholar]
- 10.Battista S, Bar F, Mengozzi G, Zanon E, Grosso M, Molino G. Hyperdynamic circulation in patients with cirrhosis: direct measurement of nitric oxide levels in hepatic and portal veins. J Hepatol. 1997;26:75–80. doi: 10.1016/s0168-8278(97)80012-8. [DOI] [PubMed] [Google Scholar]
- 11.Such J, Francés R, Muñoz C, Zapater P, Casellas JA, Cifuentes A, Rodríguez-Valera F, Pascual S, Sola-Vera J, Carnicer F, et al. Detection and identification of bacterial DNA in patients with cirrhosis and culture-negative, nonneutrocytic ascites. Hepatology. 2002;36:135–141. doi: 10.1053/jhep.2002.33715. [DOI] [PubMed] [Google Scholar]
- 12.Francés R, Benlloch S, Zapater P, González JM, Lozano B, Muñoz C, Pascual S, Casellas JA, Uceda F, Palazón JM, et al. A sequential study of serum bacterial DNA in patients with advanced cirrhosis and ascites. Hepatology. 2004;39:484–491. doi: 10.1002/hep.20055. [DOI] [PubMed] [Google Scholar]
- 13.Guarner C, Soriano G, Such J, Teixidó M, Ramis I, Bulbena O, Roselló J, Guarner F, Gelpi E, Balanzó J. Systemic prostacyclin in cirrhotic patients. Relationship with portal hypertension and changes after intestinal decontamination. Gastroenterology. 1992;102:303–309. [PubMed] [Google Scholar]
- 14.Arroyo V, Jiménez W. Complications of cirrhosis. II. Renal and circulatory dysfunction. Lights and shadows in an important clinical problem. J Hepatol. 2000;32:157–170. doi: 10.1016/s0168-8278(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 15.Knepper MA, Wade JB, Terris J, Ecelbarger CA, Marples D, Mandon B, Chou CL, Kishore BK, Nielsen S. Renal aquaporins. Kidney Int. 1996;49:1712–1717. doi: 10.1038/ki.1996.253. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen S, Marples D, Frøkiaer J, Knepper M, Agre P. The aquaporin family of water channels in kidney: an update on physiology and pathophysiology of aquaporin-2. Kidney Int. 1996;49:1718–1723. doi: 10.1038/ki.1996.254. [DOI] [PubMed] [Google Scholar]
- 17.King LS, Agre P. Pathophysiology of the aquaporin water channels. Annu Rev Physiol. 1996;58:619–648. doi: 10.1146/annurev.ph.58.030196.003155. [DOI] [PubMed] [Google Scholar]
- 18.Knepper MA. Molecular physiology of urinary concentrating mechanism: regulation of aquaporin water channels by vasopressin. Am J Physiol. 1997;272:F3–12. doi: 10.1152/ajprenal.1997.272.1.F3. [DOI] [PubMed] [Google Scholar]
- 19.Fushimi K, Uchida S, Hara Y, Hirata Y, Marumo F, Sasaki S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature. 1993;361:549–552. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- 20.Agre P, Nielsen S. The aquaporin family of water channels in kidney. Nephrologie. 1996;17:409–415. [PubMed] [Google Scholar]
- 21.Oliet SH, Bourque CW. Mechanosensitive channels transduce osmosensitivity in supraoptic neurons. Nature. 1993;364:341–343. doi: 10.1038/364341a0. [DOI] [PubMed] [Google Scholar]
- 22.Epstein M. Derangements of renal water handling in liver disease. Gastroenterology. 1985;89:1415–1425. doi: 10.1016/0016-5085(85)90664-x. [DOI] [PubMed] [Google Scholar]
- 23.Arroyo V, Clària J, Saló J, Jiménez W. Antidiuretic hormone and the pathogenesis of water retention in cirrhosis with ascites. Semin Liver Dis. 1994;14:44–58. doi: 10.1055/s-2007-1007297. [DOI] [PubMed] [Google Scholar]
- 24.Ginès P, Abraham WT, Schrier RW. Vasopressin in pathophysiological states. Semin Nephrol. 1994;14:384–397. [PubMed] [Google Scholar]
- 25.Vaamonde CA. Renal water handling in liver disease, Epstein M, ed. The Kidney in Liver Disease, 4th ed. Philadelphia: Hanley & Belfus, Inc; 1996. pp. 33–74. [Google Scholar]
- 26.Arroyo V, Rodés J, Gutiérrez-Lizárraga MA, Revert L. Prognostic value of spontaneous hyponatremia in cirrhosis with ascites. Am J Dig Dis. 1976;21:249–256. doi: 10.1007/BF01095898. [DOI] [PubMed] [Google Scholar]
- 27.Bichet D, Szatalowicz V, Chaimovitz C, Schrier RW. Role of vasopressin in abnormal water excretion in cirrhotic patients. Ann Intern Med. 1982;96:413–417. doi: 10.7326/0003-4819-96-4-413. [DOI] [PubMed] [Google Scholar]
- 28.Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med. 2006;119:S47–S53. doi: 10.1016/j.amjmed.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Warner L, Skorecki K, Blendis LM, Epstein M. Atrial natriuretic factor and liver disease. Hepatology. 1993;17:500–513. [PubMed] [Google Scholar]
- 30.Hébert RL, Jacobson HR, Breyer MD. PGE2 inhibits AVP-induced water flow in cortical collecting ducts by protein kinase C activation. Am J Physiol. 1990;259:F318–F325. doi: 10.1152/ajprenal.1990.259.2.F318. [DOI] [PubMed] [Google Scholar]
- 31.Orloff J, Handler JS, Bergstrom S. Effect of prostaglandin (pge-1) on the permeability response of toad bladder to vasopressin, theophylline and adenosine 3’,5’-monophosphate. Nature. 1965;205:397–398. doi: 10.1038/205397a0. [DOI] [PubMed] [Google Scholar]
- 32.Anderson RJ, Berl T, McDonald KD, Schrier RW. Evidence for an in vivo antagonism between vasopressin and prostaglandin in the mammalian kidney. J Clin Invest. 1975;56:420–426. doi: 10.1172/JCI108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker LA, Frölich JC. Dose-dependent stimulation of renal prostaglandin synthesis by deamino-8-D-arginine vasopressin in rats with hereditary diabetes insipidus. J Pharmacol Exp Ther. 1981;217:87–91. [PubMed] [Google Scholar]
- 34.Pérez-Ayuso RM, Arroyo V, Camps J, Rimola A, Gaya J, Costa J, Rivera F, Rodés J. Evidence that renal prostaglandins are involved in renal water metabolism in cirrhosis. Kidney Int. 1984;26:72–80. doi: 10.1038/ki.1984.136. [DOI] [PubMed] [Google Scholar]
- 35.Solis-Herruzo JA, Gonzalez-Gamarra A, Castellano G, Muñoz-Yagüe MT. Metabolic clearance rate of arginine vasopressin in patients with cirrhosis. Hepatology. 1992;16:974–979. doi: 10.1002/hep.1840160420. [DOI] [PubMed] [Google Scholar]
- 36.Kim JK, Summer SN, Howard RL, Schrier RW. Vasopressin gene expression in rats with experimental cirrhosis. Hepatology. 1993;17:143–147. [PubMed] [Google Scholar]
- 37.Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342:1581–1589. doi: 10.1056/NEJM200005253422107. [DOI] [PubMed] [Google Scholar]
- 38.Ginès P, Guevara M. Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management. Hepatology. 2008;48:1002–1010. doi: 10.1002/hep.22418. [DOI] [PubMed] [Google Scholar]
- 39.Solà E, Watson H, Graupera I, Turón F, Barreto R, Rodríguez E, Pavesi M, Arroyo V, Guevara M, Ginès P. Factors related to quality of life in patients with cirrhosis and ascites: relevance of serum sodium concentration and leg edema. J Hepatol. 2012;57:1199–1206. doi: 10.1016/j.jhep.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 40.Guevara M, Baccaro ME, Torre A, Gómez-Ansón B, Ríos J, Torres F, Rami L, Monté-Rubio GC, Martín-Llahí M, Arroyo V, et al. Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: a prospective study with time-dependent analysis. Am J Gastroenterol. 2009;104:1382–1389. doi: 10.1038/ajg.2009.293. [DOI] [PubMed] [Google Scholar]
- 41.Llach J, Ginès P, Arroyo V, Rimola A, Titó L, Badalamenti S, Jiménez W, Gaya J, Rivera F, Rodés J. Prognostic value of arterial pressure, endogenous vasoactive systems, and renal function in cirrhotic patients admitted to the hospital for the treatment of ascites. Gastroenterology. 1988;94:482–487. doi: 10.1016/0016-5085(88)90441-6. [DOI] [PubMed] [Google Scholar]
- 42.Ginés P, Quintero E, Arroyo V, Terés J, Bruguera M, Rimola A, Caballería J, Rodés J, Rozman C. Compensated cirrhosis: natural history and prognostic factors. Hepatology. 1987;7:122–128. doi: 10.1002/hep.1840070124. [DOI] [PubMed] [Google Scholar]
- 43.Cosby RL, Yee B, Schrier RW. New classification with prognostic value in cirrhotic patients. Miner Electrolyte Metab. 1989;15:261–266. [PubMed] [Google Scholar]
- 44.Fernández-Esparrach G, Sánchez-Fueyo A, Ginès P, Uriz J, Quintó L, Ventura PJ, Cárdenas A, Guevara M, Sort P, Jiménez W, et al. A prognostic model for predicting survival in cirrhosis with ascites. J Hepatol. 2001;34:46–52. doi: 10.1016/s0168-8278(00)00011-8. [DOI] [PubMed] [Google Scholar]
- 45.Shear L, Kleinerman J, Gabuzda GJ. Renal failure in patients with cirrhosis of the liver. i. clinical and pathologic characteristics. Am J Med. 1965;39:184–198. doi: 10.1016/0002-9343(65)90041-0. [DOI] [PubMed] [Google Scholar]
- 46.Arroyo V, Bosch J, Gaya-Beltrán J, Kravetz D, Estrada L, Rivera F, Rodés J. Plasma renin activity and urinary sodium excretion as prognostic indicators in nonazotemic cirrhosis with ascites. Ann Intern Med. 1981;94:198–201. doi: 10.7326/0003-4819-94-2-198. [DOI] [PubMed] [Google Scholar]
- 47.Ruf AE, Kremers WK, Chavez LL, Descalzi VI, Podesta LG, Villamil FG. Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone. Liver Transpl. 2005;11:336–343. doi: 10.1002/lt.20329. [DOI] [PubMed] [Google Scholar]
- 48.Heuman DM, Abou-Assi SG, Habib A, Williams LM, Stravitz RT, Sanyal AJ, Fisher RA, Mihas AA. Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death. Hepatology. 2004;40:802–810. doi: 10.1002/hep.20405. [DOI] [PubMed] [Google Scholar]
- 49.Follo A, Llovet JM, Navasa M, Planas R, Forns X, Francitorra A, Rimola A, Gassull MA, Arroyo V, Rodés J. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: incidence, clinical course, predictive factors and prognosis. Hepatology. 1994;20:1495–1501. doi: 10.1002/hep.1840200619. [DOI] [PubMed] [Google Scholar]
- 50.Biggins SW, Kim WR, Terrault NA, Saab S, Balan V, Schiano T, Benson J, Therneau T, Kremers W, Wiesner R, et al. Evidence-based incorporation of serum sodium concentration into MELD. Gastroenterology. 2006;130:1652–1660. doi: 10.1053/j.gastro.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Londoño MC, Guevara M, Rimola A, Navasa M, Taurà P, Mas A, García-Valdecasas JC, Arroyo V, Ginès P. Hyponatremia impairs early posttransplantation outcome in patients with cirrhosis undergoing liver transplantation. Gastroenterology. 2006;130:1135–1143. doi: 10.1053/j.gastro.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 52.Dawwas MF, Lewsey JD, Neuberger JM, Gimson AE. The impact of serum sodium concentration on mortality after liver transplantation: a cohort multicenter study. Liver Transpl. 2007;13:1115–1124. doi: 10.1002/lt.21154. [DOI] [PubMed] [Google Scholar]
- 53.Rose BD. New approach to disturbances in the plasma sodium concentration. Am J Med. 1986;81:1033–1040. doi: 10.1016/0002-9343(86)90401-8. [DOI] [PubMed] [Google Scholar]
- 54.McCormick PA, Mistry P, Kaye G, Burroughs AK, McIntyre N. Intravenous albumin infusion is an effective therapy for hyponatraemia in cirrhotic patients with ascites. Gut. 1990;31:204–207. doi: 10.1136/gut.31.2.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bosch-Marcé M, Jiménez W, Angeli P, Leivas A, Clària J, Graziotto A, Arroyo V, Rivera F, Rodés J. Aquaretic effect of the kappa-opioid agonist RU 51599 in cirrhotic rats with ascites and water retention. Gastroenterology. 1995;109:217–223. doi: 10.1016/0016-5085(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 56.Moreau R, Cailmail S, Hamon G, Lebrec D. Renal and haemodynamic responses to a novel kappa opioid receptor agonist, niravoline (RU 51,599), in rats with cirrhosis. J Gastroenterol Hepatol. 1996;11:857–863. doi: 10.1111/j.1440-1746.1996.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 57.Gadano A, Moreau R, Pessione F, Trombino C, Giuily N, Sinnassamy P, Valla D, Lebrec D. Aquaretic effects of niravoline, a kappa-opioid agonist, in patients with cirrhosis. J Hepatol. 2000;32:38–42. doi: 10.1016/s0168-8278(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 58.Inoue T, Ohnishi A, Matsuo A, Kawai B, Kunihiro N, Tada Y, Koizumi F, Chau T, Okada K, Yamamura Y, et al. Therapeutic and diagnostic potential of a vasopressin-2 antagonist for impaired water handling in cirrhosis. Clin Pharmacol Ther. 1998;63:561–570. doi: 10.1016/S0009-9236(98)90107-2. [DOI] [PubMed] [Google Scholar]
- 59.Gerbes AL, Gülberg V, Ginès P, Decaux G, Gross P, Gandjini H, Djian J. Therapy of hyponatremia in cirrhosis with a vasopressin receptor antagonist: a randomized double-blind multicenter trial. Gastroenterology. 2003;124:933–939. doi: 10.1053/gast.2003.50143. [DOI] [PubMed] [Google Scholar]
- 60.Wong F, Blei AT, Blendis LM, Thuluvath PJ. A vasopressin receptor antagonist (VPA-985) improves serum sodium concentration in patients with hyponatremia: a multicenter, randomized, placebo-controlled trial. Hepatology. 2003;37:182–191. doi: 10.1053/jhep.2003.50021. [DOI] [PubMed] [Google Scholar]
- 61.Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, Orlandi C. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–2112. doi: 10.1056/NEJMoa065181. [DOI] [PubMed] [Google Scholar]
- 62.Ginès P, Wong F, Watson H, Milutinovic S, del Arbol LR, Olteanu D. Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: a randomized trial. Hepatology. 2008;48:204–213. doi: 10.1002/hep.22293. [DOI] [PubMed] [Google Scholar]
- 63.Thuluvath PJ, Maheshwari A, Wong F, Yoo HW, Schrier RW, Parikh C, Steare S, Korula J. Oral V2 receptor antagonist (RWJ-351647) in patients with cirrhosis and ascites: a randomized, double-blind, placebo-controlled, single ascending dose study. Aliment Pharmacol Ther. 2006;24:973–982. doi: 10.1111/j.1365-2036.2006.03088.x. [DOI] [PubMed] [Google Scholar]
- 64.Berl T, Quittnat-Pelletier F, Verbalis JG, Schrier RW, Bichet DG, Ouyang J, Czerwiec FS. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21:705–712. doi: 10.1681/ASN.2009080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int. 2006;69:2124–2130. doi: 10.1038/sj.ki.5000432. [DOI] [PubMed] [Google Scholar]
- 66.Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, Horie S, Nutahara K, Ouyang J, Krasa HB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol. 2011;6:2499–2507. doi: 10.2215/CJN.03530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong F, Watson H, Gerbes A, Vilstrup H, Badalamenti S, Bernardi M, Ginès P. Satavaptan for the management of ascites in cirrhosis: efficacy and safety across the spectrum of ascites severity. Gut. 2012;61:108–116. doi: 10.1136/gutjnl-2011-300157. [DOI] [PubMed] [Google Scholar]
- 69.Miller PD, Linas SL, Schrier RW. Plasma demeclocycline levels and nephrotoxicity. Correlation in hyponatremic cirrhotic patients. JAMA. 1980;243:2513–2515. [PubMed] [Google Scholar]
- 70.Solà E, Lens S, Guevara M, Martín-Llahí M, Fagundes C, Pereira G, Pavesi M, Fernández J, González-Abraldes J, Escorsell A, et al. Hyponatremia in patients treated with terlipressin for severe gastrointestinal bleeding due to portal hypertension. Hepatology. 2010;52:1783–1790. doi: 10.1002/hep.23893. [DOI] [PubMed] [Google Scholar]