

Abstract
DNMT1, a key enzyme mediating DNA methylation, is known to be elevated in various cancers, including the mouse lung tumors induced by the tobacco-specific carcinogen NNK. However, it is not known whether DNMT1 expression is induced right after NNK treatment and how DNMT1 expression varies throughout lung tumorigenesis. In the present study, we found that administration of NNK to A/J mice caused elevation of DNMT1 in bronchial epithelial cells at days 1, 3 and 14 after NNK treatment. DNMT1 elevation at day 1 was accompanied by an increase in γ-H2AX and p-AKT. At weeks 5 to 20, NNK-induced DNMT1 in lung tissues was in lower levels than the early stages, but was highly elevated in lung tumors at week 20. In addition, the early induction of p-AKT and γ-H2AX as well as cleaved caspase-3 in NNK-treated lung tissues was not detected at weeks 5 to 20, but was elevated in lung tumors. In concordance with DNMT1 elevation, promoter hypermethylation of tumor suppressor genes Cdh13, Prdm2 and Runx3 was observed in lung tissues at day 3 and in lung tumors. Treatment by EGCG attenuated DNMT1, p-AKT and γ-H2AX inductions at days 1 and 3 and inhibited lung tumorigenesis.
Introduction
Lung cancer is the leading cause of cancer mortality in the United States and worldwide (1). The majority of lung cancer are caused by tobacco smoking and second-hand smoke exposure (2). Lung cancer development in both humans and rodents is known to be associated with the activation of oncogenes and inactivation of tumor suppressor genes (TSGs) (3, 4). An important mechanism for inactivating TSGs is gene silencing by aberrant hypermethylation of CpG islands in the promoter region (5, 6). DNA methyltransferase 1 (DNMT1) is a key enzyme mediating DNA methylation. The activation of DNMT1 has been reported in numerous cancers, including lung cancer (7), liver cancer (8), gastric cancer (9), breast cancer (10), prostate cancer (11), and retinoblastoma (12).
Experimentally, lung tumors can be readily induced in mice, rats and hamsters by the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a nicotine-derived nitrosamine ketone (13, 14). Mice treated with NNK developed pulmonary hyperplasia after 6–14 weeks, which progressed morphologically to adenoma at 16–20 weeks and adenocarcinoma after 30 weeks (15–17). In addition to gene mutation, an increase in DNMT1 expression and hypermethylation of multiple TSGs, such as cyclin-dependent kinase inhibitor 2A (p16INK4a), death-associated-protein kinase 1 (Dapk1), retinoic acid receptor β (Rarb), and runt-related transcription factor 3 (Runx3), have been observed in NNK-exposed rodent lung tumors (6, 18, 19). In human lung tumors, many TSGs such as O6-methylguanine-DNA methyltransferase (MGMT), cadherin-13 (CDH13), paired box protein Pax-5 (PAX5) and Ras associated domain-containing protein 1 (RASSF1) as well as p16INK4a, DAPK1, RARB, and RUNX3 have been shown to be hypermethylated, especially in samples from smokers (20, 21). In lung cancer samples, DNMT1 is found to be highly expressed and has a strong binding capacity to hypermethylated promoter regions of TSGs; this overexpression and region-specific binding are shown to be related to tobacco smoking (22). However, most of these human and animal studies have been carried out on tumor samples. Whether DNMT1 overexpression and epigenetic changes occur at the initiation stage of lung carcinogenesis remains to be uncovered.
(−)-Epigallocatechin-3-gallate (EGCG), the most abundant and active polyphenol in green tea, has been known to have cancer preventive activities, including inhibition of NNK-induced lung tumorigenesis (23). EGCG has also been reported to inhibit DNMT1 activity and reactivate TSGs in esophageal, colon, prostate, mammary, breast and skin cancer cells (24–26).
The present study investigated changes in DNMT1 expression and related molecular events in lung tissues following NNK administration and during the progression of lung tumorigenesis in A/J mice. Our results demonstrate the elevation of DNMT1 and activation of AKT in bronchial epithelia as a possible stress response after NNK treatment as well as the inhibitory effects of EGCG on these changes.
Materials and methods
Animal studies
Animal experimentation was conducted in accordance to the ethical federal guidelines and institutionally approved protocol. The protocol was approved by the Institutional Animal Care and Use Committee at Rutgers, the State University of New Jersey (Protocol No. 91-024). Female A/J mice (4-week old) were purchased from the Jackson Laboratory (Bar Harbor, ME). NNK was from Chemsyn Science Laboratories (Lenexa, KS). EGCG (94% pure) was a gift from Dr. Yukihiko Hara (Mitsui Norin Co., Ltd, Shizuoka, Japan). The animals were fed the AIN-93M diet and maintained at room temperature (20 ± 2 °C) with a relative humidity of 50 ± 10% and with an alternating 12 h light/dark cycle throughout the duration of the study. In the short-term experiment, after one week of acclimation, the mice were administered a single dose of NNK (100 mg/kg body weight, i.p.) or saline on day 0, and were sacrificed at days 1, 3, 7 and 14 after NNK administration by CO2 asphyxiation. For the EGCG-treated group, mice were given EGCG (0.5% in diet) starting at one week before NNK administration and ad libitum after until sacrificed. The lungs were removed, inflated and fixed in 10% buffered formalin. To study the progression of NNK-induced lung tumorigenesis, the mice were administered two doses of NNK at 1 week apart (100 and 75 mg/kg body weight, i.p.) or saline, and sacrificed at weeks 5, 10, 15 and 20 after NNK administration. The mice in EGCG treatment group received EGCG (0.5% in diet) starting at one week before the first NNK administration and ad libitum after until sacrificed. Visible tumors (> 0.1 mm in diameter) in the lungs were counted and the sizes were measured under a dissecting microscope. The tumors were fixed in 10% buffered formalin, frozen in −80°C directly, or stored in RNAlater solution (Qiagen, Valencia, CA). Tumor volumes (mm3) were calculated using the formula , where r is the radius of the tumor. Liver and spleen were also removed, weighed and examined for abnormalities and then fixed in 10% buffered formalin.
Immunohistochemistry (IHC)
Deparaffinized sections were unmasked in antigen-unmasking solution (DAKO, Copenhagen, Denmark) in a microwave oven for antigen retrieval. Endogenous peroxidase was quenched with 3% H2O2. Non-specific bindings were blocked by 3% serum. The slides were stained with the primary antibody overnight at 4°C, biotin-conjugated secondary antibody (1:200) for 45 min at room temperature, and avidin-biotin peroxidase complex (1:100) for 45 min at room temperature. Antibodies against DNMT1 (1:200; Abcam Inc, Cambridge, MA), phosphor-AKT (p-AKT) (1:200; Cell Signaling Technology, Danvers, MA), phospho-histone H2AX (γ-H2AX) (1:200; Cell Signaling Technology) and O6-methylguanine (O6-mG) (1:200; Cell Signaling Technology) were used for the analyses. Negative controls were processed in the absence of the primary antibody. For each slide, 5 representative ×200 power photomicrographs were taken and these images were quantified for positive-stained bronchial epithelial cells using the ImagePro Plus Image Processing System (Media Cybernetics, Silver Spring, MD).
Western blot analysis
Lung tissue and tumor lysates were prepared using Omni Bead Ruptor 24 (Omni International, Kennesaw, GA) in T-Per tissue extraction buffer (Pierce, Rockford, IL) with protease and phosphatase inhibitors (Sigma, St. Louis, MO). Protein concentration was determined by BCA reagent kit (Thermo Scientific, Rockford, IL). Equal amount of samples were used for Western blot analysis to determine the levels of DNMT1, p-AKT, AKT, γ-H2AX, O6-mG, and cleaved caspase-3 using antibodies against DNMT1 (Abcam), p-AKT (Cell Signaling Technology), AKT (Cell Signaling Technology), γ-H2AX (Cell Signaling Technology), O6-mG (Cell Signaling Technology), and cleaved caspase-3 (Cell Signaling Technology). Sample loadings were also monitored by β-actin levels using anti-β-actin antibody (Sigma). Western blots were carried out with the fluorescent secondary antibodies (Li-Cor Biotechnologies, Lincoln, NE) and the results were collected by Odyssey® Infrared Imaging System (Li-Cor Biotechnologies). Western blot results were quantified by the intensities of the fluorescent bands detected.
DNA methylation analysis
Genomic DNA was extracted from lung tumors and its matching normal lung tissue from 2 mice treated with NNK using AllPrep DNA/RNA Mini Kit (Qiagen). Since the tumors were small, ~10 tumors from each mouse were combined as one sample for DNA extraction. These DNA samples (2 tumors and 2 matching controls) were used for the methylation-sensitive, methylation-dependent and double restriction digestions using EpiTect Methyl DNA Restriction Kit (Qiagen) according to manufacturer’s protocol. The digested products were then used for quantitative PCR (qPCR) to analyze the DNA methylation of 24 mouse lung cancer signature genes in the Mouse Lung Cancer DNA Methylation PCR Array (Qiagen). The real-time PCR was run on ABI 7900 HT Fast Real Time PCR system (Life Technologies, Beverly, MA) according to the manufacturer recommended protocol using POWER SYBR Green PCR Master Mix (Life Technologies). The methylation status of the 24 signature genes was determined by analyzing the qPCR result with Data Analysis templates available on Qiagen/SABioscience website.
To quantify the level of DNA methylation in our lung tissues and tumors, a modified methylation-sensitive, restriction enzyme-based quantitative PCR (MSRE-qPCR) method was used (27, 28). In brief, genomic DNA was extracted from tumor or lung samples and subjected to digestion by methylation-sensitive HpaII and methylation-insensitive Isoschizomer MspI (New England Biolabs, Beverly, MA). Typically, 0.125 μg of DNA was incubated with 10 units of restriction enzyme in a 30 μl final reaction volume for 6 h according to manufacturer’s suggested conditions. Real-time PCR reactions were carried out in the ABI 7900 system using POWER SYBR Green PCR Master Mix and specific primer set that brackets 2–4 HpaII/MspI recognition sites for each gene [Cdh13: 5′-AGGCGACTCCTAGGGATTGT-3′/5′-GGGCGAAGAGAGGACGAG-3′ and 5′-CCTCTCTTCGCCCAGCTC-3′/5′-TGCTCAGGCCCCTTTCAG-3′; Prdm2: 5′-CTTCCCGTCTCCCTCCTCT-3′/5′-GGCTGTTTGCGAACTTGTG-3′ and 5′-GTTCGGAGGAAGCTCGAAG-3′/5′-GGAATGTCCCTTGGTGTCAG-3′; Runx3: 5′-CTACGCTGCAGAGCCTCAC-3′/5′-GGAGGTAGGTGTGGTGGAAG-3′ and 5′-GCTTCCACCACACCTACCTC-3′/5′-GCCATGGAGAACTGGTAGGA-3′]. PCR reaction was performed at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 65°C for 60 sec. Methylation of a given site is determined from the change in Ct value by using the basic principle that each successive round of PCR amplification results in approximately 2-fold increase in the amount of product (ΔCt of 1.0 indicates the 50% of the template has been cleaved, 2.0 equals 75% cleavage, etc.) Thus, the relationship of ΔCt to percent methylation can then be described using the formula , where ΔCt = (CtHpaII − CtMspI). Methylation fold-change is calculated by normalizing the percent methylation to adjacent lung or day 0 saline control.
Statistical analysis
All data was presented as means ± SD. Student’s t-test was used to determine the difference between two groups. Statistical significance was indicated by * (P < 0.05) and † (P < 0.01) in one-tailed or two-tailed comparison.
Results
Induction of DNMT1 and pAKT in the lung by NNK treatment
To study the early effect of NNK on DNMT1, 6-week old female A/J mice were treated with one dose of NNK. Mice were sacrificed at 1, 3 and 14 days after the NNK treatment, and DNMT1 expression was determined by IHC (Figure 1A–D). While lung tissue was histologically normal, DNMT1 protein level was found to be significantly elevated in bronchial epithelial cells at days 1, 3 and 14 after NNK treatment. The percentage of cells with positive nuclear staining for DNMT1 increased to 77.9%, 88.7% and 71.3%, respectively, from 4.4% of the saline control group (P < 0.01). The elevations of DNMT1 at days 1, 3, and 14 were significantly attenuated by dietary EGCG treatment (by 70.0%, 54.5% and 25.0%, respectively, P < 0.05) (Figure 1C and D). Elevated levels of DNMT1 in lung tissues at days 1, 3, 7 and 14 were verified by Western blot analysis, showing increases by 11-, 14-, 9- and 7-fold, respectively, compared to the saline control group (P < 0.05) (Figure 1I and J).
Fig. 1.

NNK-induced elevation of DNMT1 and p-AKT in lung tissues of NNK-treated mice and inhibitory effect of EGCG. (A–C) Representative IHC microphotographs for DNMT1 staining in lung tissues from day 0 saline control group (A), day 1 NNK-treated group (B) and day 1 NNK + 0.5% EGCG group (C). (D) Quantification of DNMT1-positive cells in the bronchial epithelial cells of NNK-treated and NNK + EGCG-treated mice at days 1, 3 and 14. (E–G) Representative IHC microphotographs for p-AKT staining in lung tissues from day 0 saline control group (E), day 1 NNK-treated group (F) and day 1 NNK + 0.5% EGCG group (G). (H) Quantification of p-AKT staining intensity in the bronchial epithelial cells of NNK-treated and NNK + EGCG-treated mice at days 1, 3 and 14. (I) Representative Western Blot of protein levels of DNMT1, p-AKT and AKT in lung tissues of saline- or NNK-treated mice at days 1, 3, 7 and 14 after NNK treatment. (J) Quantification of Western blot results. (K) mRNA level of DNMT1 in lung tissues of lung tissues of saline- or NNK-treated mice at days 1 and 3 after NNK treatment by qPCR. (L) Quantification of qPCR results. Values shown in the bar graphs are means ± SD (n = 25 (5 slides × 5 fields) for IHC, n = 3 for Western blot and n=4 for qPCR); * (P < 0.05) and † (P < 0.01) indicate statistical significance by student’s t-test for NNK + 0.5% EGCG treatment group as compared with same day NNK treatment group or NNK treatment groups at days 1, 3 and 14. Scale bar represents 50μm.
The phosphorylation of AKT has been reported to stabilize DNMT1 at the protein level by inhibiting the ubiquitination of DNMT1 (29). Since increased p-AKT has been previously observed in both NNK-treated lung cancer cells and NNK-induced mouse lung tumors (7), we reasoned that the DNMT1 elevation detected at such the earlier stage was also the result of AKT activation. Indeed, we found that the p-AKT level in the bronchial epithelial cells of NNK-treated mice were significantly higher at days 1 and 3 (Figure 1E, F and H); the percentage of the total area that stained for p-AKT increased to 67.5% and 44.8%, respectively, from 11.4% (saline controls) (P < 0.05) (Figure 1H). The level of p-AKT also appeared to be induced by NNK at day 14, but the change was not statistically significant. The NNK-induced p-AKT at days 1 and 3 were markedly reduced by EGCG (by 71.9% and 49.8%, respectively, P < 0.01) (Figure 1G and H). The induction p-AKT in lung tissues at days 1, 3 and 14 was verified by Western blot analysis showing increases by 6-, 8- and 4-fold, respectively (P < 0.05) (Figure 1I and J); whereas, the total AKT protein level was not changed by NNK treatment.
To determine whether the elevated level of DNMT1 was resulted from an increase in the transcription of DNMT1, we determined the mRNA levels of DNMT1 by real-time qPCR. No significant change on DNMT1 mRNA level was found at days 0, 1 and 3 (Figure 1K and H). This result suggests that the up-regulation of DNMT1 is not at the transcription level, but likely through DNMT1 protein stabilization mediated by p-AKT.
Induction of γ-H2AX, O6-mG and cleaved caspase-3 formation in the lung by NNK treatment
To assess the DNA damage induced by NNK, we determined the formation of γ-H2AX foci, an early event of DNA double-strand breakage and repair, by IHC (Figure 2A–D). NNK treatment was found to markedly induce γ-H2AX foci formation in bronchial epithelial cells with the percentage of cells with positive nuclear staining increased to 79.3%, 42.8% and 12.3% on days 1, 3 and 14, respectively, from 0% (saline control) (P < 0.01). The induction of γ-H2AX foci was significantly inhibited by EGCG at days 1, 3 and 14, showing reductions by 91.3%, 68.5% and 74.0%, respectively (P < 0.01) (Figure 2C and D). The IHC result was found to be consistent with the Western blot analysis (Figure 2I and J), showing a 4-fold increase of γ-H2AX protein level in lung tissues at days 1 and 3, compared to the saline control group (P < 0.05).
Fig. 2.
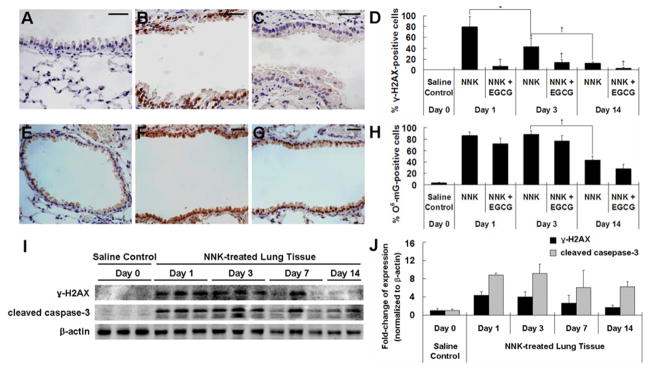
NNK-induced elevation of γ-H2AX and O6-mG in lung tissues of NNK-treated mice and inhibitory effect of EGCG. (A–C) Representative IHC microphotographs for γ-H2AX staining in lung tissues from day 0 saline control group (A), day 1 NNK-treated group (B) and day 1 NNK + 0.5% EGCG group (C). (D) Quantification of γ-H2AX-positive cells in bronchial epithelial cells of NNK-treated and NNK + EGCG-treated mice at days 1, 3 and 14. (E–G) Representative IHC microphotographs for O6-mG staining in lung tissues from day 0 saline control group (E), day 1 NNK-treated group (F), and day 1 NNK + 0.5% EGCG group (G). (H) Quantification of O6-mG-positive cells in bronchial epithelial cells of NNK-treated and NNK + EGCG-treated mice at days 1, 3 and 14. (I) Representative Western Blot of protein levels of γ-H2AX and cleaved caspase-3 in lung tissues of saline- or NNK-treated mice at days 1, 3, 7 and 14 after NNK treatment. (J) Quantification of Western blot results. Values shown in the bar graphs are means ± SD (n = 25 (5 slides × 5 fields) for IHC and n = 3 for Western blot); * (P < 0.05) and † (P < 0.01) indicate statistical significance by student’s t-test for NNK + 0.5% EGCG treatment group as compared with same day NNK treatment group or NNK treatment groups at days 1, 3 and 14. Scale bar represents 50μm.
O6-mG, generated by carbocation formed through the metabolic activation of NNK, has been shown to be critical in the initiation of lung carcinogenesis by NNK (30). Measured by IHC, a higher level of O6-mG was detected in bronchial epithelial cells at days 1, 3 and 14 after NNK treatment. The percentage of cells with positive nuclear staining for O6-mG increased to 86.4%, 88.3% and 43.3%, respectively, from 3.5% (saline controls) (P < 0.01) (Figure 2E, F and H). In mice fed with EGCG, the NNK-induced O6-mG formation was slightly attenuated by EGCG, but not statistically significant (Figure 2G and H). This result suggests that EGCG did not affect the metabolic activation of NNK, and the inhibitory effects of EGCG on DNMT1 and p-AKT up-regulations are due to other mechanism(s).
To assess the possible induction of apoptosis by NNK, we determined the level of cleaved caspase-3 in NNK-treated lung tissues and found cleaved caspase-3 was elevated by 9-fold at days 1 and 3 (P < 0.01), and 6-fold at days 7 (P < 0.01) and 14 (P = 0.08) (Figure 2I and J). This result showed that the NNK treatment induces cell death, possibly due to NNK-induced oxidative stress.
DNMT1 protein levels during the progression of lung tumorigenesis and the inhibitory effects of EGCG
An up-regulation of DNMT1 has been reported in NNK-treated lung cancer cells and NNK-induced mouse lung tumors previously (7). Our results (Figure 1) demonstrated the elevation of DNMT1 in the first two weeks after NNK treatment. However, the levels of DNMT1 during the progression of lung tumorigenesis were not known. In order to determine whether the NNK-induced changes in DNMT1, p-AKT and other cellular events persist over time, we attained and analyzed lung tissue samples from mice at 5, 10, 15 and 20 weeks after NNK treatment.
Lung tumor was observed starting from week 10 after NNK treatment, with a tumor incidence of 12.5% (1/8) (Table 1). The incidence of lung tumor increased to 100% at week 15 (8/8) and week 20 (30/30), whereas no tumor was found in the control mice that received saline. The tumors were diagnosed as lung adenomas based on our previous criteria (15). Lung adenomas observed were in a solid, papillary or mixed growth pattern and generally composed of well-differentiated cells. The tumor multiplicity (number of tumors/mouse) was 0.25 at week 10, but increased to 12.0 ± 0.76 and 22.0 ± 1.47 at weeks 15 and 20, respectively. Similarly, tumor size also increased as time progressed (Table 1). Treatment with 0.5% EGCG in the diet starting one week before the first dose of NNK treatment until the end of the experiment significantly reduced tumor multiplicity and tumor volume by 19% and 40%, respectively (P < 0.05), but the tumor morphology was not affected.
Table 1.
The inhibitory effect of EGCG on NNK-induced lung tumorigenesis*
| Group # | Treatment | Duration (weeks) | Mice (n) | Incidence | Tumors/mouse | Volume (mm3/mouse) |
|---|---|---|---|---|---|---|
| 1 | Saline | 5, 10, 15, 20 | 16 | 0/16 | 0 | 0 |
| 2 | NNK | 5 | 8 | 0/8 | 0 | 0 |
| 3 | NNK | 10 | 8 | 1/8 | 0.25 | 0.21 |
| 4 | NNK | 15 | 8 | 8/8 | 12.0 ± 0.76 | 2.27 ± 0.31 |
| 5 | NNK | 20 | 30 | 30/30 | 22.0 ± 1.47 | 4.72 ± 0.37 |
| 6 | NNK + 0.5% EGCG | 20 | 30 | 30/30 | 17.9 ± 0.92† | 2.84 ± 0.28† |
One week before the first NNK injection, mice were treated with 0.5% EGCG in diet and sacrificed at different time points. Under a dissecting microscope, tumors > 0.1 mm were scored. Tumor volumes (mm3) were measured using the formula , where r is the radius of the tumor determined by the mean values of the longest and shortest diameters.
Indicating significant difference as compared with 20 weeks NNK treatment group using student’s t-test.
DNMT1 was found to be elevated in the bronchial epithelial cells at weeks 5 to 20, but the level was lower than those observed in the first two weeks after NNK treatment (Figure 3A–E). The percentages of cells with positive nuclear staining for DNMT1 were 20–30% during weeks 5 to 20, which is markedly higher than the 1.9% found in the saline controls at the same time points (Figure 3G). Interestingly, EGCG treatment starting one week before the first dose of NNK treatment significantly decreased the DNMT1 protein level in bronchial epithelial cells at week 20 after NNK treatment (Figure 3F). The percentage of DNMT1-positive cells at week 20 was significantly reduced in the EGCG-treated group (Figure 3G). Western blot analysis confirmed that the levels of DNMT1 in NNK treated groups at weeks 5 to 20 were much lower than the level on day 3, but still remained 3- to 7-fold higher than the saline control group (P < 0.05) (Figure 3H and I). The induction of p-AKT, γ-H2AX and cleaved caspase-3 in the lung tissues at weeks 5 to 20 dropped markedly from their day 3 level, with some reduced to the level of the control group as determined by Western blots (Figure 3H and I).
Fig. 3.

Increased DNMT1 expression during the progression of NNK-induced lung tumorigenesis and inhibitory effect of EGCG. (A–F) Representative IHC microphotographs for DNMT1 staining in lung tissues from saline control group at week 5 (A) and NNK-treated groups at weeks 5 (B), 10 (C), 15 (D) and 20 (E), and NNK + 0.5% EGCG group at week 20 (F). (G) Quantification of the percentage of DNMT1-positive cells in bronchial epithelial cells. (H) Representative Western Blot of protein levels of DNMT1, p-AKT, γ-H2AX and cleaved caspase-3 in lung tissues of saline- or NNK-treated mice at day 3, weeks 5, 10, 15 and 20, or NNK-induced lung tumors at week 20. (I) Quantification of Western blot results. Values shown in the bar graphs are means ± SD (n = 25 (5 slides × 5 fields) for IHC, n = 2 for Western blot); † (P < 0.01) indicate statistical significance by student’s t-test for NNK + 0.5% EGCG treatment group as compared to the same day NNK treatment group. Scale bar represents 50μm.
We further examined these changes in NNK-induced lung tumors and determined the level of DNMT1 protein to be much higher than the lung tissues in the control group (by 16-fold, P < 0.01). In fact, DNMT1 elevation in tumors was equivalent to the lung tissues at day 3 after NNK treatment (Figure 3H and I). The levels of p-AKT, γ-H2AX and cleaved caspase-3 were also significantly higher in NNK-induced lung tumors (6- to 7-fold higher than the lung tissues of the control group, P < 0.01) and similar to the levels found in day 3 samples.
Together, the DNMT1 at week 5 and 20 were lower than those detected at days 1, 3 and 14, suggesting the possibility that at these time points, the slight DNMT elevation may represent the residue of the up-regulation induced by NNK at the earlier stage. However, given that doses of NNK administered for early study differs from the progression study, further investigation is need to support the possible connection in the upregulated DNMT1 expression.
Increase of gene-specific promoter hypermethylation in lung tissues and lung tumors of NNK-treated mice
Promoter hypermethylation of several genes in NNK-induced lung tumors has been previously described (6, 31, 32). In this study, we investigated whether similar events occurred in our NNK-induced tumors harvested at week 20 as well as in the first two weeks after NNK treatment. To determine the methylation status, the Mouse Lung Cancer DNA Methylation PCR Array was used to measure 24 different TSGs whose promoter CpG islands hypermethylation has been reported in lung tumors. The promoter methylation of Cdh13, Rassf1 and PR domain zinc finger protein 2 (Prdm2) was increased (by 200%, 200% and 150%, respectively, P < 0.05) in the lung tumor samples compared to the adjacent normal lung tissues (Figure 4A). No hypermethylation was detected in the DNA of saline-treated controls (data not shown). Further investigation using MSRE-qPCR confirmed the hypermethylation of Cdh13 and Prdm2 promoters (Figure 4B, Rassf1 was not investigated). In addition, a significant increase in promoter methylation of Runx3 was also detected in lung tumors. While promoter hypermethylation of Cdh13, Rassf1 and Runx3 has already been reported in literature (31), Prdm2 hypermethylation is new in NNK-induced lung carcinogenesis.
Fig. 4.

Increased gene-specific promoter hypermethylation in NNK-induced lung tumors and lung tissues of NNK-treated mice. (A and B) The fold-change of methylation of promoter regions of TSGs in NNK-induced lung tumors and the adjacent lung tissues by microarray (Cdh13, Prdm2 and Rassf1) (A) and qPCR (Cdh13, Prdm2 and Runx3) (B). (C) Fold-change of methylation of promoter regions of Cdh13, Prdm2 and Runx3 in lung tissues treated with saline or NNK at days 1, 3 and 14 after NNK treatment. Values shown in the bar graphs are means ± SD (n = 4, 2 mice × 2 repeat). * (P < 0.05) and † (P < 0.01) indicate statistical significance by student’s t-test for NNK-induced lung tumors as compared with the adjacent lung tissues or day 3 NNK treatment group as compared with day 0 saline control group.
To determine whether hypermethylation of genes occurred in the first two weeks after NNK treatment, samples from days 1, 3 and 14 were analyzed using MSRE-qPCR. The results indicated there was a slight, but non-significant change in promoter methylation of Cdh13, Prdm2 and Runx3 at day 1 after NNK treatment, compared to the saline control (Figure 4C). However, a significant increase in methylation was observed for Cdh13 (6.4-fold), Prdm2 (7.9-fold) and Runx3 (3.6-fold) on day 3. On day 14, the level of methylation has decreased approximately to the baseline level of the saline control. Similar results were observed in a repeated experiment, and the results indicate that gene hypermethylation is associated with the up-regulation of DNMT1 at day 3.
Discussion
In the present study, we demonstrated the elevation of DNMT1 in the bronchial epithelial cells of A/J mice at 1, 3 and 14 days following NNK administration. To our knowledge, this is the first demonstration of DNMT1 up-regulation by NNK in the early stage after administration and may be related to NNK-induced oxidative stress and activation of AKT. While the elevated level of DNMT1 is lower at weeks 5 to 20 than the early stage, the level was still significantly higher than the saline control group. In addition, the levels of both DNMT1 and p-AKT were highly elevated in the lung tumors indicating similarities with the early stage events.
The importance of DNMT1 in cancer has been demonstrated in many recent publications (33–35). Aberrant promoter hypermethylation of TSGs occurs frequently in different types of cancers in humans and animals. Increased levels of DNMT1 and hypermethylation of multiple TSGs have been reported in NNK-induced mouse lung tumors (6, 31, 32). Consistent with previous reports, our study detected the elevation of DNMT1 and gene promoter hypermethylation of several TSGs (i.e., Cdh13, Rassf1 and Runx3) in NNK-induced lung tumors. In addition, we made the new finding that Prdm2 is also hypermethylated in the lung tumors. Surprisingly, we observed the hypermethylation of Cdh13, Prdm2 and Runx3 as early as day 3 after NNK treatment, correlating with the elevation of DNMT1 and the high level of bronchial epithelial cell proliferation reported previously by our lab (15). It would be interesting to determine whether the gene hypermethylations persist during the progression of lung tumorigenesis. However, the difference in NNK dosage in the early stage study with the progression study limited our effort and ability to substantiate the possible connection with further animal experimentation. Overall, the close association between DNMT1 elevation, gene hypermethylations and lung cancer seems to indicate that the early stage up-regulation of DNMT1 may play an important functional role in NNK-induced lung carcinogenesis.
Our result that DNMT1 mRNA levels were not significantly affected at days 1 and 3 indicates that the elevation of the DNMT1 protein was not due to increased transcription. We hypothesize that NNK treatment induces the phosphorylation of AKT to form p-AKT, which mediates DNMT1 protein stabilization akin to recent reports in cell lines (7, 29). Our result on the increase of p-AKT in bronchial epithelial cells at 1 and 3 days is consistent with the reported PI3K/AKT pathway activation in in vitro lung cancer and airway epithelial cell line studies (36, 37) as well as in normal and premalignant bronchial epithelial cells of smokers with airway lesions (38). Another possible mechanism of NNK-induced DNMT1 protein stabilization is through disrupting APC-mediated DNMT1 degradation as demonstrated in breast and colon cancer cell line studies (39).
Time- and dose-dependent induction of γ-H2AX by tobacco smoke condensate have been previously shown in A549 lung cancer cells and normal human bronchial epithelial cells (40). γ-H2AX foci formation is an early molecular event in the repair of DNA breaks and is commonly used as marker for oxidative DNA damage. Our results on the similar induction of γ-H2AX and p-AKT in bronchial epithelial cells at days 1, 3 and 14 suggest that AKT is phosphorylated in response to NNK-induced oxidative stress. On the other hand, there could be a functional link between DNA repair (as indicated by the elevated γ-H2AX) and DNMT1. The recruitment of DNMT1 to DNA damage sites is responsible for the maintenance of DNA methylation pattern in the newly synthesized DNA during the repair process (41). In vitro studies have demonstrated the maintenance of DNA methylation pattern in DNA damage repair is critical because the loss of DNA methylation during the process leads to epigenetic deregulation and genomic instability (42, 43).
As reviewed previously (23), many mechanisms have been proposed for the inhibitory actions of EGCG against cancer. Our lab and others have demonstrated the inhibition of DNMT1 activity and re-activation of methylation silenced genes by EGCG in esophageal, colon, prostate and other cell lines (24, 25, 44). The present study is the first study demonstrating the inhibitory effects of dietary EGCG on the NNK-induced elevation of DNMT1 in vivo. Accompanying this inhibition was the marked decrease of p-AKT and γ-H2AX, but the levels of O6-mG were not significantly inhibited. The formation of O6-mG is a result of the metabolic activation of NNK. Our results suggest that the inhibitory action of EGCG against the up-regulation of DNMT1, p-AKT and γ-H2AX is not due to inhibition of the metabolic activation of NNK, but rather, the antioxidant activities of EGCG or other mechanisms.
In our study, dietary EGCG treatment reduced the expression of DNMT1 in the lung tissues and also, decreased the lung tumor multiplicity and volume at week 20. While EGCG effect on tumor multiplicity is modest, the inhibition on tumor volume is considerable and almost to the same degree as the DNMT1 inhibition. Inhibition of DNMT, especially DNMT1, has been reported to reverse the hypermethylation and the re-expression of the silenced genes (45–47), and studies using DNMT inhibitors such as 5-aza-2-deoxycytidine and Zebularine have been shown to inhibit cancer cell growth and reduce tumor volume in mice (48–50). Thus, it would be interesting to propose that the attenuation of DNMT1 and promoter hypermethylation of TSGs by EGCG contributed to the inhibition of lung tumorigenesis. Unfortunately, we did not save tumor samples from the EGCG-treated mice for the DNA methylation analysis. More work is needed to substantiate this proposal.
In summary, the present study demonstrated the NNK-induced elevation of DNMT1 in lung tissues of A/J mice at days 1, 3 and 14 following NNK administration and during the progression of lung tumorigenesis. The induction of DNMT1 is likely due to its protein stabilization by the activation of AKT in response to NNK-induced oxidative stress. The inhibitory effects of EGCG in the induction of DNMT1, p-AKT and γ-H2AX levels could be due to its antioxidant activities. Promoter hypermethylation of several TSGs was observed in lung tissues at day 3 and in lung tumors at week 20 after NNK treatment. The functional importance of early stage induction of DNMT1 and gene hypermethylation in NNK-induced lung carcinogenesis remains to be further investigated.
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (NIH) (RO1 CA120915, RO1 CA122474 and RO1 CA133021) as well as the shared facilities funded by National Cancer Institute Cancer Center Support grant (CA72720) and National Institute of Environmental Health Center grant (ES05022).
Abbreviations
- NNK
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone
- EGCG
(−)-epigallocatechin-3-gallate
- DNMT1
DNA methyltransferase 1
- p-AKT
phospho-AKT
- γ-H2AX
phospho-histone H2AX
- O6-mG
O6-methylguanine
- TSGs
tumor suppressor genes
- p16INK4a
cyclin-dependent kinase inhibitor 2A
- Dapk1
death-associated-protein kinase 1
- Rarb
retinoic acid receptor β
- Runx3
runt-related transcription factor 3
- MGMT
O6-methylguanine-DNA methyltransferase
- CDH13
cadherin-13
- PAX5
paired box protein Pax-5
- RASSF1
Ras associated domain-containing protein 1
- Prdm2
PR domain zinc finger protein 2
- IHC
immunohistochemistry
- qPCR
quantitative-PCR
- MSRE-qPCR
restriction enzyme-based quantitative PCR
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA: a cancer journal for clinicians. 2011 doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Henley SJ, Eheman CR, Richardson LC, Plescia M, Asman KJ, et al. State-Specific Trends in Lung Cancer Incidence and Smoking. Morbidity and mortality weekly report. 2011;60:1243–1247. [PubMed] [Google Scholar]
- 3.Riely GJ, Marks J, Pao W. KRAS mutations in non–small cell lung cancer. Proceedings of the American Thoracic Society. 2009;6:201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 4.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 5.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nature Reviews Cancer. 2004;4:707–717. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 6.Belinsky SA. Silencing of genes by promoter hypermethylation: key event in rodent and human lung cancer. Carcinogenesis. 2005;26:1481–1487. doi: 10.1093/carcin/bgi020. [DOI] [PubMed] [Google Scholar]
- 7.Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, et al. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. The Journal of clinical investigation. 2010;120:521. doi: 10.1172/JCI40706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyake T, Endo K, Honjo S, Hirooka Y, Ikeguchi M. Expression of DNA Methyltransferase (DNMT) 1, 3a and 3b Proteins in Human Hepatocellular Carcinoma. Yonago Acta medica. 2010;53:1–7. [Google Scholar]
- 9.Etoh T, Kanai Y, Ushijima S, Nakagawa T, Nakanishi Y, et al. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. The American journal of pathology. 2004;164:689–699. doi: 10.1016/S0002-9440(10)63156-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girault I, Tozlu S, Lidereau R, Bièche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clinical cancer research. 2003;9:4415–4422. [PubMed] [Google Scholar]
- 11.Patra SK, Patra A, Zhao H, Dahiya R. DNA methyltransferase and demethylase in human prostate cancer. Molecular carcinogenesis. 2002;33:163–171. doi: 10.1002/mc.10033. [DOI] [PubMed] [Google Scholar]
- 12.Qu Y, Mu G, Wu Y, Dai X, Zhou F, et al. Overexpression of DNA methyltransferases 1, 3a, and 3b significantly correlates with retinoblastoma tumorigenesis. American journal of clinical pathology. 2010;134:826–834. doi: 10.1309/AJCPHGQ69FXDFWII. [DOI] [PubMed] [Google Scholar]
- 13.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chemical research in toxicology. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 14.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. The lancet oncology. 2002;3:461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 15.Yang G, Wang ZY, Kim S, Liao J, Seril DN, et al. Characterization of early pulmonary hyperproliferation and tumor progression and their inhibition by black tea in a 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis model with A/J mice. Cancer Research. 1997;57:1889. [PubMed] [Google Scholar]
- 16.Hecht SS, Morse MA, Amin S, Stoner GD, Jordan KG, et al. Rapid single-dose model for lung tumor induction in A/J mice by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the effect of diet. Carcinogenesis. 1989;10:1901–4. doi: 10.1093/carcin/10.10.1901. [DOI] [PubMed] [Google Scholar]
- 17.Belinsky SA, Devereux TR, Foley JF, Maronpot RR, Anderson MW. Role of the alveolar type II cell in the development and progression of pulmonary tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the A/J mouse. Cancer Res. 1992;52:3164–73. [PubMed] [Google Scholar]
- 18.Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Research. 2004;64:3844. doi: 10.1158/0008-5472.CAN-03-2119. [DOI] [PubMed] [Google Scholar]
- 19.Pulling LC, Klinge DM, Belinsky SA. p16INK4a and beta-catenin alterations in rat liver tumors induced by NNK. Carcinogenesis. 2001;22:461–466. doi: 10.1093/carcin/22.3.461. [DOI] [PubMed] [Google Scholar]
- 20.Feng Q, Hawes SE, Stern JE, Wiens L, Lu H, et al. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiology Biomarkers & Prevention. 2008;17:645–654. doi: 10.1158/1055-9965.EPI-07-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pesek M, Kopeckova M, Benesova L, Meszarosova A, Mukensnabl P, et al. Clinical Significance of Hypermethylation Status in NSCLC. Evaluation of a 30-Gene Panel in Patients with Advanced Disease. Anticancer research. 2011;31:4647–4652. [PubMed] [Google Scholar]
- 22.Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT, et al. Alteration of DNA methyltransferases contributes to 5′ CpG methylation and poor prognosis in lung cancer. Lung Cancer. 2007;55:205–213. doi: 10.1016/j.lungcan.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 23.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nature Reviews Cancer. 2009;9:429–439. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, et al. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Research. 2003;63:7563. [PubMed] [Google Scholar]
- 25.Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Molecular pharmacology. 2005;68:1018–1030. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 26.Nandakumar V, Vaid M, Katiyar SK. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011;32:537–544. doi: 10.1093/carcin/bgq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hua D, Hu Y, Wu YY, Cheng ZH, Yu J, et al. Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma. Experimental and molecular pathology. 2011;91:455–460. doi: 10.1016/j.yexmp.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Oakes CC, La Salle S, Robaire B, Trasler JM. Research Paper Evaluation of a Quantitative DNA Methylation Analysis Technique Using Methylation-Sensitive/Dependent Restriction Enzymes and Real-Time PCR. Epigenetics. 2006;1:146–152. doi: 10.4161/epi.1.3.3392. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Zhao H, Xu Z, Liu Q, Liang Y, et al. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cellular signalling. 2007;19:2255–2263. doi: 10.1016/j.cellsig.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Peterson LA, Hecht SS. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–64. [PubMed] [Google Scholar]
- 31.Vuillemenot BR, Hutt JA, Belinsky SA. Gene promoter hypermethylation in mouse lung tumors. Molecular cancer research. 2006;4:267. doi: 10.1158/1541-7786.MCR-05-0218. [DOI] [PubMed] [Google Scholar]
- 32.Belinsky SA, Nikula KJ, Baylin SB, Issa J. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proceedings of the National Academy of Sciences. 1996;93:4045. doi: 10.1073/pnas.93.9.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamaluddin MS, Yang X, Wang H. Hyperhomocysteinemia, DNA methylation and vascular disease. Clinical Chemical Laboratory Medicine. 2008;45:1660–1666. doi: 10.1515/CCLM.2007.350. [DOI] [PubMed] [Google Scholar]
- 34.Kanai Y. Alterations of DNA methylation and clinicopathological diversity of human cancers. Pathology international. 2008;58:544–558. doi: 10.1111/j.1440-1827.2008.02270.x. [DOI] [PubMed] [Google Scholar]
- 35.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 36.Tsurutani J, Castillo SS, Brognard J, Granville CA, Zhang C, et al. Tobacco components stimulate Akt-dependent proliferation and NFκB-dependent survival in lung cancer cells. Carcinogenesis. 2005;26:1182–1195. doi: 10.1093/carcin/bgi072. [DOI] [PubMed] [Google Scholar]
- 37.West KA, Brognard J, Clark AS, Linnoila IR, Yang X, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. Journal of Clinical Investigation. 2003;111:81–90. doi: 10.1172/JCI16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafson AM, Soldi R, Anderlind C, Scholand MB, Qian J, et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Science translational medicine. 2010;2:26ra25–26ra25. doi: 10.1126/scitranslmed.3000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agoston AT, Argani P, De Marzo AM, Hicks JL, Nelson WG. Retinoblastoma pathway dysregulation causes DNA methyltransferase 1 overexpression in cancer via MAD2-mediated inhibition of the anaphase-promoting complex. The American journal of pathology. 2007;170:1585. doi: 10.2353/ajpath.2007.060779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albino A, Huang X, Jorgensen E, Yang J, Gietl D, et al. Report Induction of H2AX Phosphorylation in Pulmonary Cells by Tobacco Smoke. Cell Cycle. 2004;3:1062–1068. [PubMed] [Google Scholar]
- 41.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8905. doi: 10.1073/pnas.0501034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nature genetics. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 43.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455–455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 44.Fang M, Chen D, Yang CS. Dietary polyphenols may affect DNA methylation. The Journal of nutrition. 2007;137:223S. doi: 10.1093/jn/137.1.223S. [DOI] [PubMed] [Google Scholar]
- 45.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–77. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 46.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, et al. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 47.Clark SJ, Melki J. DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene. 2002;21:5380–7. doi: 10.1038/sj.onc.1205598. [DOI] [PubMed] [Google Scholar]
- 48.Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95–101. [PubMed] [Google Scholar]
- 49.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 50.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]