

Abstract
Retinitis pigmentosa (RP), a heterogeneous group of inherited ocular diseases, is a genetic condition that causes retinal degeneration and eventual vision loss. Though some genes have been identified to be associated with RP, still a large part of the clinical cases could not be explained. Here we reported a four-generation Chinese family with RP, during which 6 from 9 members of the second generation affected the disease. To identify the genetic defect in this family, whole-exome sequencing together with validation analysis by Sanger sequencing were performed to find possible pathogenic mutations. After a pipeline of database filtering, including public databases and in-house databases, a novel missense mutation, c. 424 C > T transition (p.R142W) in OR2W3 gene, was identified as a potentially causative mutation for autosomal dominant RP. The mutation co-segregated with the disease phenotype over four generations. This mutation was validated in another independent three-generation family. RT-PCR analysis also identified that OR2W3 gene was expressed in HESC-RPE cell line. The results will not only enhance our current understanding of the genetic basis of RP, but also provide helpful clues for designing future studies to further investigate genetic factors for familial RP.
Retinitis pigmentosa (RP, MIM#268000), a heterogeneous group of inherited ocular diseases, results in 1 from 3,000 to 5,000 people affecting progressive retinal degeneration1. It is clinically characterized by some degenerative symptoms including progressive night blindness, tunnel vision and bone-spicule pigmentation in retina, then cause severe vision impairment and often blindness2. The disease is of highly clinical and genetic heterogeneity and could be inherited in autosomal dominant (about 30–40% of total cases), autosomal recessive (50–60%), and X-linked models (5–15%)3. Presently, 57 genes/loci have been identified to be associated with RP (https://sph.uth.tmc.edu/RetNet/home.htm), 32 of which were associated with autosomal recessive RP, 20 with autosomal dominant RP, and 5 with X-linked RP. However, a large part of the clinical cases still could not be explained by these genes.
So far, whole-exome sequencing has become a powerful strategy in the detection of rare causal variants of Mendelian disorders, including RP, because disease-causing mutations usually change an encoded protein4,5,6,7,8,9,10,11,12,13,14,15,16,17. However, many of these studies focus only on simplex families or one affected child from multiplex families. In this study, we selected a large four-generation family, in which six out of nine members in the second generation were affected by RP. Prescreen has excluded known causing mutations for RP. We aimed to identify possible causal genes of RP in this Chinese family using a whole-exome sequencing approach, together with validation by another independent three-generation family.
Methods
Subjects and clinical evaluation
We recruited a four-generation Chinese family from Chongqing in Southwest China. Six of nine members in the second generation affected RP (Figure 1-A). All participants underwent a full ophthalmologic examinations, including slit-lamp biomicroscopy, fundus examination, visual field test, and full-field flash electroretinography (ERG). Blood-derived DNA was available from five cases II-1, II-2, II-3, II-4, II-7 and from twelve healthy family members including II-8, II-9, III-1, III-2, III-3, III-15, III-16, IV-1, IV-2, IV-4, IV-5, IV-6. The study was approved by Ethics Review Committee of Third Military Medical University and carried out in accordance with the Declaration of Helsinki. Peripheral venous blood samples were derived after a signed informed consent.
Figure 1. Pedigree of the two familys and fundus photography of an affected case and a normal subject.

(A) Pedigree of the four-generation Chinese family with RP: Squares represent males, and circles represent females. Solid symbols indicate affected individuals, while open symbols indicate unaffected individuals. Slash indicates the deceased. (B) Fundus photography of an affected case: attenuation of retinal vascular, bone-spicule pigmentation, Chorioretinal degeneration with peripapillary atrophy, waxy-pale discs, and enlarged optic cups; (C) Normal fundus; (D) Pedigree of the second independent family.
DNA extraction, mutation screening
Genomic DNA was extracted from peripheral leukocytes using the TIANamp Blood DNA Kit (Tiangen Biotech Co. Ltd, Beijing, China). To identify whether RP patients in this family were caused by unknown genes, the previously known genes (Supplementary Table 1) shown to be mutated in RP patients were first screened among one RP case (II-1) and one healthy control (II-8) using a targeted gene capture chip developed by BGI, Shenzhen, China. Sanger sequencing was then used to replicate the positive findings.
Whole-exome sequencing
The whole-exome sequencing approach was employed to identify the disease-associated genes in five subjects, including four RP cases (II-2, II-3, II-4, and II-7) and one healthy control (II-9) by BGI, Shenzhen, China. Thirty microgram (μg) human genomic DNA was extracted from peripheral venous blood samples of each participant. Qualified genomic DNA sample was randomly fragmented by Covaris Acoustic System. Then adapters were ligated to both ends of the resulting fragments. Extracted DNA was then amplified by ligation-mediated PCR (LM-PCR), purified, and hybridized to the Nimblegen SeqCap EZ Library v3.0 (Roche/NimbleGen, Madison, WI) for enrichment. Both non-captured and captured LM-PCR products were subjected to quantitative PCR to estimate the magnitude of enrichment. Each captured library was then loaded on Hiseq2500 platform (Illumina, San Diego, CA). We performed high-throughput sequencing for each captured library to ensure that each sample meets the desired average sequencing depth (90×). Raw image files were processed by Illumina base calling Software 1.7 for base-calling with default parameters and the sequences of each individual were generated as 90 bp pair-end reads.
Bioinformatics analysis
The clean reads were aligned to the human reference genome (GRCh37, UCSC hg19) by SOAPaligner (soap2.21)18. Based on the results from SOAPaligner, software SOAPsnp (version 1.03) was used to assemble the consensus sequence and call genotypes in target regions19. When analyzing indel, BWA was used to map reads onto the reference 20, then we passed the alignment result to the Genome Analysis Toolkit (GATK) to identify the breakpoints21. Only mapped reads were used for subsequent analysis. Coverage and depth calculations were based on all mapped reads and the exome region. All variants were first filtered against several public databases for the minor allele frequency (MAF) > 0.5%, including dbSNP135, 1000 genomes data (pilot1, 2, 3), hapmap (release 24), YH project22, then against two in-house databases (sample size were 7,000 from Vanderbilt Epidemiology Center and 1,414 from BGI, respectively; samples of both databases come from Chinese population, which have the similar genetic background with the subjects in current study).
Mutation validation
To determine whether any of the remaining variants co-segregated with the disease phenotype in this family, the mutations were then confirmed in all other family members that DNA samples were available by Sanger sequencing. Direct polymerase chain reaction (PCR) products were sequenced using ABI 3730 Genetic Analyzer. Sequencing data were compared pair-wisely with the Human Genome database (GRCh37, UCSC hg19) to detect mutations. The possible causative mutation was further confirmed using RP pedigree database of GBI.
Cell culture, differentiation and identification
The HESC line H1 was induced to differentiate into retinal pigmented epithelium cells (HESC-RPE) as described previously23. Immunofluorescence analysis was performed according previous methods24. In brief, the HESC-RPE cells were fixed with 4% paraformaldehyde for 20 min, permeabilized using 0.1% Triton X-100 in PBS for 15 min and blocked for 30 min in 3% BSA. The following primary antibody were used: Mitf (Abcam, 1:50), Pax6 (Abcam, 1:50), zonula occludens-1 (ZO-1, Invitrogen, 1:400).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA of HESC-RPE on 60 day, 80 d and 100 d were extracted using an RNAprep Pure Cell Kit (Sangon Biotech, CHN) according to the manufacturer's instructions. Total RNA (approximately 1–2 μg per 20 μl reaction) was reverse transcribed using a PrimeScript® RT Reagent Kit (Takara, JPN). PCR amplification of OR2W3 gene (primers: F- TGGTGTTTATCCTGCTCTCTTAC; R- CTCTGTTTCTGAGGGTGTAGATG) was performed by the CFX96 Real-Time PCR System (Bio-Rad, USA) using a PCR Mix (Dongsheng Biotech, CHN) according to the manufacturer's instructions.
Results
Clinical characteristics
Figure 1-A presents the pedigree of the four-generation Chinese family, which was consistent with autosomal dominant inheritance. Totally there are 7 members in this family affected RP, including two deceased members (I-1 and II-5). The two deceased members showed similar clinical symptoms and pathogenesis with other 5 alive members (II-1, II-2, II-3, II-4, II-7). Night blindness appeared first, followed by progressive reduction of the visual field, and finally complete blindness in later life. Table 1 presents the clinical data of 5 alive affected individuals. All patients had a progressive bilateral decrease of visual acuity, peripheral visual field, and photophobia. Fundus photography revealed similar clinical features for the affected individuals, including attenuation of retinal vascular, bone-spicule pigmentation, chorioretinal degeneration with peripapillary atrophy, optic disc pallor, and enlarged optic cups, comparing with the normal subject (Figure 1-B, 1-C). ERG records showed no detectable cone or rod responses in the patients.
Table 1. Characteristics of 5 alive affected individuals from RP pedigree.
| Characteristics | II-1 | II-2 | II-3 | II-4 | II-7 |
|---|---|---|---|---|---|
| Age (years) | 64 | 60 | 58 | 54 | 46 |
| Gender | Female | Male | Male | Male | Female |
| Age of night blindness onset (years) | 20 | 30 | 21 | 20 | 30 |
| Visual field | None | None | None | None | None |
| Optic disc | pallor | pallor | pallor | pallor | pallor |
| Artery attenuation | Yes | Yes | Yes | Yes | Yes |
| Pigment deposits | Yes | Yes | Yes | Yes | Yes |
| Electroretinography | non-detectable | non-detectable | non-detectable | non-detectable | non-detectable |
Mutation screening
To find the causative mutations and exclude the known genes, we sequenced all exons and the flanking intronic splicing sites of the previously known causative genes of RP (Supplementary Table 1) among one RP case (II-1) and one healthy control (II-8), and confirmed by Sanger sequencing. All genes showed no pathogenic mutations, indicating the possibility of the familial cases in current study were caused by mutations in unknown genes.
Whole-exome sequencing
Whole-exome sequencing was performed upon five subjects, including four RP cases (II-2, II-3, II-4, and II-7) and one healthy control (II-9). An average of 11,747 MB raw data was generated with a mean depth of 101.74-fold for the target regions. Approximately 98.64% of the targeted bases (64,482,551 bp in length) were covered sufficiently to pass our thresholds for calling SNPs and indels. We identified 144,701-150,367 SNPs and 15368-16173 indels for the five sequenced subjects. For rare inherited diseases, the frequency of the possible pathogenic mutations in healthy population should be very low. Therefore, as shown in Table 2 and Table 3, the results were then filtered against several public variation databases, removing all previously reported variants. We focused only on non-synonymous (NS) variants, variants in splicing sites, and short, frame-shift coding insertions or deletions (Indels). After filtering against these databases, we found 72 SNPs and 15 indels were shared by affected patients and absent in healthy controls. Furthermore, two in-house databases were used to filter the remaining variants, which resulted that 10 SNPs were left (OR2W3 R142W, DNM2 R297H, ROBO2 P1106S, CSMD3 K3075Q, ZHX2 G799R, PALM3 E658Q, HAP1 E269Q, BRIP1 N775S, INTS2 I775L, and TSSC4 H81R).
Table 2. Number of candidate variants filtered against several public variation databases.
| Feature_SNP | control:II-9 | case:II-2 | case:II-3 | case:II-4 | case:II-7 |
|---|---|---|---|---|---|
| Total_SNPs1 | 150367 | 146587 | 149036 | 144701 | 147040 |
| Functional_SNPs2 | 15982 | 15944 | 15897 | 15814 | 15847 |
| Filtered_DBsnp | 13286 | 13229 | 13202 | 13084 | 13195 |
| Filtered_DBsnp_1000gene | 2857 | 2834 | 2804 | 2716 | 2709 |
| Filtered_DBsnp_1000gene_Hapmap | 2817 | 2793 | 2763 | 2678 | 2670 |
| Filtered_DBsnp_1000gene_Hapmap_YH | 2664 | 2638 | 2609 | 2524 | 2515 |
| Filtered_DBsnp_1000gene_Hapmap_YH_II-93 | 0 | 870 | 840 | 786 | 850 |
| Share_all_cases | 72 | ||||
| Filtered _Housedatabase | 10 | ||||
| Genotype & phenotype coseparation | 1 (OR2W3 R142W) |
1Total-SNPs detection were performed on the targeted exome regions and flanking regions within 200 bp. SNP types include variants of nonsense, missense, splicing site, 5-UTR, 3-UTR, NR_exon, synonymous-coding, intron, intergenic.
2Functional_SNPs include variants of nonsense, missense, splicing site.
3In this step, variants were filtered by mutations of healthy control: II-9.
Table 3. Number of candidate Indels filtered against several public variation databases.
| Feature_Indel | control:II-9 | case:II-2 | case:II-3 | case:II-4 | case:II-7 |
|---|---|---|---|---|---|
| Total_Indels1 | 16173 | 15403 | 16053 | 15368 | 15772 |
| Functional_Indels2 | 2053 | 1976 | 2058 | 1996 | 2089 |
| Filtered_DBsnp | 586 | 576 | 582 | 570 | 565 |
| Filtered_DBsnp_1000gene | 337 | 346 | 323 | 325 | 327 |
| Filtered_DBsnp_1000gene_Hapmap | 337 | 346 | 323 | 325 | 327 |
| Filtered_DBsnp_1000gene_Hapmap_YH | 335 | 344 | 321 | 324 | 325 |
| Filtered_DBsnp_1000gene_Hapmap_YH_ II-93 | 0 | 159 | 147 | 146 | 151 |
| Share_all_cases | 15 | ||||
| Housedatabase_filter | 0 |
1Total-Indels detection were performed on the targeted exome regions and flanking regions within 100 bp. Indel types include variants of frameshift, cds-Indel, spliceSite, 5-UTR, 3-UTR, intron, promoter, intergenic.
2Functional_Indels include variants of frameshift, cds-Indel, spliceSite.
3In this step, variants were filtered by mutations of healthy control: II-9.
Phenotype & genotype co-segregation and validation of the mutations
The ten remaining mutations were then confirmed in other twelve family members that DNA samples were available by Sanger sequencing to co-segregate with the disease phenotype (Figure 2). Genetic analysis demonstrated that only OR2W3 (Olfactory receptor 2, W3) R142W was carried by affected patients and absent in healthy controls. Then, OR2W3 R142W mutation was also observed in another three-generation RP family (Figure 1-D), including 3 cases (II-1, II-2, III-1) and 1 control (I-1); three RP cases were found to carry the same mutation and one healthy control does not. Furthermore, immunofluorescent analysis of HESC-RPE revealed the expression of RPE cells markers (Mitf, PAX6, and ZO-1), while RT-PCR analysis showed that HESC-RPE expressed OR2W3 (Figure 3).
Figure 2. Sanger sequencing of OR2W3 R142W mutation.

Figure 3. Identification of HESC-RPE cells.
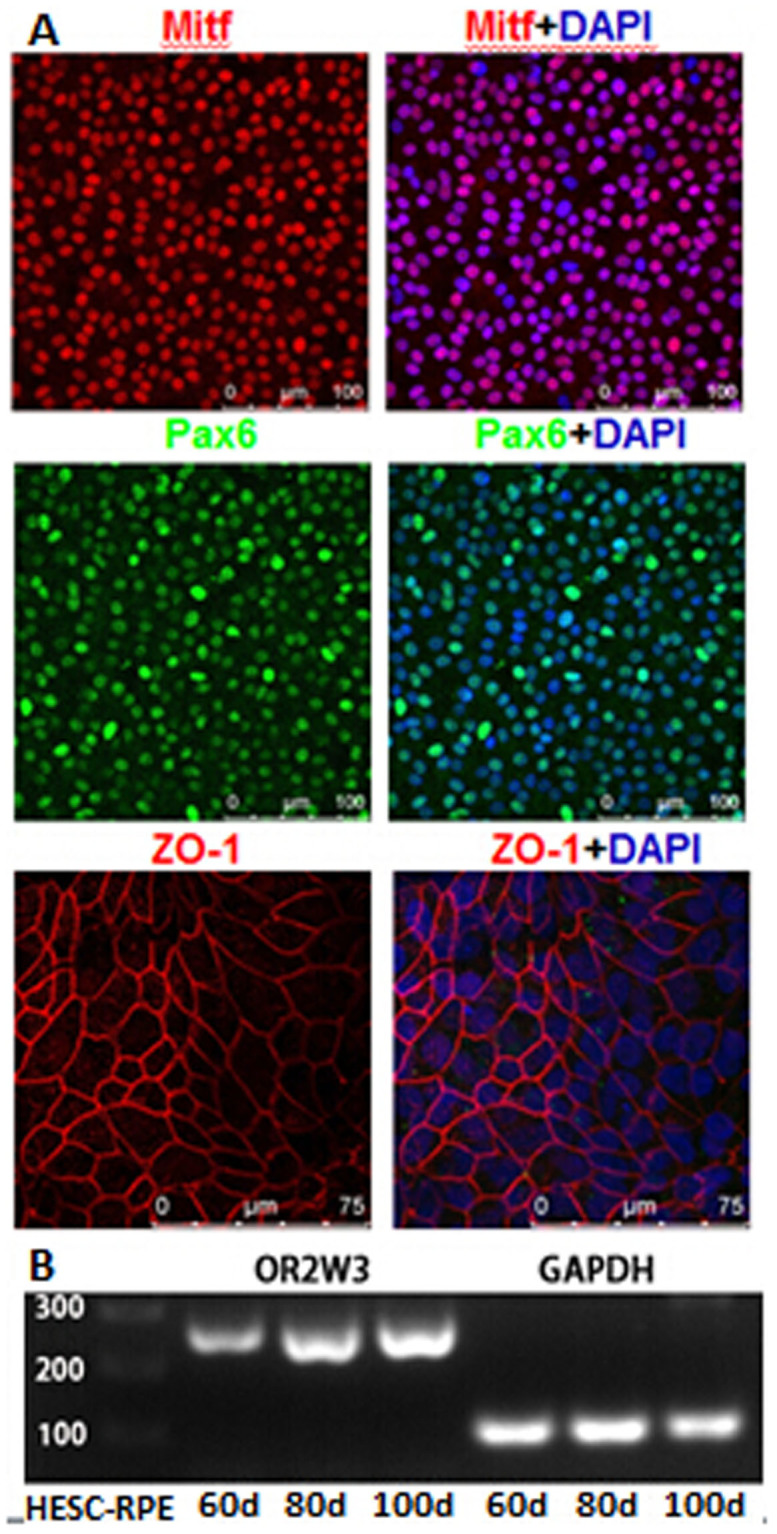
(A) Immunocytochemistry of HESC-RPE cells demonstrating the expression of Mitf, Pax6 and ZO-1. (B) RT-PCR analysis of OR2W3 in HESC-RPE. Cropped gel has been run under the same experimental conditions. Full-length blot is presented in Supplementary Figure S1.
Conservation of R142W in OR2W3 gene
Pathogenicity assessment of OR2W3 R142W mutation was undertaken by evaluation of amino acid evolutionary conservation and in-silico prediction studies. Using UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway), we found the variant was highly conserved in nine primate species, including human, chimp, gorilla, orangutan, bibbon, rhesus, ab-eating_macaque, baboon, green_monkey, and bushbaby, although not conserved in non-primate mammals. According to two web-based topology prediction package: TMPred (http://www.ch.embnet.org/software/TMPRED_form.html) and TopPred (http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::toppred)25, OR2W3 R142W mutation is located in a transmembrane domain of OR2W3 gene. The variants was also predicted to have a deleterious effect by Mutation Taster26. Exome Variant Server (EVS) database retrieval didn't find this variant.
Discussion
RP, the most frequent inherited retinal degeneration, has become one of the commonest causes of genetic visual dysfunction27. Since RP1 identified by linkage study in 199128, 56 susceptibility genes/loci for RP have been subsequently discovered by different approaches. However, due to the enormous heterogeneity of the disease pathogenesis, a large part of the familial cases still could not be explained. In this study, using a whole exome sequencing approach, we identified a novel missense mutation, c. 424 C > T transition (p.R142W) in OR2W3 gene, associated with autosomal dominant RP in a large Chinese family. This mutation was validated in another independent three-generation family. RT-PCR analysis also identified that OR2W3 gene was expressed in HESC-RPE cell line. To the best of our knowledge, OR2W3 gene was identified to be associated with RP for the first time.
The olfactory receptors (ORs), including OR2W3, were first defined as a supergene family that encodes G-protein coupled receptor proteins (GPCRs) in olfactory epithelium of the rat in 199129,30. Zhao et al. explored the physiological function of ORs in initiating transduction in olfactory receptor neurons31. However, ORs were not exclusively expressed in the olfactory epithelium. Recent studies have demonstrated ORs were expressed in a broad variety of other tissues, including autonomic nervous system, brain, tongue, erythroid cells, prostate, placenta, gut and kidney32. Furthermore, RNA sequencing of 16 different human tissues by Next Generation Sequencing (NGS) revealed OR2W3 gene were expressed in 9 different tissue samples, and most highly expressed in thyroid33. These indicated the different potential functions of OR2W3 gene in different human biological process.
OR2W3 gene, which was located in 1q44, has an intron-free reading frame of 942 nucleotides that encodes 314 amino acids. UCSC Genome Browser34 showed that OR2W3 shares exons with Trim58 (Tripartite motif-containing protein 58). When we used SWISS-MODEL server35 to model the structure of OR2W3 protein, JAGGED-1 (PDB ID: 2vj2B)36, which was also associated with one kind of autosomal dominant inherited disease - Alagille syndrome37, showed the biggest sequence identity with OR2W3. Recent studies also revealed that the biological functions of OR2W3 gene was not only restricted to olfactory system, like G-protein coupled receptor activity and olfactory receptor activity. Aston et al.38 and Plaseski et al.39 found OR2W3 rs11204546 was associated with both azoospermia and oligozoospermia risk; a mutation in OR2W3 gene (chr1:248059606, p.T240P) was associated with the metastasis of pancreatic ductal adenocarcinoma40; expression of OR2W3 was also identified to be associated with long-tern schizophrenia41, variability in response tob-blockers42, and the changes in global gene-expression profiles in human cervical cancer HeLa cells exposed to non-activated Dendrimers and Dendriplexes43. However, through epidemiological survey and Medical record retrieval, all the subjests in current study don't have related diseases and mutations.
Vision and olfaction are two of the major sensory systems, which coordinate and integrate the information to provide us a unified perception of our environment. Studies showed that they share many links and common points in different aspects, including neuroanatomical pathways44, cross-modal links and the extension of this notion to goal-directed actions45, pathogenic or biological genes46,47,48. Woodard et al.47 found rdgB (retinal degeneration B), a gene required for normal visual system physiology, was shown to be necessary for olfactory response of both adult flies and larvae, indicating that rdgB was required for both visual and olfactory physiology. Loss of olfactory receptor genes were also found to coincide with the acquisition of full trichromatic vision46. In this study, we revealed a novel missense mutation in OR2W3 gene, was associated with autosomal dominant RP. This finding may indicate the essential links between Vision and olfaction, and strongly suggested an exchange in the importance of these two senses.
As we mentioned above, RP refers to a highly clinical and genetic heterogeneous group of inherited ocular diseases. Inheritance patterns included autosomal dominant, autosomal recessive, and X-linked models. In this study, we presumed autosomal dominant to be the inheritance pattern of this family basing on two reasons. First, both the first two generations have affected patients. We excluded the possibility of intermarriage through intensive epidemiologic survey. Second, high prevalence rate (6/9 = 66.7%) in the second generation. Nevertheless, we also analyzed the data based on the autosomal recessive model, including homozygous inheritance model and compound heterozygous model, but no promising mutations were detected. One limitations of this study is that due to patient's refusal for retinal biopsy, the results could not be strengthened by RNA analysis of this gene or immune-localisation of the protein using multiple tissues including the retina and retinal pigment epithelium(RPE) cells.
Conclusion
A novel missense mutation (OR2W3 R142W) was identified to be associated with RP by whole-exome sequencing. Our findings expand the phenotypic and mutation spectrum of RP and provide helpful clues for designing future studies to further investigate genetic factors for familial RP.
Author Contributions
L.Y., X.X., X.H., M.X. and G.L. designed the experiments; M.X., G.L., W.W., Z.Y., W.N., W.L., X.Y. and X.B. performed the investigations and experiments; M.X. and G.L. analyzed the data; L.L., Z.W., G.Y., L.J., S.M., C.Y., W.Y., Y.Y. and L.Y. provided technical and material support. M.X. and G.L. wrote the manuscript; all authors reviewed the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
This research was supported by grants from National Natural Science Foundation of China (Grant No. 81171903 and 81302498); The State Key Development Program for Basic Research of China—973 Program (NO.2011CB809201). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Veltel S., Gasper R., Eisenacher E. & Wittinghofer A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nature structural & molecular biology 15, 373–380, 10.1038/nsmb.1396 (2008). [DOI] [PubMed] [Google Scholar]
- Busskamp V. et al. Genetic Reactivation of Cone Photoreceptors Restores Visual Responses in Retinitis Pigmentosa. Science 329, 413–417, 10.1126/science.1190897 (2010). [DOI] [PubMed] [Google Scholar]
- Hartong D. T., Berson E. L. & Dryja T. P. Retinitis pigmentosa. Lancet 368, 1795–1809, 10.1016/S0140-6736(06)69740-7 (2006). [DOI] [PubMed] [Google Scholar]
- Bamshad M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature Reviews Genetics 12, 745–755, 10.1038/Nrg3031 (2011). [DOI] [PubMed] [Google Scholar]
- Bowne S. J. et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 19, 1074–1081, 10.1038/ejhg.2011.86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon-Camacho O. F., Jitskii S., Buentello-Volante B., Quevedo-Martinez J. & Zenteno J. C. Exome sequencing identifies RDH12 compound heterozygous mutations in a family with severe retinitis pigmentosa. Gene 528, 178–182, 10.1016/j.gene.2013.07.021 (2013). [DOI] [PubMed] [Google Scholar]
- Davidson A. E. et al. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 93, 321–329, 10.1016/j.ajhg.2013.06.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A. E. et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat 34, 506–514, 10.1002/humu.22264 (2013). [DOI] [PubMed] [Google Scholar]
- Goldenberg-Cohen N. et al. Genetic heterogeneity and consanguinity lead to a “double hit”: homozygous mutations of MYO7A and PDE6B in a patient with retinitis pigmentosa. Mol Vis 19, 1565–1571 (2013). [PMC free article] [PubMed] [Google Scholar]
- Khateb S. et al. Exome sequencing identifies a founder frameshift mutation in an alternative exon of USH1C as the cause of autosomal recessive retinitis pigmentosa with late-onset hearing loss. PLoS One 7, e51566, 10.1371/journal.pone.0051566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Vidal C. et al. Whole-exome sequencing identifies novel compound heterozygous mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa. Mol Vis 19, 2187–2195 (2013). [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi K. M. et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci U S A 110, 16139–16144, 10.1073/pnas.1308243110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemiatkowska A. M. et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology 120, 2697–2705, 10.1016/j.ophtha.2013.07.052 (2013). [DOI] [PubMed] [Google Scholar]
- Tucker B. A. et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci U S A 108, E569–576, 10.1073/pnas.1108918108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A. L. et al. F45L Allele Does Not Cause Autosomal Dominant Retinitis Pigmentosa in a Large Caucasian Family. Translational vision science & technology 2, 4, 10.1167/tvst.2.2.4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with retinitis pigmentosa. PLoS One 7, e33673, 10.1371/journal.pone.0033673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S. et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am J Hum Genet 88, 201–206, 10.1016/j.ajhg.2011.01.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967, 10.1093/bioinformatics/btp336 (2009). [DOI] [PubMed] [Google Scholar]
- Li R. et al. SNP detection for massively parallel whole-genome resequencing. Genome Res 19, 1124–1132, 10.1101/gr.088013.108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. & Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, 10.1093/bioinformatics/btp698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, 10.1101/gr.107524.110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. et al. The YH database: the first Asian diploid genome database. Nucleic Acids Res 37, D1025–1028, 10.1093/nar/gkn966 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vugler A. et al. Elucidating the phenomenon of HESC-derived RPE: anatomy of cell genesis, expansion and retinal transplantation. Exp Neurol 214, 347–361, 10.1016/j.expneurol.2008.09.007 (2008). [DOI] [PubMed] [Google Scholar]
- Duan P., Xu H., Zeng Y., Wang Y. & Yin Z. Q. Human bone marrow stromal cells can differentiate to a retinal pigment epithelial phenotype when co-cultured with pig retinal pigment epithelium using a transwell system. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 31, 601–613, 10.1159/000350080 (2013). [DOI] [PubMed] [Google Scholar]
- Claros M. G. & von Heijne G. TopPred II: an improved software for membrane protein structure predictions. Computer applications in the biosciences: CABIOS 10, 685–686 (1994). [DOI] [PubMed] [Google Scholar]
- Schwarz J. M., Rodelsperger C., Schuelke M. & Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7, 575–576, 10.1038/nmeth0810-575 (2010). [DOI] [PubMed] [Google Scholar]
- Chacon-Camacho O. F., Jitskii S., Buentello-Volante B., Quevedo-Martinez J. & Zenteno J. C. Exome sequencing identifies RDH12 compound heterozygous mutations in a family with severe retinitis pigmentosa. Gene, 10.1016/j.gene.2013.07.021 (2013). [DOI] [PubMed] [Google Scholar]
- Blanton S. H. et al. Linkage mapping of autosomal dominant retinitis pigmentosa (RP1) to the pericentric region of human chromosome 8. Genomics 11, 857–869 (1991). [DOI] [PubMed] [Google Scholar]
- Buck L. & Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65, 175–187 (1991). [DOI] [PubMed] [Google Scholar]
- Malnic B., Godfrey P. A. & Buck L. B. The human olfactory receptor gene family. Proc Natl Acad Sci U S A 101, 2584–2589 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. et al. Functional expression of a mammalian odorant receptor. Science 279, 237–242 (1998). [DOI] [PubMed] [Google Scholar]
- Feldmesser E. et al. Widespread ectopic expression of olfactory receptor genes. BMC Genomics 7, 121, 10.1186/1471-2164-7-121 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegel C., Manteniotis S., Osthold S., Hatt H. & Gisselmann G. Expression profile of ectopic olfactory receptors determined by deep sequencing. PLoS One 8, e55368, 10.1371/journal.pone.0055368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R. M. et al. The UCSC Genome Browser Database: update 2009. Nucleic Acids Res 37, D755–761, 10.1093/nar/gkn875 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordoli L. et al. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4, 1–13, 10.1038/nprot.2008.197 (2009). [DOI] [PubMed] [Google Scholar]
- Cordle J. et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nature structural & molecular biology 15, 849–857, 10.1038/nsmb.1457 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T. et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16, 235–242, 10.1038/ng0797-235 (1997). [DOI] [PubMed] [Google Scholar]
- Aston K. I., Krausz C., Laface I., Ruiz-Castane E. & Carrell D. T. Evaluation of 172 candidate polymorphisms for association with oligozoospermia or azoospermia in a large cohort of men of European descent. Hum Reprod 25, 1383–1397, 10.1093/humrep/deq081 (2010). [DOI] [PubMed] [Google Scholar]
- Plaseski T., Noveski P., Popeska Z., Efremov G. D. & Plaseska-Karanfilska D. Association study of single-nucleotide polymorphisms in FASLG, JMJDIA, LOC203413, TEX15, BRDT, OR2W3, INSR, and TAS2R38 genes with male infertility. J Androl 33, 675–683, 10.2164/jandrol.111.013995 (2012). [DOI] [PubMed] [Google Scholar]
- Zhou B. et al. Exome sequencing and digital PCR analyses reveal novel mutated genes related to the metastasis of pancreatic ductal adenocarcinoma. Cancer Biol Ther 13, 871–879, 10.4161/cbt.20839 (2012). [DOI] [PubMed] [Google Scholar]
- Narayan S. et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Research 1239, 235–248, 10.1016/j.brainres.2008.08.023 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli U. et al. Change in mRNA Expression after Atenolol, a Beta-adrenergic Receptor Antagonist and Association with Pharmacological Response. Arch Drug Inf 2, 41–50, 10.1111/j.1753-5174.2009.00020.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo J. H., Liou M. J. & Chiu H. C. Evaluating the gene-expression profiles of HeLa cancer cells treated with activated and nonactivated poly(amidoamine) dendrimers, and their DNA complexes. Mol Pharm 7, 805–814, 10.1021/mp900303s (2010). [DOI] [PubMed] [Google Scholar]
- Cooper H. M., Parvopassu F., Herbin M. & Magnin M. Neuroanatomical pathways linking vision and olfaction in mammals. Psychoneuroendocrinology 19, 623–639 (1994). [DOI] [PubMed] [Google Scholar]
- Castiello U., Zucco G. M., Parma V., Ansuini C. & Tirindelli R. Cross-modal interactions between olfaction and vision when grasping. Chemical senses 31, 665–671, 10.1093/chemse/bjl007 (2006). [DOI] [PubMed] [Google Scholar]
- Gilad Y., Przeworski M. & Lancet D. Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates. PLoS Biol 2, E5, 10.1371/journal.pbio.0020005 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard C., Alcorta E. & Carlson J. The rdgB gene of Drosophila: a link between vision and olfaction. Journal of neurogenetics 21, 291–305, 10.1080/01677060701695441 (2007). [DOI] [PubMed] [Google Scholar]
- Colbert H. A., Smith T. L. & Bargmann C. I. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J Neurosci 17, 8259–8269 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information