

Abstract
Calcium-independent phospholipase A2 group VIa (iPLA2β) preferentially releases docosahexaenoic acid (DHA) from the sn-2 position of phospholipids. Mutations of its gene, PLA2G6, are found in patients with several progressive motor disorders, including Parkinson disease. At 4 months, PLA2G6 knockout mice (iPLA2β−/−) show minimal neuropathology but altered brain DHA metabolism. By 1 year, they develop motor disturbances, cerebellar neuronal loss, and striatal α-synuclein accumulation. We hypothesized that older iPLA2β−/− mice also would exhibit inflammatory and other neuropathological changes. Real-time polymerase chain reaction and Western blotting were performed on whole brain homogenate from 15 to 20-month old male iPLA2β−/− or wild-type (WT) mice. These older iPLA2β−/− mice compared with WT showed molecular evidence of microglial (CD-11b, iNOS) and astrocytic (glial fibrillary acidic protein) activation, disturbed expression of enzymes involved in arachidonic acid metabolism, loss of neuroprotective brain derived neurotrophic factor, and accumulation of cytokine TNF-α messenger ribonucleic acid, consistent with neuroinflammatory pathology. There was no evidence of synaptic loss, of reduced expression of dopamine active reuptake transporter, or of accumulation of the Parkinson disease markers Parkin or Pink1. iPLA2γ expression was unchanged. iPLA2β deficient mice show evidence of neuroinflammation and associated neuropathology with motor dysfunction in later life. These pathological biomarkers could be used to assess efficacy of dietary intervention, antioxidants or other therapies on disease progression in this mouse model of progressive human motor diseases associated with a PLA2G6 mutation.
Keywords: Calcium-independent phospholipase A2 (iPLA2β) knockout, Brain, Parkinson disease, Arachidonic and docosahexaenoic acid, Motor disturbances, Neuropathology
Introduction
In vertebrates, the polyunsaturated fatty acid (PUFA) docosahexaenoic acid (DHA, 22:6n-3) is thought to play antioxidant, neuroprotective, and neurotransmission roles in brain [1–4]. DHA must be obtained directly from the diet or by liver synthesis from its dietary essential precursor, α-linolenic acid (α-LNA, 18:3 n-3) [5]. It is found in high concentrations in the sn-2 position of brain membrane phospholipids, from where it can be released preferentially by a calcium-independent phospholipase A2, iPLA2β or iPLA2γ [6, 7].
iPLA2β accounts for 70 % of cytosolic PLA2 activity in rat brain [6, 8]. Mutations in its gene, PLA2G6, occur in infantile neuroaxonal dystrophy (INAD) [9, 10], neurodegeneration with brain iron accumulation (NBIA) [10, 11], early-onset dystonia-parkinsonism [12–14], and Parkinson disease [15]. iPLA2β knockout mice have been developed to understand the consequences of these mutations [16, 17].
iPLA2β plays a role in motor behavior, since injection of the iPLA2β inhibitor bromoenol lactone (BEL) into the rat striatum, thalamus, or motor cortex produces chewing movements [18]. Furthermore, iPLA2β−/− mice exhibit age-dependent motor dysfunction associated with axonal degeneration and spheroids in the central nervous system by 1 year of age [16, 19]. Similar axonal swelling with spheroids occurs in the central and peripheral nervous systems of INAD and NBIA patients. Thirteen month-old iPLA2β−/− mice also show cerebellar atrophy with Purkinje cell loss, increased levels of inflammatory cytokines, glial cell activation, and striatal accumulation of α-synuclein [16, 20].
To characterize progressive age-related neuropathology in iPLA2β−/− mice, we first studied brain PUFA metabolism in 4-month old mice with normal motor function and minimal brain spheroid accumulation [4, 21]. Compared to wild-type (WT) controls, these mice exhibited disturbances in brain DHA metabolism that included reduced esterified brain DHA concentration in ethanolamine and serine glycerophospholipids, reduced DHA incorporation rate from plasma into brain phospholipids at rest or during cholinergic activation, and disturbed fatty acid concentrations in lysophospholipids. In addition, their brains showed increased expression of enzymes involved in arachidonic acid (AA, 20:4n-6) metabolism, including cytosolic cPLA2-IVα (cPLA2α), secretory sPLA2-V and cyclooxygenase (COX)-2 [8, 22].
In the present follow-up study, we measured expression of a number of brain parameters in 15 to 20-month old iPLA2-β−/− mice and in WT mice. These included: (1) enzymes involved in AA metabolism [cPLA2α, sPLA2-V, COX-2, COX-1, and microsomal prostaglandin synthase (mPGES) [22] ]; (2) enzymes involved in DHA metabolism [acyl-CoA synthetase (Acsl)-6, membrane-bound O-acyltransferase (MBOAT)-7, acyl-CoA:lysophosphatidylethanolamine acyltransferase (LPEAT)-2, and 15-lipoxygenase (15-LOX)]; (3) inflammatory cytokines [tumor necrosis factor (TNF)-α and interleukin (IL)-1β]; (4) markers of microglial and astrocytic activation [CD11b, glial fibrillary acidic protein (GFAP), and inducible nitrite oxide synthase (iNOS)]; (5) presynaptic synaptophysin, postsynaptic drebrin and postsynaptic density protein (PSD-95); (6) brain derived neurotrophic factor (BDNF); and 7) Parkinson disease markers [dopamine active reuptake transporter (DAT), α-synuclein, PTEN induced kinase 1 (Pink1) and Parkin].
An abstract of part of this work has been published (Society for Neuroscience Meeting, New Orleans, LA, 2012).
Materials and Methods
Animals
Experiments were conducted in accord with the National Institutes of Health guidelines for the Animal Care and Use Committee (Publication no. 86-23) and followed a protocol approved by the Animal Care and Use Committee of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Male iPLA2β−/− mice and their WT littermates [23] were housed in an animal facility having regulated temperature, humidity and light cycle until they reached 15–20 months of age. They had free access to water and to a diet (PicoLab® Rodent Diet 20, 5053, LabDiet, Richmond, IN) containing (as % total fatty acid): 20.0 % saturated, 22.2 % monounsaturated, 47.7 % linoleic acid, 5.1 % linolenic acid, 0.1 % AA, 0.2 % eicosapentaenoic acid and 0.9 % DHA. Mice (n = 8 per group) were asphyxiated by CO2 inhalation and decapitated, and their brains were rapidly excised and frozen in 2-methylbutane at −50 °C and stored at −80 °C for subsequent analyses.
Gene Expression
Half brains were homogenized with an Ultraturax homogenizer (IKA Works, Wilmington, DE) in QIAzol lysis reagent (Qiagen, Valencia, CA) and mRNA was isolated with a phenol–chloroform extraction method using the RNeasy Lipid tissue kit (Qiagen). Total mRNA was reverse-transcribed to cDNA according to the manufacturer's instructions with a high capacity cDNA Archive kit (Applied Biosystems, Carlsbad, CA). Gene expression was determined by real time-polymerase chain reaction (RT-PCR) using the Taqman® Universal PCR Mastermix and specific Taqman® primers and probe (Applied Biosystems). The PCR reaction was performed in duplicate, using ABI Prism 7000 sequence detection system (Applied Biosystems) as follows: 2 min at 50 °C, 5 min at 95 °C, 40 cycles of 10 s at 95 °C, and 1 min at 60 °C. Relative gene expression was determined using the ΔΔCt method, using the 18S gene expression for normalization.
Protein Extraction
Half-brains were homogenized in a buffer containing 10 mM HEPES, pH 7.5, 1 mM EDTA, 0.34 M sucrose and protease inhibitors (Complete EDTA-free, Roche Applied Science, Indianapolis, IN), using a Dounce glass homogenizer (Thomas Scientific, Swedesboro, NJ). After ultra-centrifugation (100,000g, 1 h, 4 °C), the supernatant was used as the cytosolic fraction. The pellet was resuspended in Tris 20 mM, pH 7.4 containing 0.2 % Triton X-100 for 1 h at 4 °C under agitation. The sample was centrifuged (100,000g, 1 h, 4 °C) and the resulting supernatant contained the membrane fraction. Protein concentrations in the cytosolic and membrane fractions were determined with a Bradford assay [24] and samples were stored at −80 °C until analyzed.
Protein Analysis
Cytosolic or membrane proteins (20 lg) were separated by SDS-PAGE in 4–20 % Tris–HCl polyacrylamide gels (Bio-Rad, Hercules, CA) and transferred on a nitrocellulose membrane (Bio-Rad) for immunoblotting. Membranes were blocked for 90 min at room temperature with a casein-based blocking buffer (Sigma Aldrich, St Louis, MO) before incubation overnight at 4 °C with primary antibodies. Cytosolic proteins were probed with specific antibodies for cPLA2α (1:500), sPLA2-V (1:500), iNOS (1:200), COX-1 (1:200), Il-1β (1:200), 15-LOX (1:200), drebrin (1:500) and Pink1 (1:200) (Santa Cruz Biotechnology, Santa Cruz, CA), for COX-2 (1:200), GFAP (1:50,000), PSD-95 (1:1,000), BDNF (1:1,000) (AbCam, Cambridge, MA), for Acsl-6 (Novus Biochemical, Littleton, MO) or for α-synuclein (1:5,000) (Cell signaling, Danvers, MA). Membrane fractions were blotted with antibodies targeting phosphor-ylated-cPLA2α (P- cPLA2α) (1:500, Santa Cruz), mPGES (1:500, Cayman Chemical, Ann Arbor, MI), CD11b (1:500), synaptophysin (1:15,000) (AbCam), MBOAT7 (Aviva Systems Biology, San Diego, CA), LPEAT2 (1:1,500) (ProteinTech, Chicago, IL) and DAT (1:1,000) (Millipore, Billerica, MA). All samples were probed with a β-actin antibody (Sigma Aldrich). Parkin expression was determined in the cytosol and membrane fractions (1:2,000, AbCam). Membranes then were incubated for 90 min at room temperature with the appropriate horseradish peroxidase-conjugated secondary antibodies (Bio-Rad), and peroxidase activity was determined by chemiluminescence using the SuperSignal West Pico reagents (Thermo Fisher Scientific, Rockford, IL). Relative protein expression was determined after normalization with the β-actin expression using ImageJ software [25]. TNF-α protein was quantified from 100 μg of brain cytosolic extract using an enzyme-linked immunosorbent assay (ELISA) method according to the manufacturer's instruction (Thermo Fisher Scientific).
Statistical Analyses
Data are expressed as mean ± SEM (n = 7–8 per group). A two-tailed, unpaired t test using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA) was used to determine statistical significance, when P< 0.05. Cohen's d effect size was used to interpret differences between the means when the P value was between 0.05 and 0.2, since this suggested increased risk of type II error associated with the small number of animals available for the study. An effect size is considered small if 0.3 <d < 0.5, medium if 0.5 <d < 0.8 and high if d > 0.8 [26]. We considered effect sizes greater than 0.5 likely to be significant. Outliers were identified using Grubb's test and removed from the statistical analysis.
Results
Brain AA and DHA Metabolism
iPLA2β protein expression was not measured in this study but iPLA2β deficiency was confirmed in the iPLA2β−/− mice by the virtual absence of brain iPLA2β mRNA compared to WT mice (P < 0.001; Fig. 1), whereas the iPLA2γ mRNA level did not differ between genotypes (Fig. 2c). cPLA2α mRNA and protein levels were similar between groups, but the protein level of the activated phosphorylated form of cPLA2α in the membrane fraction was significantly higher for iPLA2β−/− compared to WT mice (51 %, P = 0.04) (Fig. 2a). Although sPLA2-V mRNA also was higher (64 %, P = 0.02), sPLA2-V protein did not differ significantly between genotypes (Fig. 2b). COX-1 mRNA and protein and COX-2 mRNA also did not differ between genotypes (Fig. 3a), while COX-2 protein was reduced in iPLA2β−/− mice (12 %, P = 0.03) (Fig. 3b). Brain mPGES mRNA was significantly higher in iPLA2β−/− mice, but mPGES protein did not differ between genotypes (Fig. 3c). Protein levels of Acsl-6, MBOAT7, LPEAT2 and 15-LOX also did not differ between iPLA2β−/− and WT mice (Fig. 4).
Fig. 1.
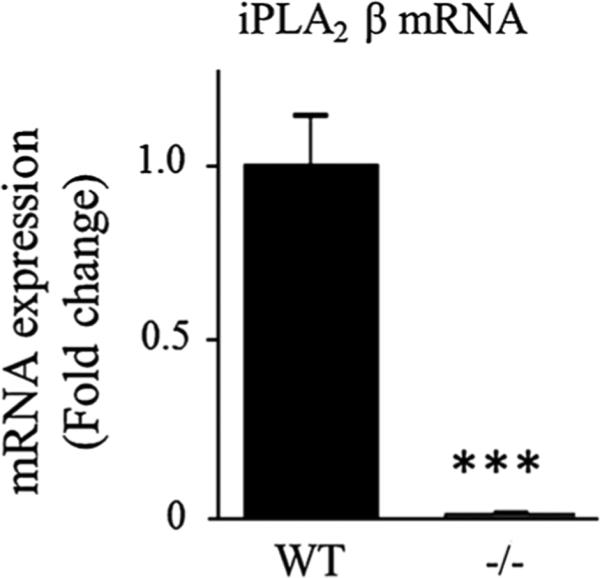
mRNA expression of iPLA2β in WT and iPLA2β−/− mice. mRNA was quantified in duplicate with Taqman® RT-PCR using the DDCt method and normalization to 18S expression. Results are expressed as mean ± SEM (n = 8). ***P < 0.001 (t test)
Fig. 2.

Expression of cPLA2α (a), sPLA2-V (b) and iPLA2c in WT and iPLA2β−/− mice. mRNA was quantified in duplicate with Taqman® RT-PCR using the ΔΔCt method and normalization to 18S expression. Results are expressed as mean ± SEM (n = 8). cPLA2α and sPLA2-V protein was measured in brain cytosol and P-cPLA2α was quantified in the membrane fraction by Western blot. Results are normalized to β-actin expression (representative immunoblot is displayed) and expressed as % of the WT mice as mean ± SEM (n = 8, except n = 7 for P-cPLA2 in iPLA2β−/− because an outlier value of 7.4 was removed). Numbers in italics indicate molecular weight markers (in kDa), NS : non-specific band. *P < 0.05 (t-test)
Fig. 3.

Expression of COX-2 (a), COX-1 (b) and mPGES (c) in WT and iPLA2β−/− mice. mRNA was quantified in duplicate with Taqman® RTPCR using the ΔΔCt method and normalization to 18S expression. Results are expressed as mean ± SEM (n = 8). COX-2, COX-1 were measured in brain cytosol and mPGES in the membrane fraction by western blot and normalized to β-actin expression (representative immunoblot is displayed). Results are expressed as % of the WT mice as mean ± SEM (n = 8, except n = 7 for COX-1 protein in WT due to an outlier value of 198−/−). Numbers in italics indicate molecular weight markers (in kDa). *P < 0.05, **P < 0.01 (t test)
Fig. 4.

Expression of brain DHA-metabolizing enzymes in WT and iPLA2β−/− mice. Acsl-6 and 15-LOX protein was measured in brain cytosol, MBOAT7 and LPEAT2 in the membrane fraction by Western blot and normalized to β-actin expression (representative immunoblot is displayed). Results are expressed as % of the WT mice as mean ± SEM (n = 8). Numbers in italics indicate molecular weight markers (in kDa), NS non-specific band
Neuroinflammation and Synaptic Loss
TNF-α mRNA was increased 2.1-fold (P < 0.001) in iPLA2β−/− mice compared to WT, but TNF-α protein levels were similar in both groups (Fig. 5a), as were levels of IL-1b mRNA and protein (Fig. 5b). iNOS mRNA and protein did not differ significantly on t tests (P = 0.07 and 0.14 respectively), but their effect sizes measured as Cohen's d were large, 0.99 and 0.98 respectively (Fig. 5c).
Fig. 5.

Expression of the pro-inflammatory cytokines TNF-α (a) and Il-1β (b) and the iNOS (c) in WT and iPLA2β−/− mice. mRNA was quantified in duplicate with Taqman® RT-PCR using the ΔΔCt method and normalization to 18S expression. Results are expressed as mean ± SEM (n = 8). An ELISA assay was used to quantify cytosolic TNF-α. Il-1β and iNOS protein was measured in brain cytosol by Western blot and normalized to β-actin expression (representative immunoblot is displayed). Results are expressed as % of the WT mice as mean ± SEM (n = 8, iNOS n = 6 for WT and n = 7 for iPLA −/−2 due to unquantifiable signal). Numbers in italics indicate molecular weight markers (in kDa), NS non-specific band. ***P < 0.001 (t test) and ##d > 0.8 (Cohen's d test)
CD11b mRNA and protein levels were significantly higher in iPLA2β−/− mice compared to WT (60 and 30 %, P < 0.05, respectively) (Fig. 6a), suggesting microglial activation [27]. GFAP mRNA was elevated 2.2-fold (P < 0.01) in iPLA2β−/− mice, but its protein level did not differ significantly between genotypes on t-tests (P = 0.19); nevertheless, Cohen's d test suggested a modest increase (d = 0.68) in iPLA2β−/− mice (Fig. 6b). Protein levels of pre-synaptic synaptophysin and post-synaptic drebrin and PSD95 did not differ between genotypes (Fig. 6c), while BDNF protein was reduced in iPLA2β−/− mice (22 %, P = 0.01) (Fig. 6d).
Fig. 6.

Expression of microglial, astrocytic, synaptic markers and BNDF in WT and iPLA2β−/− mice. mRNA was quantified in duplicate with Taqman® RT-PCR using the ΔΔCt method and normalization to 18S expression. Results are expressed as mean ± SEM (n = 8). CD11b, synaptophysin and PSD-95 expression was measured in brain membrane fractions by Western blot and GFAP, drebrin and BDNF were quantified in the cytosol. Results are normalized to β-actin expression (representative immunoblot is displayed) and expressed as % of the WT mice as mean ± SEM (n = 8 except n = 7 for BDNF in WT and iPLA2β−/− , respectively due to the removal of an outlier value of 222 and sample loss). Numbers in italics indicate molecular weight markers (in kDa). *P < 0.05, **P < 0.01 (t test) and #0.5 < d > 0.8 (Cohen's d test)
Parkinson Disease Markers
Protein levels of both reduced DAT (50 kDa) and non-reduced DAT (75 kDa) did not differ between iPLA2β−/− mice and WT mice (Fig. 7). Alpha-synuclein mRNA was significantly higher (53 %, P = 0.01), but α-synuclein protein (Fig. 8a), Pink1 (Fig. 8b) and Parkin (Fig. 8c) mRNA and protein did not differ between groups.
Fig. 7.

Expression of DAT in WT and iPLA2β−/− mice. DAT protein was measured in brain membrane fractions by Western blot. Results are normalized to β-actin expression (representative immunoblot is displayed) and expressed as % of WT mice as mean ± SEM (n = 8). Numbers in italics indicate molecular weight markers (in kDa)
Fig. 8.

Expression of α-synuclein (a), PINK1 (b) and Parkin (c) in WT and iPLA2β−/− mice. mRNA was quantified in duplicate with Taqman® RT-PCR using the ΔΔCt method and normalization to 18S expression. Results are expressed as mean ± SEM (n = 8). α-synuclein (a) and PINK1 (b) were quantified in the cytosolic fraction. Both cytosolic (c-i) and membrane (c-ii) parkin protein levels were determined. After normalization to β-actin expression (representative immunoblot is displayed), results are expressed as % of the WT mice as mean ± SEM (n = 8, n = 7 for α-synuclein WT (outlier value of 241 was removed) and for n = 7 for Pink1 due to an unquantifiable signal). Numbers in italics indicate molecular weight markers (in kDa). *P < 0.05 (t test)
Discussion
These results indicate that long-term disruption of brain DHA metabolism in aged mice due to the absence of PLA2G6 results in microglial and astrocytic activation, increased TNF-α and α-synuclein, disturbed expression of enzymes involved in AA metabolism, and a reduced BDNF level. The results also show that iPLA2β and iPLA2γ play independent roles in brain, because brain iPLA2γ expression was unchanged iPLA2β−/− mice. The large effect sizes of increments in iNOS mRNA and protein and in TNF-α mRNA (and protein by Cohen's d), and increased CD11b mRNA and protein, suggest microglial activation by age 15–20 months [27]. Motor function was not assessed in the present study, but these neuropathological markers might be associated with appearance of age-dependent motor abnormalities in iPLA2β−/− mice reported by others [16, 19]. The lack of change in IL-1β levels suggests that full-blown global inflammation is not reached at this age and may require more time to evolve. On the other hand, Purkinje cell loss, glial cell activation and elevated TNF-α and IL-1b expression have been reported in the cerebellum of iPLA2-β−/− mice at age 13 months [20].
The many late-appearing neuropathological changes in the iPLA2β−/− mice suggest that early defective DHA metabolism can contribute to human diseases that result from PLA2G6 mutations, such as INAD, neurodegeneration with brain iron accumulation, and early onset-dystonia parkinsonism (see ‘Introduction’). Thus, this mouse model might be used to develop therapy for these diseases. Since iPLA2β has an anti-oxidant function that protects mitochondria from peroxidation damage [17], one therapeutic approach with the model would be to administer antioxidants, as recommended for Parkinson disease [17, 28, 29]. Feeding DHA has been considered in this regard [2], but the mice already were on a high DHA diet. On the other hand, an AA-free diet low in linoleic acid (18:2n-6) might be help to dampen the upregulated AA-metabolizing enzymes in the iPLA2β−/− mouse brain [30–32].
Unesterified DHA is preferentially acylated by Acsl-6 to DHA-CoA [33], which can be reacylated into a lysophospholipid by an acyl-CoA lysophospholipid acyltransferase [34]. Within this latter enzyme family, we analyzed expression of MBOAT7, which recognizes AA-CoA as a substrate [35], although its activity toward DHA-CoA remains to be tested. We also measured LPEAT2, which preferentially acylates ethanolamine glycerophospholipid enriched with DHA [36, 37]. The incorporation rate of unesterified DHA into brain ethanolamine glycerophospholipids and their DHA content are reduced in 4-month old iPLA2β−/− mice [4, 21], but we found no significant difference between genotypes in brain expression of these acyltransferases or of 15-LOX, which is also involved in DHA metabolism [38].
Reduced DHA incorporation from plasma into brain in iPLA2β−/− mice would be expected to reduce production of DHA-derived resolvin D1 and neuroprotectin D1, which have potent anti-inflammatory properties [39, 40]. DHA also may reduce pro-inflammatory AA metabolite production by inhibiting COX-2 [1, 41], consistent with the suggested role of iPLA2β in regulating prostaglandin E2 formation [7]. It would be worthwhile to test whether iPLA2β−/− mice have reduced DHA-derived metabolites and increased AA-derived products in future studies.
The elevated brain levels in iPLA2β−/− mice of sPLA2-V mRNA and P-cPLA2α protein, which represents the catalytically active form of cPLA2α, suggests compensatory upregulation of brain AA metabolism [42]. Upregulation also occurs when DHA metabolism is reduced by dietary n-3 fatty acid deprivation [43]. Increased P-cPLA2α protein suggests accelerated post-translational phosphorylation of cPLA2α serine residues [44]. COX-2 expression was reduced, although it was reported to be elevated at 4 months [21]. Expression of COX-1, which can be coupled to iPLA2β [45], was unchanged.
Similar protein levels of drebrin, PSD95, synaptophysin and DAT in the 15-20 month old iPLA2β−/− and WT mice suggest absence of synaptic loss. On the other hand, presynaptic DAT protein is reduced in Parkinson disease [46], and following ablation of the substantia nigra in rodent models of Parkinson disease [47]. Thus, at 15–20 months, the model does not have all the classical features of Parkinson disease. This also applies to the absence of differences in expression of Parkin (Park2) [48] and of Pink1 (Park6) [49], which regulate mitochondrial integrity [50]. Regional changes potentially identifiable by immunocytochemistry cannot yet be excluded, especially in the substantia nigra, which is specifically impacted by dopaminergic neuronal loss in Parkinson disease.
Activated microglia produce NO via iNOS, reactive oxygen species, and pro-inflammatory cytokines that might contribute to brain pathology in Parkinson disease [51, 52]. Reduced BDNF expression in the older iPLA2β−/− mice also suggests increased susceptibility to neuronal death [53], which is observed in the substantia nigra of Parkinson disease patients [54].
Brain α-synuclein mRNA was increased in the older iPLA2β−/− mice, consistent with reported α-synuclein-positive spheroids in the striatum of iPLA2β−/− mice [16]. Alpha-synuclein can activate glia [55] and is thought to promote a self-perpetuating neurotoxic process in Parkinson disease [52]. It impacts PUFA metabolism [56] and may bind long chain fatty acids [57]. On the other hand, dietary DHA supplementation was reported to promote asynuclein accumulation in a mouse model of Parkinson disease [58].
An iPLA2β preference for DHA-containing phospholipid substrates is well established [4, 6, 7, 59], but esterified AA also can be hydrolyzed by iPLA2β [60, 61]. Thus, an iPLA2β deficiency also might influence AA release and eicosanoid formation.
In summary, life-long distortion of brain DHA content and metabolism in iPLA2β−/− mice is associated with late-appearing motor disturbances accompanied by microglial and astrocytic activation, disturbed expression of enzymes involved in arachidonic acid metabolism, loss of neuroprotective BDNF, and increased neuroinflammatory cytokine expression. Using these biomarkers, efficacy of dietary interventions or suppressors of oxidative stress might be tested on disease progression in this animal model, which mimics many aspects of human progressive motor diseases, including Parkinson disease.
Acknowledgments
The authors thank the NIH Fellow Editorial Board and Ms. Mairi Stevens for editorial assistance and Dr. Dede Greenstein for statistical support. Research was supported by the Intramural Research Program of the National Institute on Aging and, for JT, by United States Public Health Service Grants R37-DK34388, P41-RR00954, P60-DK20579, and P30-DK56341.
Footnotes
Conflict of interest Authors declare no competing financial interests.
Contributor Information
Helene Blanchard, Brain Physiology and Metabolism Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes of Health, Bldg. 9, Room 1S126, 9000 Rockville Pike, Bethesda, MD 20892, USA.
Ameer Y. Taha, Brain Physiology and Metabolism Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes of Health, Bldg. 9, Room 1S126, 9000 Rockville Pike, Bethesda, MD 20892, USA
Yewon Cheon, Brain Physiology and Metabolism Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes of Health, Bldg. 9, Room 1S126, 9000 Rockville Pike, Bethesda, MD 20892, USA.
Hyung-Wook Kim, Brain Physiology and Metabolism Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes of Health, Bldg. 9, Room 1S126, 9000 Rockville Pike, Bethesda, MD 20892, USA; College of Life Sciences, Sejong University, 98 Gunja-dong, Gwangjin-Gu, Seoul 143–747, Korea.
John Turk, Division of Endocrinology, Medicine Department Metabolism and Lipid Research, Washington University School of Medicine, St. Louis, MO 63110, USA.
Stanley I. Rapoport, Brain Physiology and Metabolism Section, Laboratory of Neurosciences, National Institute on Aging, National Institutes of Health, Bldg. 9, Room 1S126, 9000 Rockville Pike, Bethesda, MD 20892, USA
References
- 1.Corey EJ, Shih C, Cashman JR. Docosahexaenoic acid is a strong inhibitor of prostaglandin but not leukotriene biosynthesis. Proc Natl Acad Sci USA. 1983;80(12):3581–3584. doi: 10.1073/pnas.80.12.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yavin E, Brand A, Green P. Docosahexaenoic acid abundance in the brain: a biodevice to combat oxidative stress. Nutr Neurosci. 2002;5(3):149–157. doi: 10.1080/10284150290003159. [DOI] [PubMed] [Google Scholar]
- 3.Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell Signal. 2005;17(9):1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Basselin M, Rosa AO, Ramadan E, Cheon Y, Chang L, Chen M, Greenstein D, Wohltmann M, Turk J, Rapoport SI. Imaging decreased brain docosahexaenoic acid metabolism and signaling in iPLA(2)beta (VIA)-deficient mice. J Lipid Res. 2010;51(11):3166–3173. doi: 10.1194/jlr.M008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domenichiello AF, Chen CT, Trepanier MO, Stavro PM, Bazinet RP. Whole body synthesis rates of DHA from alpha-linolenic acid are greater than brain DHA accretion and uptake rates in adult rats. J Lipid Res. 2014;55(1):62–74. doi: 10.1194/jlr.M042275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang HC, Mosior M, Ni B, Dennis EA. Regional distribution, ontogeny, purification, and characterization of the Ca2 + -independent phospholipase A2 from rat brain. J Neurochem. 1999;73(3):1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- 7.Strokin M, Sergeeva M, Reiser G. Prostaglandin synthesis in rat brain astrocytes is under the control of the n-3 docosahexaenoic acid, released by group VIB calcium-independent phospholipase A2. J Neurochem. 2007;102(6):1771–1782. doi: 10.1111/j.1471-4159.2007.04663.x. [DOI] [PubMed] [Google Scholar]
- 8.Balboa MA, Varela-Nieto I, Killermann Lucas K, Dennis EA. Expression and function of phospholipase A(2) in brain. FEBS Lett. 2002;531(1):12–17. doi: 10.1016/s0014-5793(02)03481-6. [DOI] [PubMed] [Google Scholar]
- 9.Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, Verga L, Angelini L, Savoiardo M, Bugiani O. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology. 1999;52(7):1472–1478. doi: 10.1212/wnl.52.7.1472. [DOI] [PubMed] [Google Scholar]
- 10.Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, Gitschier J, Hayflick SJ. Neurodegeneration associated with genetic defects in phospholipase A(2) Neurology. 2008;71(18):1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, Hayflick SJ. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38(7):752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, Hashimoto T, Takashima S, Hattori N. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. 2010;75(15):1356–1361. doi: 10.1212/WNL.0b013e3181f73649. [DOI] [PubMed] [Google Scholar]
- 13.Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, Chang HC, Wu-Chou YH, Yeh TH. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(2):183–191. doi: 10.1002/ajmg.b.32012. [DOI] [PubMed] [Google Scholar]
- 14.Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, Chataway J, Bhatia KP, Lees AJ, Hardy J, Revesz T, Houlden H. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33(4):814–823. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gui YX, Xu ZP, Wen L, Liu HM, Zhao JJ, Hu XY. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Relat Disord. 2013;19(1):21–26. doi: 10.1016/j.parkreldis.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 16.Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, Kotzbauer PT. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol. 2008;172(2):406–416. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Z, Zhang X, Zhao C, Choi J, Shi J, Song K, Turk J, Ma ZA. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology. 2010;151(7):3038–3048. doi: 10.1210/en.2010-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee LY, Ong WY, Farooqui AA, Burgunder JM. Role of calcium-independent phospholipase A2 in cortex striatum thalamus cortex circuitry-enzyme inhibition causes vacuous chewing movements in rats. Psychopharmacology. 2007;195(3):387–395. doi: 10.1007/s00213-007-0912-y. [DOI] [PubMed] [Google Scholar]
- 19.Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci. 2008;28(9):2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Z, Wang J, Zhao C, Bi W, Yue Z, Ma ZA. Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy characterized by Purkinje cell loss and glial cell activation. Plos One. 2011;6(10):e26991. doi: 10.1371/journal.pone.0026991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheon Y, Kim HW, Igarashi M, Modi HR, Chang L, Ma K, Greenstein D, Wohltmann M, Turk J, Rapoport SI, Taha AY. Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A(2)-VIA (iPLA(2)beta)-knockout mice. Biochim Biophys Acta. 2012;1821(9):1278–1286. doi: 10.1016/j.bbalip.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rapoport SI. Brain arachidonic and docosahexaenoic acid cascades are selectively altered by drugs, diet and disease. Prostaglandins Leukot Essent Fatty Acid. 2008;79(3–5):153–156. doi: 10.1016/j.plefa.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279(37):38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics Int. 2004;11(7):36–42. [Google Scholar]
- 26.Cohen J. A power primer. Psychol Bull. 1992;112(1):155–159. doi: 10.1037//0033-2909.112.1.155. [DOI] [PubMed] [Google Scholar]
- 27.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17(4):942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chao J, Leung Y, Wang M, Chang RC. Nutraceuticals and their preventive or potential therapeutic value in Parkinson’s disease. Nutr Rev. 2012;70(7):373–386. doi: 10.1111/j.1753-4887.2012.00484.x. [DOI] [PubMed] [Google Scholar]
- 29.Sutachan JJ, Casas Z, Albarracin SL, Stab BR, 2nd, Samudio I, Gonzalez J, Morales L, Barreto GE. Cellular and molecular mechanisms of antioxidants in Parkinson’s disease. Nutr Neurosci. 2012;15(3):120–126. doi: 10.1179/1476830511Y.0000000033. [DOI] [PubMed] [Google Scholar]
- 30.Igarashi M, Gao F, Kim HW, Ma K, Bell JM, Rapoport SI. Dietary n-6 PUFA deprivation for 15 weeks reduces arachidonic acid concentrations while increasing n-3 PUFA concentrations in organs of post-weaning male rats. Biochim Biophys Acta. 2009;1791(2):132–139. doi: 10.1016/j.bbalip.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HW, Rao JS, Rapoport SI, Igarashi M. Dietary n-6 PUFA deprivation downregulates arachidonate but upregulates docosahexaenoate metabolizing enzymes in rat brain. Biochim Biophys Acta. 2011;1811(2):111–117. doi: 10.1016/j.bbalip.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramsden CE, Mann JD, Faurot KR, Lynch C, Imam ST, Mac-Intosh BA, Hibbeln JR, Loewke J, Smith S, Coble R, Suchindran C, Gaylord SA. Low omega-6 versus low omega-6 plus high omega-3 dietary intervention for chronic daily headache: protocol for a randomized clinical trial. Trials. 2011;12:97. doi: 10.1186/1745-6215-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marszalek JR, Kitidis C, Dirusso CC, Lodish HF. Long-chain acyl-CoA synthetase 6 preferentially promotes DHA metabolism. J Biol Chem. 2005;280(11):10817–10826. doi: 10.1074/jbc.M411750200. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita A, Sugiura T, Waku K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J Biochem. 1997;122(1):1–16. doi: 10.1093/oxfordjournals.jbchem.a021715. [DOI] [PubMed] [Google Scholar]
- 35.Gijon MA, Riekhof WR, Zarini S, Murphy RC, Voelker DR. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J Biol Chem. 2008;283(44):30235–30245. doi: 10.1074/jbc.M806194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeGeorge JJ, Nariai T, Yamazaki S, Williams WM, Rapoport SI. Arecoline-stimulated brain incorporation of intravenously administered fatty acids in unanesthetized rats. J Neurochem. 1991;56(1):352–355. doi: 10.1111/j.1471-4159.1991.tb02603.x. [DOI] [PubMed] [Google Scholar]
- 37.Cao J, Shan D, Revett T, Li D, Wu L, Liu W, Tobin JF, Gimeno RE. Molecular identification of a novel mammalian brain isoform of acyl-CoA:lysophospholipid acyltransferase with prominent ethanolamine lysophospholipid acylating activity, LPEAT2. J Biol Chem. 2008;283(27):19049–19057. doi: 10.1074/jbc.M800364200. [DOI] [PubMed] [Google Scholar]
- 38.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115(10):2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells autacoids in anti-inflammation. J Biol Chem. 2003;278(17):14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 40.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swamy MV, Cooma I, Patlolla JM, Simi B, Reddy BS, Rao CV. Modulation of cyclooxygenase-2 activities by the combined action of celecoxib and decosahexaenoic acid: novel strategies for colon cancer prevention and treatment. Mol Cancer Ther. 2004;3(2):215–221. [PubMed] [Google Scholar]
- 42.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2 +)-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65(6):1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 43.Kim HW, Rao JS, Rapoport SI, Igarashi M. Regulation of rat brain polyunsaturated fatty acid (PUFA) metabolism during graded dietary n-3 PUFA deprivation. Prostaglandins Leukot Essent Fatty Acids. 2011;85(6):361–368. doi: 10.1016/j.plefa.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buschbeck M, Ghomashchi F, Gelb MH, Watson SP, Borsch-Haubold AG. Stress stimuli increase calcium-induced arachidonic acid release through phosphorylation of cytosolic phospholipase A2. Biochem J. 1999;344(Pt 2):359–366. [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami M, Kambe T, Shimbara S, Kudo I. Functional coupling between various phospholipase A2 s and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways. J Biol Chem. 1999;274(5):3103–3115. doi: 10.1074/jbc.274.5.3103. [DOI] [PubMed] [Google Scholar]
- 46.Scherfler C, Schwarz J, Antonini A, Grosset D, Valldeoriola F, Marek K, Oertel W, Tolosa E, Lees AJ, Poewe W. Role of DAT-SPECT in the diagnostic work up of parkinsonism. Mov Disord. 2007;22(9):1229–1238. doi: 10.1002/mds.21505. [DOI] [PubMed] [Google Scholar]
- 47.Bhattacharjee AK, Meister LM, Chang L, Bazinet RP, White L, Rapoport SI. In vivo imaging of disturbed pre- and post-synaptic dopaminergic signaling via arachidonic acid in a rat model of Parkinson’s disease. NeuroImage. 2007;37(4):1112–1121. doi: 10.1016/j.neuroimage.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 49.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 50.Pilsl A, Winklhofer KF. Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathol. 2012;123(2):173–188. doi: 10.1007/s00401-011-0902-3. [DOI] [PubMed] [Google Scholar]
- 51.Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson’s disease. Ann NY Acad Sci. 2003;991:214–228. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 52.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 53.Barde YA. Neurotrophins: a family of proteins supporting the survival of neurons. Prog Clin Biol Res. 1994;390:45–56. [PubMed] [Google Scholar]
- 54.Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol. 2000;166(1):127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 55.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 56.Golovko MY, Rosenberger TA, Feddersen S, Faergeman NJ, Murphy EJ. Alpha-synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J Neurochem. 2007;101(1):201–211. doi: 10.1111/j.1471-4159.2006.04357.x. [DOI] [PubMed] [Google Scholar]
- 57.Golovko MY, Rosenberger TA, Faergeman NJ, Feddersen S, Cole NB, Pribill I, Berger J, Nussbaum RL, Murphy EJ. Acyl-CoA synthetase activity links wild-type but not mutant alpha-synuclein to brain arachidonate metabolism. Biochemistry. 2006;45(22):6956–6966. doi: 10.1021/bi0600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yakunin E, Loeb V, Kisos H, Biala Y, Yehuda S, Yaari Y, Selkoe DJ, Sharon R. Alpha-synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson’s disease. Brain Pathol. 2012;22(3):280–294. doi: 10.1111/j.1750-3639.2011.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramadan E, Rosa AO, Chang L, Chen M, Rapoport SI, Basselin M. Extracellular-derived calcium does not initiate in vivo neurotransmission involving docosahexaenoic acid. J Lipid Res. 2010;51(8):2334–2340. doi: 10.1194/jlr.M006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lehman JJ, Brown KA, Ramanadham S, Turk J, Gross RW. Arachidonic acid release from aortic smooth muscle cells induced by [Arg8]vasopressin is largely mediated by calcium-independent phospholipase A2. J Biol Chem. 1993;268(28):20713–20716. [PubMed] [Google Scholar]
- 61.Wolf MJ, Gross RW. Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J Biol Chem. 1996;271(48):30879–30885. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]