

Abstract
With recent technical advances, important signaling pathways have continuously been discovered and dissected in many biological events. The vast majority of these signaling pathways involve reversible protein phosphorylation, and the dynamics of phosphorylation provides important mechanisms on how signaling networks function and interact. With a variety of research projects using lab models or clinical samples, a simple and reliable phosphorylation assay is highly desirable for routine detection of phosphorylation in sample mixtures. The protocols in this article describe the general procedure for a new non-antibody strategy for phosphorylation assay, termed pIMAGO (phospho-imaging). This novel design takes advantage of not only the unique properties of the soluble nanoparticles, but also of the multiple functionality of the molecule, allowing for highly selective, sensitive and quantitative assessment of protein phosphorylation without the use of either radioactive isotopes or limited phosphospecific antibodies. It also offers the capability for multiplexed detection of phosphorylation and total protein amount simultaneously. The described procedures allow for straightforward and routine detection and quantitation of general phosphorylation on any site of any protein in Western Blot and ELISA formats.
Keywords: Phosphoprotein detection, phosphorylation, ELISA, Western Blot, kinase assay, high-throughput screening
INTRODUCTION
Protein phosphorylation is a crucial post-translational modification that regulates a broad range of cellular activities including the cell cycle, differentiation, metabolism, and signaling (Hunter, 2000; Pawson, 2004). Abnormal phosphorylation events are implicated in many disease states (Blume-Jensen and Hunter, 2001). Therefore, assessing the phosphorylation status of an individual protein or classes of proteins, qualitatively or quantitatively, has become a routine but extremely important step in a majority of research labs in life sciences. Common methods for phosphorylation analyses include the use of phospho-specific antibodies, 32P radioactive labeling, and mass spectrometry. The method of choice may vary depending on many factors including the specific question being asked and availability of specialized equipment or reagents. Almost every existing method may associate with certain drawbacks, such as low sensitivity, poor specificity, undesirable reproducibility, intensive labor, or safety issues. A simple and reliable phosphorylation assay for routine detection of phosphorylation in complex and typically heterogeneous biological samples is urgently needed to evaluate the dynamics of phosphorylation, which provides important mechanisms on how the signaling networks function and interact.
This article describes the procedural details for a non-antibody-based universal detection of protein phosphorylation, termed pIMAGO (phospho-imaging). The technology is based on water-soluble, globular nanopolymers (i.e., dendrimers) that are multi-functionalized with reactive groups (e.g., Ti(IV) ions) for the site-specific recognition of phosphate groups, and with ‘reporting’ groups that facilitate detection (Iliuk, et al., 2012; Iliuk, et al., 2011). A simplified representative illustration of pIMAGO is shown in Figure 1. pIMAGO can be primarily utilized for selective detection of phosphorylated proteins bound to solid-phase formats such as 96-well plates or PVDF membranes. Each pIMAGO molecule contains multiple functional groups, thus improving the detection efficiency to achieve the analysis of low abundant phosphoproteins in biological samples. The general protocols detailed here enable phosphoprotein detection after immobilization on a membrane (Western Blot format) or on a 96-well plate (Enzyme-Linked ImmunoSorbent Assay – ELISA format).
Figure 1.

Graphic illustration and proposed schematic structure of the pIMAGO molecule.
BASIC PROTOCOL 1
pIMAGO-based phosphoprotein detection in Western Blot format
Detection of protein phosphorylation using on-membrane format (i.e. Western Blotting) has remained the most common approach in a regular biological laboratory, typically through the use of a phospho-specific antibody. Despite this decades-long practice, there are multiple shortcomings that limit its applications. First, the availability of phospho-specific antibody requires prior knowledge of the site of phosphorylation of interest, thus limiting the analysis to only well-characterized phosphorylation events. Second, an effective phosphosite-specific antibody has to be made for every single phosphosite and phosphoprotein, making assays highly expensive. Third and finally, while much progress has been made in the field of antibody generation, development of phosphosite-specific antibodies faces the challenges of poor selectivity, inconsistent quality, and high cost. Phosphosite-specific antibodies remain extremely valuable for analysis of a specific phosphorylation event, but they are sub-optimal for discovery-based experiments, where the site is not known or it is difficult to make a high-quality antibody. Most often, the first step in molecular signaling studies is to identify whether a protein of interest demonstrates any change in phosphorylation under a specific condition, a positive identification of which can then be subsequently followed by an in-depth examination of modification sites. pIMAGO-based detection offers such initial screen of phosphorylation.
pIMAGO-based phosphoprotein detection on a membrane follows a very simple protocol, similar to a typical Western Blotting procedure (a procedural workflow is demonstrated in Figure 2A). The primary difference lies in the substitution of a primary antibody with biotin-linked pIMAGO reagent, and of a secondary antibody with an avidin-peroxidase or avidin-fluorophore conjugate for detection. A control protein sample should also be analyzed side-by-side to detect any change in phosphorylation (e.g. in vitro kinase assays with and without ATP).
Figure 2.
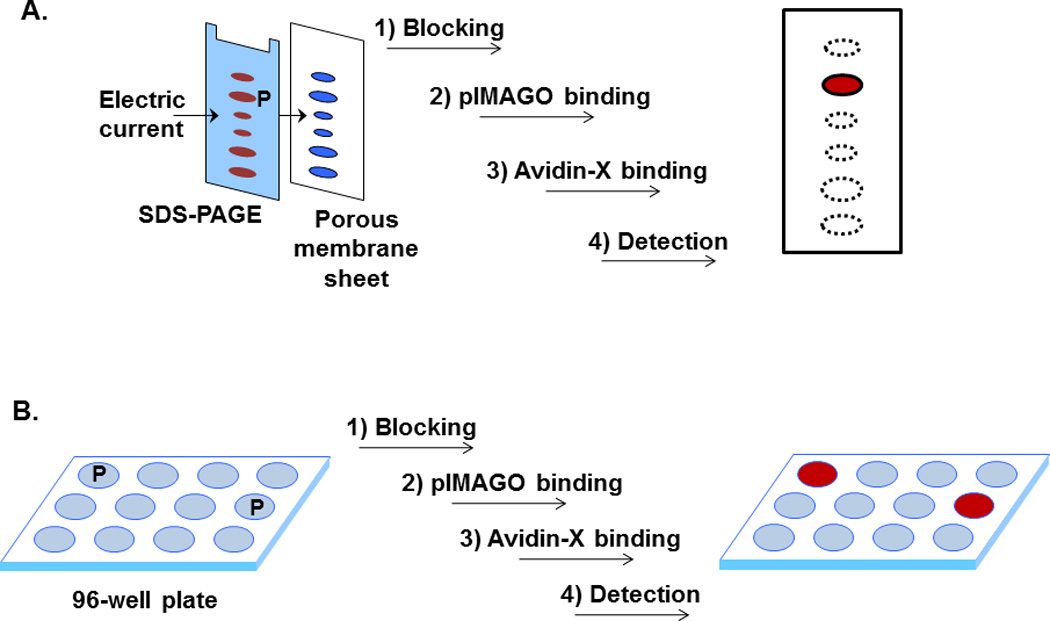
Procedural workflows of pIMAGO-based detection of phosphorylated proteins on membrane (A) or 96-well plate (B). X = horseradish peroxidase or fluorophore conjugated to avidin. P denotes phosphoprotein.
Materials
Phosphorylated protein (e.g. β-casein) in LDS/SDS sample loading buffer as a control
4× LDS/SDS sample loading buffer
200mM dithiothreitol solution (prepare fresh)
Blocking buffer for Western Blot (see recipe)
pIMAGO buffer (see recipe)
Washing buffer (see recipe)
400mM iodoacetamide solution (prepare fresh and protect from light)
pIMAGO reagent (Tymora Analytical, cat. no. PMGO)
Avidin-peroxidase conjugate, if ECL-based detection (Sigma, cat. no. A3151)
Avidin-fluorophore conjugate, if fluorescence-based detection (multiple suppliers with various fluorophores available)
1× TBST (see recipe)
Run SDS-PAGE and transfer
Before running the gel, boil the samples for 5 minutes in 1× LDS/SDS sample loading buffer supplemented with 20mM dithiothreitol, let them cool down to room temperature and add 400mM iodoacetamide solution to a 80mM final concentration directly to the samples. Incubate in the dark for 15 minutes, then load the samples onto a gel Load one well with 10–100 ng of the control phosphoprotein.
-
Run SDS-PAGE with a typical procedure following manufacturer’s instructions.
Before transfer, cut off the part of the gel with the dye front to reduce background.
-
Transfer the proteins onto a PVDF or nitrocellulose membrane using your standard procedure following manufacturer’s instructions.
Any transfer buffer may be used, but Tris-glycine transfer buffer result sin lowest background. If it is desired to do fluorescent-based detection, use a special membrane with low autofluorescence.
Important Note: In many cases, the transfer system itself might contain contaminants, increasing the nonspecific background signal. To reduce this, we strongly recommend including a second piece of PVDF membrane before the gel to bind any of these contaminants (suggested set-up: filter-membrane-gel-membrane-filter). This step is not necessary for nitrocellulose.
Detect phosphoproteins using pIMAGO
-
3
Block the membrane with the Blocking buffer for 1 hour with slight shaking (e.g. 10 mL for a mini blot; this step can also be carried out overnight at 4°C).
-
4
Prepare 1:1,000 mixture of pIMAGO reagent in pIMAGO buffer (e.g. 10 µL pIMAGO in 10 mL pIMAGO buffer for mini gel). Mix well, add to the membrane, and incubate 1 hour with slight shaking.
-
5
Wash the membrane 3 times with 10–20mL of Washing buffer and once with 1× TBST (5 min each wash).
-
6
Prepare 1:1,000 mixture of avidin-peroxidase or avidin-fluorophore in the Blocking buffer (e.g. 10 µL avidin conjugate reagent in 10 mL of Blocking buffer for mini gel). Mix and add to the membrane, incubate 1 hour with slight agitation.
-
7
Wash the membrane 3 times with 1× TBST (5 min each wash).
-
8
Detect the signal as usual using fluorescence scanner or peroxidase chemiluminescence substrate and film according to manufacturer’s instructions.
Typically, do not need to expose the film for more than 1–2 min to avoid high background. There is no need to dry the membrane for fluorescence detection.
-
9
Signal can be quantified using band density analyzing software for chemiluminescence (for example, ImageJ) or fluorescence quantitation software typically supplied with fluorescence scanner instruments.
BASIC PROTOCOL 2
pIMAGO-based phosphoprotein detection in ELISA format
As with the on-membrane detection, pIMAGO can be easily adapted for phosphoprotein detection on 96- or 384-well plates (in ELISA format). Similarly, this procedure also permits general screening to determine whether or not one sample or a set of samples are phosphorylated and the extent of phosphorylation. However, dissimilar to Western Blot, microplate-based detection can be accomplished with a much higher throughput, often enabling generation of thousands of data points in one day’s experiment. As a result, microplate-based detection using pIMAGO can be the procedure of choice during high-throughput kinase activity profiling and inhibitor screening.
pIMAGO-based phosphoprotein detection on a microplate follows a protocol similar to the standard ELISA procedure (procedural workflow is demonstrated in Figure 2B). After protein immobilization on a plate using passive adsorption, pIMAGO-biotin is used instead of primary antibody for binding to phosphoproteins, and avidin-peroxidase or avidin-linked fluorophore can then be added for signal detection and quantitation. Similarly, it is recommended to include a control sample to quantitatively measure the phosphorylation changes (e.g. in vitro kinase assays with and without ATP).
Materials
Phosphorylated protein (e.g. β-casein) as a control
Carbonate buffer for protein binding (see recipe)
Blocking buffer for microplate (see recipe)
pIMAGO buffer (see recipe)
2% Oxalic acid (protect from light)
pIMAGO reagent (Tymora Analytical, cat. no. PMGO)
Avidin-peroxidase conjugate, if colorimetry-based detection (Sigma, cat. no. A3151)
Avidin-fluorophore conjugate, if fluorescence-based detection (multiple suppliers with various fluorophores available)
1× TBST (see recipe)
Colorimetric TMB peroxidase substrate kit, if colorimetry-based detection (Bio-Rad, cat. no. 172-1066)
96-well clear High Bind polystyrene plate (Sigma, cat. no. CLS3590)
Analyze phosphoproteins on a 96-well plate using pIMAGO
-
Prepare a protein solution of your sample (phosphoprotein or substrate of interest) in the carbonate buffer. A control sample (e.g. alkaline phosphatase-treated or without ATP) should also be prepared as a reference. If protein amount is known, use 10 to 500 ng of the protein or mixture of proteins per 100 µl of carbonate buffer per well. Add 100 µl of the mixture into each well on a 96-well plate. Incubate overnight at 4°C at 400–600 rpm to bind the proteins to the plate.
As a positive control, in a separate well mix 10–100 ng of the prepared phosphoprotein in 100 µL of carbonate buffer and bind overnight.
-
Remove the solution from wells, add 150 µl of the blocking buffer into each well and incubate at room temperature for 2–3 minutes. Remove the solution and add 150 µl of the blocking buffer again and incubate at room temperature for 30 min while shaking at 400–600 rpm.
At this stage, any additional manipulations can be carried out (e.g. kinase/phosphatase assay, inhibitor screening, etc.). Make sure to wash the wells 3× with the 1× TBST after each treatment.
In a clean tube, prepare a 1:100 mixture of the pIMAGO reagent in the pIMAGO buffer (1 µl of the reagent for every 100 µl of buffer per well). Empty the wells and add 100 µl per well of the prepared pIMAGO solution. Incubate at room temperature for 1 hour at 400–600 rpm.
Empty the wells and add 150 µl of the pIMAGO buffer into each well; incubate 2–3 minutes at 400–600rpm. Remove the buffer and repeat the washing step two more times with the pIMAGO buffer for a total of 3 washes. Remove the solution and incubate the wells with 150 µl of the blocking buffer for 15 min at 400–600 rpm at room temperature.
In a clean tube, prepare 1:100 mixture of avidin-peroxidase or avidin-fluorophore in the blocking buffer (1 µl of the avidin conjugate in 100 µl of Blocking buffer per well). Empty the wells and add 100 µl per well of the prepared avidin conjugate solution in blocking buffer. Incubate the plate for 1 hour at 400–600 rpm at room temperature.
-
Empty the wells and add 150 µl of 1× TBST into each well; incubate 2–3 minutes at 400–600 rpm and remove the solution. Repeat the washing step with TBST two more times for a total of three washes. Empty the wells.
At this point, if avidin-flurophore was used, the fluorescence signal may be detected using a fluorescence scanner.
-
For colorimetry-based detection (chromogenic), use the TMB peroxidase substrate kit. Prepare 9 to 1 mixture of the Colorimetric Substrates A and B (has to be made fresh each time before detection), and add 100 µl to each well. Shake the plate until satisfied with signal – solution will turn green if signal is present (usually 1–2 min), and then add 150 µl of the 2% oxalic solution to stop the peroxidase-substrate reaction. Read the plate at 415 nm in a plate reader.
Alternatively, any other peroxidase substrate can be used (chromogenic or chemiluminescent). However, for chemiluminescence-based detection, a different plate with non-transparent walls must be used.
Signal can be quantified using absorbance analysis software for chromogenic detection or fluorescence quantitation software typically supplied with scanning instruments.
REAGENTS AND SOLUTIONS
Use Milli-Q water or equivalent in all solutions and protocol steps
Blocking buffer for Western blot
Prepare the solution containing 0.5% Bovine Serum Albumin, 0.1% Tween 20, and 0.1% PAMAM generation 4 dendrimer (Sigma, cat. no. 412449). Store at 4°C for up to 1 year.
Blocking buffer for microplate
Prepare the solution containing 1% Bovine Serum Albumin in 1× TBST (see recipe for 1× TBST). Store at 4°C for up to 1 year.
pIMAGO buffer
Prepare the solution containing 500mM glycolic acid in 1% trifluoroacetic acid. Store indefinitely at room temperature.
Washing buffer
Prepare the solution containing 50mM 2,5-dihydrobenzoic acid in 0.1% trifluoroacetic acid. Store indefinitely at room temperature in a dark bottle.
1× TBST
Prepare the solution containing 50mM Tris base and 150mM NaCl in water. Adjust the pH to 7.5 with HCl and supplement with 0.1% Tween 20. Store indefinitely at room temperature.
10× TBS (tris-buffered saline) buffer solution can be made alternatively without Tween 20; and Tween 20 can then be added later to each 1× buffer dilution.
Carbonate buffer
Prepare the solution containing 30mM sodium carbonate and 70mM sodium bicarbonate in water (pH should be ~9.2–9.6). Store at 4°C for up to 1 year.
COMMENTARY
Background Information
Titanium ion or titanium dioxide has strong affinity toward phosphorylated residues (Nawrocki, et al., 2004; Torta, et al., 2009). While such binding is mainly based on charge-charge interactions, Ti, in the presence of benzoic or α-hydroxy acids, shows remarkable specificity toward phosphate groups among other negatively-charged molecules (Jensen and Larsen, 2007; Larsen, et al., 2005). In our lab, we have previously demonstrated the highly effective utility of titanium-based enrichment of phosphopeptides for MS analysis under homogeneous conditions (PolyMAC technology; Iliuk, et al., 2010). For this application, we utilized a soluble polyamidoamine synthetic nanopolymer (e.g. dendrimer) whose hyperbranched surface can be functionalized with desired chemical groups (i.e. titanium for phosphate binding). Besides high solubility, the advantages of using dendrimers include high structural and chemical homogeneity, compact spherical shape, and controlled surface functionalities (Boas and Heegaard, 2004). The homogeneous and hyper-branched nature of Ti-functionalized nanopolymer further enhances specificity toward phosphorylated molecules.
Following the initial MS-based applications, we have expanded the concept of titanium-functionalized nanopolymer to the development of a universal phosphoprotein detection approach, which we termed pIMAGO. Each dendrimer molecule was synthesized to contain titanium metal ions for highly specific binding to phosphorylated proteins, and biotin groups for subsequent signal detection. After incubation of pIMAGO with proteins immobilized on a membrane or a plate, the phosphoprotein-bound reagent can be detected with horseradish peroxidase (HRP) conjugated to avidin. Under the strong acidic condition provided by the pIMAGO buffer, the reagent is capable of binding only to phosphorylated groups without any bias toward the amino acid sequence or different phospho-sites. Moreover, because each nanopolymer is functionalized with multiple biotin groups, the signal from each binding event is amplified, thus allowing for the detection of low abundant phosphoproteins. Using this method, the phosphorylation levels of proteins of interest under physiological conditions can be readily detected in the format of a standard Western blot or ELISA-like procedure without the need for radioactive ATP or phosphosite-specific antibodies.
Critical Parameters
Choice of detection method
Chemiluminescent and colorimetric substrates are most common and convenient methods for Western Blot and ELISA, respectively. While the protocols described here are for peroxidase-based detection using chemiluminescence for membrane and chromogenic substrate for microplate-based analysis, the signal detection is not limited to these. Any detection method can be used as long as the signaling molecule is conjugated to avidin or streptavidin. These include peroxidase, alkaline phosphatase, dyes or fluorophores across the whole wavelength range. In fact, for more accurate signal quantitation, we recommend using a fluorophore conjugate avidin reagent, which would provide more consistent signal that is not dependent on enzyme activity or substrate depletion. In all of these cases, the same described protocols can be applied with the modification on the signal detection step.
Selection of binding platform
For on-membrane phosphoprotein detection, it is necessary to ensure that a high-quality PVDF membrane is used in order to avoid potentially high background. If fluorescence-based detection is desirable, a specialty membrane with low autofluorescence should be selected. Similar properties should be kept in mind for on-plate detection as well, where a low-autofluorescence plate should be used for fluorophore-based detection. In addition, if chemiluminescence substrate will be used for on-plate detection to improve sensitivity, a black non-translucent plate needs to be used to avoid signal crossover between wells.
Troubleshooting
Anticipated Results
Basic Protocols 1 and 2 are designed to be simple and routine procedures for detection of general protein phosphorylation. We expect that any phosphoprotein sample of over 1ng will be easily detected by either avidin-conjugated detection method. However, because pIMAGO binds to any phosphosite, one cannot differentiate which sites on a protein are phosphorylated. In addition, even after a stimulus, change in phosphorylation may not be observed if it affects only one minor site but not the overall phosphorylation level of a protein of interest. Therefore, this approach should be used as a screening method, and combined with other approaches (e.g., mass spectrometry) for in-depth analysis of targeted phosphoproteins.
Time Consideration
After protein immobilization onto a membrane (protein transfer) or onto a microplate, the whole pIMAGO detection procedure will take less than 4 hours. If the procedure cannot be completed on the same day, a membrane or a microplate may be left to block overnight at 4°C prior to pIMAGO binding step.
Table 1.
Troubleshooting for pIMAGO-based detection
| Problem | Possible Cause | Solution |
|---|---|---|
| High background on membrane | Contaminants in the PAGE system | Include a second piece of PVDF membrane in front of the gel during transfer |
| Too much signal/background | Excess of avidin conjugate | Reduce the amount of avidin conjugate (e.g. peroxidase) used. |
| Too much signal/background on membrane | Exposure time is too long | Reduce the exposure time during detection to 30 seconds. |
| Potential false positives | Control is lacking | Include a negative control of your samples by either excluding ATP during kinase reaction or dephosphorylating your sample with a general phosphatase |
| No detectable signal | Procedural problems or no phosphoprotein present | Include a known phosphoprotein as a positive control (e.g. casein) to check if there are any procedural problems. |
Acknowledgement
This project has been funded in part by an NSF CAREER award CHE-0645020, National Institutes of Health grants 1R01GM088317 and R43EB15809-1, and NSF grant IIP-1256600.
Literature Cited
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Boas U, Heegaard PM. Dendrimers in drug research. Chem Soc Rev. 2004;33:43–63. doi: 10.1039/b309043b. [DOI] [PubMed] [Google Scholar]
- Hunter T. Signaling--2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Iliuk A, Liu XS, Xue L, Liu X, Tao WA. Chemical visualization of phosphoproteomes on membrane. Mol Cell Proteomics. 2012;11:629–639. doi: 10.1074/mcp.O112.018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliuk A, Martinez JS, Hall MC, Tao WA. Phosphorylation assay based on multifunctionalized soluble nanopolymer. Anal Chem. 2011;83:2767–2774. doi: 10.1021/ac2000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliuk AB, Martin VA, Alicie BM, Geahlen RL, Tao WA. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol Cell Proteomics. 2010;9:2162–2172. doi: 10.1074/mcp.M110.000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen SS, Larsen MR. Evaluation of the impact of some experimental procedures on different phosphopeptide enrichment techniques. Rapid Commun Mass Spectrom. 2007;21:3635–3645. doi: 10.1002/rcm.3254. [DOI] [PubMed] [Google Scholar]
- Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics. 2005;4:873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- Nawrocki J, Dunlap C, McCormick A, Carr PW. Part I. Chromatography using ultra-stable metal oxide-based stationary phases for HPLC. Journal of chromatography. A. 2004;1028:1–30. doi: 10.1016/j.chroma.2003.11.052. [DOI] [PubMed] [Google Scholar]
- Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- Torta F, Fusi M, Casari CS, Bottani CE, Bachi A. Titanium dioxide coated MALDI plate for on target analysis of phosphopeptides. J Proteome Res. 2009;8:1932–1942. doi: 10.1021/pr8008836. [DOI] [PubMed] [Google Scholar]