

Abstract
Over the past two decades, pattern mining techniques have become an integral part of many bioinformatics solutions. Frequent itemset mining is a popular group of pattern mining techniques designed to identify elements that frequently co-occur. An archetypical example is the identification of products that often end up together in the same shopping basket in supermarket transactions. A number of algorithms have been developed to address variations of this computationally non-trivial problem. Frequent itemset mining techniques are able to efficiently capture the characteristics of (complex) data and succinctly summarize it. Owing to these and other interesting properties, these techniques have proven their value in biological data analysis. Nevertheless, information about the bioinformatics applications of these techniques remains scattered. In this primer, we introduce frequent itemset mining and their derived association rules for life scientists. We give an overview of various algorithms, and illustrate how they can be used in several real-life bioinformatics application domains. We end with a discussion of the future potential and open challenges for frequent itemset mining in the life sciences.
Keywords: pattern mining, frequent item set, association rule, market basket analysis, biclustering
INTRODUCTION
High-throughput molecular analysis techniques nowadays yield datasets with a size and complexity at which they are no longer directly interpretable by humans. In recent years, pattern mining methods have become indispensable for life scientists to narrow down the search for relevant new knowledge instead of getting lost in the wealth of information. The term ‘pattern mining' covers a wide variety of techniques that are all designed to transform complex datasets into something more manageable. In this introductory article, we focus on a group of techniques referred to as ‘frequent itemset mining'.
Frequent itemset mining methods were developed to identify elements that often co-occur in a dataset. The archetypical usage case is the market basket problem [1], in which frequent itemset mining techniques are applied to discover which items are often bought together by customers (referred to as ‘patterns'). An example of an interesting pattern could be that beer and chips frequently co-occur in the same supermarket basket (also termed a ‘transaction'). This type of information can be of great interest for shopkeepers. For example, they could decide to place these items further apart, so the customer will follow a longer route through the store. Additionally, the pattern mining results may reveal other items that may be of use for the target population, which could then be suggestively placed in between the two co-occurring items to increase overall sales. Despite the seeming simplicity of the problem, the number of possible frequent itemsets rapidly explodes with larger datasets, making a brute-force search intractable. Nevertheless, more efficient algorithms have been developed to tackle this computationally demanding problem.
The application of frequent itemset mining is not restricted to market basket analysis. These techniques have proven their value in a wide range of knowledge extraction problems. In bioinformatics, typical applications include the interpretation of gene expression data [2], annotations [3], protein interaction networks [4] and biomolecular localization prediction [5]. Frequent itemset mining is typically used in bioinformatics to identify biologically relevant patterns that can be interpreted in a biological context.
The algorithms that have been developed for market basket type problems can often be readily applied to bioinformatics problems, as long as the biological problem is properly translated into the transactional input that the algorithms can accept. Equivalent to finding items that are often purchased together, a biological question may be to identify frequently co-occurring protein domains in a set of proteins. In this example, each protein represents a single transaction, equivalent to the market basket, with the domains being the items, equivalent to the products. The same class of algorithms can be applied to both of these analogous problems. In other cases, the conversion of biological data can be more challenging, due to for example the complex structure of many biological datasets, their often stochastic nature, the presence of missing values and scaling issues.
Methods to extract relevant frequent itemsets from transactional data have been extensively studied, and many efficient algorithms are available. They offer several advantages over other pattern detection methods, including the computational efficiency of the search and the intuitive interpretability of the extracted patterns. Frequent itemsets can furthermore be converted into rules that can be used in various downstream applications. A key factor that often hampers their application in bioinformatics lies not in the extraction of patterns itself, but rather in how they are subsequently ranked and filtered. For example, the most commonly used algorithm (Apriori [6]) is notorious for the redundancy in the itemsets it generates, and the number of patterns it finds rapidly explodes unless parameters are stringently controlled. Various metrics that define the interestingness of a pattern (support, lift, maximal entropy, etc) for subsequent ranking and filtering of retrieved patterns have been studied at theoretical and experimental level. Nevertheless, biological questions often require the definition of special task-specific interestingness metrics, in which (biological) domain knowledge is formalized.
The goal of this primer is to first explain the central concepts of frequent itemset mining and association rule generation. We then introduce a number of representative and popular algorithms and software frameworks. To conclude, we give an overview of successful bioinformatics applications and highlight the future challenges and opportunities in the use of these techniques for biological data interpretation.
DEFINITIONS
Some key terms used in frequent itemset mining have already been mentioned in the introduction. In this section, we explain and formalize these expressions to introduce the basic concepts of frequent itemset mining. A more in-depth introduction can be found in [7] and [8].
Frequent itemsets
Let 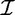 be the set of all possible items. A subset X = {i1 , … , ik} ⊆
be the set of all possible items. A subset X = {i1 , … , ik} ⊆ 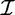 is called an itemset, or a k-itemset if it contains k items.
is called an itemset, or a k-itemset if it contains k items.
A transaction over 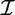 is a pair T = (tid, I), where tid is the transaction identifier and I is an itemset.
is a pair T = (tid, I), where tid is the transaction identifier and I is an itemset.
A set of transactions over 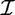 can be termed as a transaction database
can be termed as a transaction database
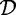 over
over 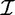 . We omit
. We omit 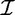 whenever it is clear from the context.
whenever it is clear from the context.
The support of an itemset X is the number of transactions that contain the itemset X:
An itemset is called frequent if its support is no less than a given minimal support threshold σ, with 0 ≤ σ ≤ |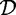 |. The collection of frequent itemsets in
|. The collection of frequent itemsets in 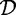 with respect to σ is denoted by:
with respect to σ is denoted by:
Frequent itemset mining is concerned with finding the set of itemsets 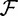 . Note that items can be any kind of attribute–value pairs; thus, they can also represent the absence of an item i2 in presence of another item i1 (negative occurrences) [9].
. Note that items can be any kind of attribute–value pairs; thus, they can also represent the absence of an item i2 in presence of another item i1 (negative occurrences) [9].
Association rules
Additionally, we can perform association rule mining. An association rule is an expression of the form X ⇒ Y, where X and Y are itemsets, and X ∩ Y = Ø. Such a rule expresses the association that if a transaction contains all items in X, then that transaction also contains all items in Y. X is called the body or antecedent, and Y is called the head or consequent of the rule.
The support of an association rule X ⇒ Y, is the support of X ∪ Y:
The confidence of an association rule X ⇒ Y is the conditional probability of having Y contained in a transaction, given that X is contained in that transaction:
The rule is called confident if its confidence exceeds a given minimal confidence threshold γ, with 0 ≤ γ ≤ 1. The collection of frequent and confident association rules in 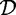 with respect to σ and γ is denoted by:
with respect to σ and γ is denoted by:
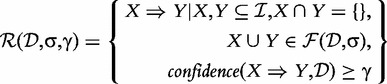 |
Association rule mining is concerned with finding the set of association rules 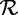 . Note that itemset mining is actually a special case of association rule mining. Every frequent itemset represents the trivial rule X ⇒ {}, which has the same support as the support of X and holds with 100% confidence. Association rule mining is typically the step conducted after the actual itemset mining, as the rules can be derived from the itemsets.
. Note that itemset mining is actually a special case of association rule mining. Every frequent itemset represents the trivial rule X ⇒ {}, which has the same support as the support of X and holds with 100% confidence. Association rule mining is typically the step conducted after the actual itemset mining, as the rules can be derived from the itemsets.
This notion of association rules is very general, and much research has been invested into constraint-association rule mining, which can efficiently limit the search to rules that satisfy constraints, such as rules having a negative consequent [10].
Interestingness measures
Some examples of interestingness measures have already been introduced, in particular the support and confidence measures. Additionally, several other interestingness measures have been proposed [11], with some potentially being better suited to handle large biological databases (e.g. [12]). However, support and confidence remain the two most widely used constraints.
Support is an important measure because a rule that has low support may occur simply by chance. Confidence, on the other hand, measures the reliability of the inference made by a rule.
Other frequently used measures include lift and coverage. The lift of an association rule X ⇒ Y is the ratio of the observed support for this association rule, to the expected support if X and Y were independent:
The coverage of an association rule X ⇒ Y measures how often the rule is applicable in the transaction database:
We can illustrate these definitions with a representative toy example. Figure 1 shows how association rules are generated out of transactions. The transactions are shown in circular boxes on the left. These transactions each support some (frequent) itemsets. The frequent itemsets with respect to a minimal support threshold of 2 are shown in squared boxes (itemsets with a lower support are omitted). Equivalently, association rules can be generated out of the frequent itemsets. The frequent and confident association rules with a support threshold of 2 and a confidence threshold of 50% are shown in octagonal boxes. Edges between the frequent itemsets and the association rules indicate which itemsets have been used to generate the association rules. Additionally, Table 1 presents an overview of interestingness measures for these association rules.
Figure 1:

Toy example to demonstrate how frequent itemsets and association rules can be derived from a series of transactions. Transactions are indicated by circular boxes, and are labeled as (tid, I), where tid is the transaction identifier and I = {i1, … , ik} is an itemset containing the items i1 to ik. Frequent itemsets are represented as a squared box, and association rules are shown as an octagonal box.
Table 1:
Measures related to the itemsets and association rules presented in Figure 1
| Rule | Support | Confidence | Lift | Coverage |
|---|---|---|---|---|
| {a} ⇒ {b} | 2 | 100% | 33% | 2 |
| {b} ⇒ {a} | 2 | 66% | 33% | 3 |
ALGORITHMS AND IMPLEMENTATIONS
Problem statement
A brute-force approach for association rule mining is to compute the support and confidence for every possible rule. This method is prohibitively expensive because the search space is exponential to the number of items occurring in the database. More specifically, for a set of items 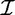 , 2
, 2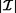 itemsets and 3
itemsets and 3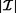 association rules can be generated [6]. Therefore, a common strategy is to divide the problem into two subtasks. First, all frequent itemsets are generated, after which all frequent and confident association rules are generated. Figure 1 illustrates these two subtasks intuitively. In the next section, we further elaborate on the algorithmic approaches to tackle both subtasks.
association rules can be generated [6]. Therefore, a common strategy is to divide the problem into two subtasks. First, all frequent itemsets are generated, after which all frequent and confident association rules are generated. Figure 1 illustrates these two subtasks intuitively. In the next section, we further elaborate on the algorithmic approaches to tackle both subtasks.
Algorithms for itemset and association rule mining
In the first subtask, all frequent itemsets are generated. Most algorithms for general itemset mining can be characterized based on two properties: their traversal of the search space, and their computation of support. In general, all itemset mining algorithms repeatedly generate relatively small collections of candidate frequent itemsets, count their supports and remove all itemsets that turn out to be infrequent. The most important property, also called the Apriori Property, is that all supersets of an infrequent itemset must also be infrequent. Hence, many itemsets can be pruned from the search space when one of their subsets is known to be infrequent.
Essentially, the search space traversal will be either a depth-first traversal of all candidate itemsets or a breadth-first traversal. In a breadth-first traversal, all itemsets of size k are iteratively generated, starting with k = 1. In a depth-first traversal, a recursive divide and conquer principle is followed. More specifically, for a selected item i, first, all frequent itemsets containing i are generated, after which all frequent itemsets not containing i are generated.
The chosen traversal strategy is typically closely connected to the size of the database and the computation of the support of all candidate itemsets. If the data do not fit in main (fast) memory, the supports are counted by considering all transactions one by one, testing for every candidate itemset whether it is contained in that transaction. Here, a breadth-first approach is typically used, such as in the standard Apriori [6] algorithm. However, many optimizations already exist for this algorithm, partitioning or sampling the data in such a way that they do fit in memory. In that case, a depth-first search is typically used. The support of an itemset is then computed by simply storing for each item the ids of transactions it is contained in, counting the size of the intersection of these sets for each item in the itemset. For example, this strategy is used in Eclat [13].
Again, a plethora of optimizations and variations exist, of which frequent pattern (FP)-growth [14] is one of the most common. It combines a depth-first search with a compressed memory-resident database.
After the generation of all frequent itemsets, the second subtask consists of the computation of all frequent and confident association rules. Essentially, each frequent itemset is divided into two parts, an antecedent and a consequent, for every possible combination, and the corresponding confidences are then computed.
Software for frequent itemset mining
A detailed discussion of each itemset mining algorithm is beyond the scope of this review. However, Table 2 presents, summarizes and compares some important characteristics of commonly used methods and provides a reference to software implementations when available.
Table 2:
Overview of popular frequent itemset mining algorithms and implementations
| Algorithm | Itemsets, subgraphs or rules | Context | License | Publication | Additional information or implementations |
|---|---|---|---|---|---|
| Anets | All (apriori), various threshold measures | Annotation mining | GNU GPL | 16 | http://sourceforge.net/projects/anets |
| AGM | All (apriori) | Subgraph mining | / | 57 | / |
| Apriori (Borgelt) | All | / | GNU GPL | 6, 94 | http://www.borgelt.net/apriori.html |
| Apriori (Goethals) | All | / | Research only | 6 | http://adrem.ua.ac.be/∼goethals/software/ |
| ARIA | All (apriori), various verifications | Annotation mining | / | 15 | http://pedant.gsf.de/ARIA/index.htm |
| Carpenter | Closed | Quantitative omics profiles | GNU GPL | 29 | http://www.borgelt.net/carpenter.html |
| CBA | All (apriori) | Classifiers | / | 67 | / |
| CMAR | All (FP-growth) | Classifiers | / | 68 | / |
| Cobbler | Closed | Quantitative omics profiles | / | 40 | / |
| CODENSE | Coherent dense subgraphs | Subgraph mining | Research only | 62 | http://zhoulab.usc.edu/CODENSE/ |
| COLL | All (apriori), chi-squared threshold pruning | Annotation mining | Open source | 3 | http://datadryad.org/resource/doi:10.5061/dryad.nr353 |
| COPS | All (FP-tree), score threshold | Biclustering | Contact authors | 18 | http://www.cos.uni-heidelberg.de/index.php/n.ha |
| CPAR | All | Classifiers | / | 69 | / |
| CPMine | All (eclat), machine learning | Structural patterns | / | 22 | / |
| DeBi | Maximal | Biclustering | Creative Commons 2.0 | 29 | http://www.molgen.mpg.de/∼serin/debi/main.html |
| Distiller | Closed | Quantitative omics profiles | Academic use only | 49 | http://homes.esat.kuleuven.be/∼kmarchal/Supplementary_Information_Lemmens_2008/startPage.html |
| Eclat (Borgelt) | All | / | GNU GPL | 13, 94 | http://www.borgelt.net/eclat.html |
| FESP | Emerging patterns | Classifiers | / | 92, 93 | / |
| Farmer | Closed | Quantitative omics profiles | Research only | 41 | http://www.sgi.com/tech/mlc/ |
| FSG | / | Subgraph mining | / | 58 | / |
| FPGrowth | All | / | GNU GPL | 14 | / |
| GenMax | Maximal | Quantitative omics profiles | Research only | 36 | http://www.cs.rpi.edu/∼zaki/www-new/pmwiki.php/ Software/Software |
| GenMiner | Closed | Quantitative omics profiles | Research only | 46 | http://keia.i3s.unice.fr/?Logiciels_et_Implémentations___GenMiner |
| gSpan | All | Subgraph mining | Internal research only | 59 | http://www.cs.ucsb.edu/∼xyan/software/gSpan.htm |
| KRIMP | Minimal descriptive length | Future work | 83 | http://adrem.ua.ac.be/∼jvreeken/prj/krimp/ | |
| MAFIA | Maximal | / | Contact authors | 95 | http://sourceforge.net/projects/himalaya-tools/files/ |
| MAGO | Multilevel association rules | Quantitative omics profiles | / | 47 | / |
| MaxConf | Closed | Quantitative omics profiles | Research only | 96 | https://bitbucket.org/tara/fpm/src/5813e782542b?at=default |
| MaxMiner | Maximal | Quantitative omics profiles | / | 35 | / |
| Min-Ex | δ-free itemsets | Quantitative omics profiles | / | 32 | / |
| MULE | Maximal frequent connected subgraph | Subgraph mining | Research only | 4 | http://compbio.case.edu/koyuturk/software/mule/ |
| NetCAR | Maximal frequent connected subgraph | Classifiers | Research only | 77 | http://bioinformatics.oxfordjournals.org/content/24/13/1523.long |
| PathFinder | Large | Subgraph mining | / | 64 | / |
| REMMAR | Shortest distance thresholding | Quantitative omics profiles | Research only | 48 | http://websystem.csie.ncku.edu.tw/REMMAR_Program.rar |
| TD-Close | Closed | Quantitative omics profiles | / | 42 | / |
| TopKRGs | Top-k | Quantitative omics profiles | / | 43 | / |
Several implementations presented in Table 2 can be run as stand-alone software. Additionally, data mining frameworks that allow frequent itemset mining exist for practical use, often with a graphical user interface and interactivity features. Table 3 shows a number of popular software frameworks, including their license and their corresponding references (when available).
Table 3:
Overview of software frameworks for frequent itemset mining
| Application name | Description | License | Publication | Available from |
|---|---|---|---|---|
| Arules | FIM toolbox in R | GNU GPL-2 | 84 | http://cran.r-project.org/web/packages/arules/index.html |
| ARtool | FIM toolbox for binary databases | GNU GPL | 97 | http://www.cs.umb.edu/∼laur/ARtool/ |
| KNIME Desktop | Data analytics platform | GNU GPL | 98 | http://www.knime.org/ |
| MIME | Interactive FIM toolbox | Research only | 99 | http://adrem.ua.ac.be/mime |
| Orange | Data analytics platform | GNU GPL-3 | 100 | http://orange.biolab.si/ |
| PyFIM | Python library | GNU LPL | 94 | http://www.borgelt.net/pyfim.html |
| Rapidminer | Data analytics platform | AGPL-3 | 101 | http://rapid-i.com/ |
| SPMF | FIM toolbox | GNU GPL-3 | / | http://www.philippe-fournier-viger.com/spmf/index.php |
| Weka | Machine learning library | GNU GPL | 102 | http://www.cs.waikato.ac.nz/ml/weka/ |
BIOINFORMATICS APPLICATIONS
Frequent itemset mining can be used to tackle a broad range of bioinformatics problems. For the purpose of providing a representative overview of potential applications, we discuss six bioinformatics subdomains in which these techniques have been successfully used.
Frequent annotation mining
Annotations of a molecular entity (such as a gene) describe certain properties (e.g. function or localization) by means of terms of a controlled vocabulary. They are crucial in many bioinformatics workflows. A useful application of frequent itemset mining is the prediction of novel annotations. Patterns of frequently co-occurring annotations derived with frequent itemset mining techniques can play an essential role in that task. Co-occurrence of annotations can be defined strictly, with each biomolecule corresponding to a transaction and each annotation term as an item. However, it can also be defined in terms of neighborhood, e.g. by considering which annotations frequently co-occur over pairs of biomolecules that undergo a physicochemical interaction (e.g. protein interactions). Figure 2 shows such an example of how frequent itemset mining can be used to extract co-occurring annotations from a network of annotated and interacting biomolecules. Derived associations could then be used to improve the unsupervised annotation of biomolecules [15].
Figure 2:

Mining for frequent co-occurrences in annotations. Annotations can be mapped to biological entities, such as interactions between biological molecules. As such, each transaction is composed of the transaction identifier (represents the interaction between both partners) and the items (the annotations corresponding to each of the biomolecules). Frequent itemset miners can then be used to uncover patterns of often co-occurring annotations and several interestingness measures can be computed (e.g. support). This information can then be interpreted by the researcher or used to create weighted protein networks [16].
Frequent itemset mining can also be used to identify relationships between various existing ontologies. For example, cross-ontology association rule mining can connect the biological process, cellular compartment and protein function subtrees within the Gene Ontology [3].
There are, however, some specific challenges in frequent annotation mining. Annotations will only frequently co-occur if the items are frequent, regardless of the hierarchical structure of the ontology. Inconsistencies in the level of specificity of the annotations of individual biomolecules can result in an apparently lower frequency in individual annotation terms, potentially leaving interesting patterns undetected. A solution for this problem is the explicit integration of the annotation structure into association networks [16].
Structural motif discovery
Structural patterns or motifs are frequently occurring combinations of structural properties in biomolecules (such as molecular sequences). Although these features are omnipresent and extremely diverse, the underlying conservation typically points to a functionally important role. As a consequence, motif discovery is an important and widely explored topic in bioinformatics. When structural features are transformed into transactions, frequent itemset mining can be used to discover combinations of structural features that occur more frequently than expected. Examples of frequent itemset mining-based motif discovery include transcription factor binding motifs [17, 18], splicing patterns [19], combinatorial patterns involved in histone modification [20] and even spatial motifs [21, 22]. Additional constraints can be used to mine for specific patterns, such as the spacing between the motifs in a sequence [23], the spatial proximity of amino acids in a 3D structure [24] and peptide binding to the major histocompatibility complex [25].
A simplified example of structural mining is the discovery of motifs in sequences surrounding a specific site, e.g. for a class of known post-translational modifications, as demonstrated in Figure 3. Biological sequences with the site of interest can be retrieved from public repositories and aligned with the common site as the central anchor point. All surrounding residues can then be given indexes to capture positional information relative to the site of interest. Each of these short sequence stretches can be considered as a transaction and the whole as a transaction database that can be mined for patterns. The resulting patterns indicate a degree of conservation and may be used to discriminate between classes.
Figure 3:

Visualization of structural pattern mining. Here the biological sequence of a domain on a biomolecule is processed with frequent itemset mining algorithms to identify conserved motifs. These motifs incorporate the underlying dependencies between the items in the form of the support value or other quality measures.
Frequent itemset mining has also been applied to aid in the alignment of 3D structures. For example, the Sequence Order Independent aLignment (SOIL) algorithm [26] uses frequent itemset mining to find subsets of amino acids that often spatially co-occur. Using frequent itemset mining in this case speeds up the protein structure alignment. This top-K itemset-based approach was competitive with other alignment methods and allowed for a more restrictive similarity measurement.
Pattern detection in quantitative ‘omics' profiles
Association rule mining has been extensively used for the analysis of quantitative molecular profiles. A popular application is biclustering, which is the discovery of sets of submatrices within a larger matrix. The stereotypical use case of biclustering in bioinformatics is the analysis of co-expressed genes (with measured expression values) from a dataset under a (sub)set of conditions. High-throughput techniques for genome-wide expression profiling have resulted in the availability of many gene expression matrices [27]. However, the analysis thereof is confounded by the size of the data. Studying gene co-expression often requires a condition selection strategy, as even genes under influence of a common regulator are not necessarily co-regulated under all conditions. The dimensionality of this problem rapidly limits the applicability of standard clustering approaches. An elegant solution is frequent itemset mining. The problem is then translated into the discovery of associations between the expression values of genes and (optionally) additional data sources [28]. While biclustering is not exclusively a frequent itemset mining problem, frequent itemset mining-based algorithms have been shown to perform equally or superior to various other methods [29]. For example, they have proven their value in the elucidation of disease mode-of-actions such as for HIV-1 [30] and exploration of protein complexes in cell lysates with blue native gel electrophoresis [31]. Before frequent itemset mining-based biclustering, continuous values are typically discretized [28], e.g. to a binary (up and down) or ternary (up, down and unchanged) format. Frequent itemset mining is applied to this converted dataset, so that each condition can be considered as a transaction containing all measured genes and their regulation direction. The problem is thus reduced to finding frequently occurring sets of genes with a specific regulation pattern [28]. A toy example is shown in Figure 4.
Figure 4:

From expression matrix to bicluster. Gene expression data are converted into a matrix and discretized into a regulation category. In this figure, there are three groups: up, down or unchanged. This matrix can then be formatted into a suitable format for frequent itemset miners (transactional layout) to generate biclusters or rules.
For more than a decade, association rule mining has been used to identify relationships in gene expression data [32, 33]. However, algorithms such as Apriori [6] have limitations: they tend to detect a large number of redundant patterns and suffer from poor scaling. These limitations have been partially addressed using post-processing methods [34] or by the introduction of modified algorithms. For example, the redundancy in itemsets can be decreased by ignoring irrelevant rules [34] or by limiting the search space to certain itemset classes, such as maximal itemsets [35, 36] or the highest scoring itemsets (top-K) [37]. Furthermore, the need for a discretization step can be circumvented, e.g. by using quantitative association rules based on half-spaces [38]. In addition, various row-enumeration strategies were found to be highly successful to find correlations in micro-array data [39–43]. Each of these methods has its advantages and issues, but makes distinct assumptions at the start. For example, some methods search for closed itemsets [39, 40], whereas others only consider the top-K results [43]. The itemset type also affects the analysis of the significance of the discovered patterns. For example, maximal itemset mining leads to a drastically reduced number of patterns but also results in the loss of information on the relative importance of the itemset subsets in relation to the dataset. As such, the support of the maximal itemset can be very close to the minimal threshold, while the relations between various items in this itemset are frequent. When in doubt, less restrictive methods such as closed itemset mining should also be explored.
The need to reduce the (often large) number of patterns to those that actually matter has led to a new generation of techniques that focus on biological importance, instead of pure database characteristics. Several methods take an integrative approach, in which correlations between co-regulated genes and external sources of information are considered. Most common are the incorporation of gene or pathway annotations [44, 45], regulatory network evidence, expression data or combinations thereof [2, 46–49]. However, defining the biological interestingness is still not trivial, and various derived measures have been proposed [50].
Frequent itemset-based exploration of single-nucleotide polymorphisms
Frequent itemset mining has also been used to identify strong associations between allelic combinations associated with diseases. An FP-based method was found to be suitable for the detection of strong interactive effects [51]. More recently, a scalable Apriori-based approach to identify discriminative patterns between high-order single-nucleotide polymorphisms (SNPs) and disease phenotypes was proposed [52]. Another method based on Apriori separates the search within the set attributes from the search between the set attributes, resulting in rules that were shown to be consistent with literature [53].
Millions of SNPs exist, with many of these showing correlated genotypes. This has led to the search for so-called tag SNPs that are subsets sufficient to infer the other SNPs from. Common methods suffer from various problems with larger chromosomes, thus becoming very memory-intensive and time-consuming [54]. FastTagger [54] incorporated frequent itemset mining to overcome several of these problems.
Subgraph mining in molecular networks
Network analysis is highly relevant for biological research. By understanding the functional interactions between processes and molecules ongoing in living organisms, a much deeper understanding of the organismal response can be obtained. It is nowadays a popular task in systems biology to identify biologically relevant subgraphs in these networks, e.g. to reveal underlying regulatory principles.
Finding structures in networks has been a long-standing question in data mining and has inspired the creation of several subgraph mining algorithms, some of which are based on frequent itemset mining. A major distinction between different approaches can be made according to whether subgraphs are searched in a single graph or in multiple graphs (Figure 5). Although algorithms to query a single biological network for its frequent subgraphs exist [55, 56], the most common and straightforward applications deal with multiple graphs. Traditional methods started as Apriori-based frequent substructure miners [57, 58]. These methods search for frequent subgraphs across multiple graphs instead in a way equivalent to searching frequent itemsets in a dataset of transactions. Although the core algorithm remains the same, the interestingness measures need to be retooled to the graph field. For example, support can be redefined as the number of graphs in the dataset containing a given subgraph. Non-Apriori-derived methods are often based on pattern growth. They iteratively attempt to add edges in every direction to frequent subgraphs, simulating a ‘grow out’ process [59–61].
Figure 5:

Gene interaction networks in mouse, human and rat as derived from String [12]. Frequent edges among these interaction networks can be extracted and presented as a frequent subgraph. Conserved subgraphs can have universal functional importance within the studied species.
The aforementioned methods are all capable of identifying substructures in a dataset, but biological networks pose additional challenges for conventional network mining approaches. Memory limitations are for example a typical issue [4]. The massive size of biological networks requires the use of heuristics to reduce the possible pattern space without information loss. Common approaches include the collapse of the different graphs to be analyzed into one or more summary graphs [62], which can then be mined for coherent dense subgraphs. Another approach is the reduction of each graph individually by collapsing nodes with identical labels into a single node [4]. Both methods can reduce noise and increase functional coherence of the patterns. To further reduce false patterns due to noise, weights can be added to the edges based on the reliability of the relation (e.g. based on experimental reproducibility) [63], or complementary types of experimental evidence (e.g. expression profiles, subcellular localization and sequence information) can be integrated [64].
Molecular interactions can also be represented as a transactional database for use with regular frequent itemset mining tools, where each edge is considered a transaction. For example, a protein-protein interaction (PPI) network can be converted into a set of transactions to detect rules that provide novel insights into the functional annotation of the network [65]. In a related analysis [66], additional features (e.g. subcellular localization information, motifs, various annotation types) were added to the items present in the transactions before rule mining.
Frequent itemsets for classification
Patterns and, in particular, association rules can be used as a foundation to construct a classifier. Several popular implementations exist [67–69]. They all rely on the philosophy that if attributes frequently appear together, there must be an underlying relation between them and this relation can be used for classification.
Machine learning techniques such as support vector machines (SVM) [70] largely function as a black box. The underlying models are often not interpretable in regard to the predictions they make. Association rule-based classifiers overcome this problem. They are more transparent about the reasoning behind their predictions, as they provide knowledge-based explanative rules and thus serve as a ‘white-box’ model [71]. Association rule-based classifiers have achieved accuracies equivalent to traditional SVM methods for common biological problems [71]. This transparency has enabled a range of studies that used frequent itemset mining to generate rules for classification [72–74].
Generating rules for classification is not a trivial task. Normally, a transactional database layout is used for mining and rules for classification are of the form X ⇒ Ci, with X being an itemset of observables and Ci being the class label. Thus, the data need to be transformed, so that each item represents a pair of attribute and value, together with a class label (e.g. P53, downregulated ⇒ cancer). A common example is the classification of sample types (e.g. tumor and healthy) with gene expression data [37, 72, 73]. For this purpose, expression values are discretized, and association rules are generated from maximal itemsets [72]. Furthermore, any other discrete or discretizable feature can be used, from cell properties [74] to protein–protein interactions [75]. Typically, only the rules that exceed a defined minimal support and confidence will be used for classification.
The combination of association rule mining with other classification methods, such as SVMs, can significantly increase their accuracy [76]. Association rule-based methods are also still being improved, e.g. by incorporation of a phylogenetic co-occurrence graph [77] or by speeding up rule detection with an ANT-based optimization [78].
FUTURE DIRECTIONS
Frequent itemset mining techniques can be powerful and elegant tools to extract meaningful patterns from biological data. Nevertheless, we would like to highlight some remaining challenges. Addressing these challenges would benefit further adoption of frequent itemset mining approaches by the bioinformatics community.
First, the definition of interestingness is very dependent on the biological problem at hand, and there are no simple guidelines to develop an appropriate interestingness metric for a new problem. Existing measures such as support, lift, coverage, occupancy [79] and entropy [80] give information about the dataset, but are not guaranteed to identify biologically relevant patterns. Another measure is the minimum improvement constraint [81], which only retains associations that have stronger correlations than their generalizations. This method rejects many unproductive and redundant rules [82]. In addition, support-based measures prioritize patterns that occur more often, but these patterns can be biologically trivial. For example, the detection of a frequent co-occurrence of the ATP-binding domain and a kinase domain while mining kinase structures offers little novel insight, whereas more interesting co-occurring domains will receive a much lower score. There is a clear need for measures that quantify biological interestingness.
A second challenge lies in the definition of a threshold that patterns need to exceed before they are considered frequent. If set too low, the number of patterns explodes, making proper interpretation impossible. If the threshold is too high, interesting less-frequent patterns might be missed. Methods such as Top-K mining avoid the problem of defining the lower threshold entirely [82].
A third problem is related to the heuristics used by the algorithms. Calculating all possible itemsets or association rules is not computationally efficient for large datasets and rarely useful for life sciences. Furthermore, generating all possible patterns often results in lists mostly comprising redundant patterns. Various heuristics have been proposed to more efficiently traverse the solution space and better capture the characteristics of the dataset in the shape of informative patterns. For example, Krimp [83] tries to find the best compression for a dataset and top-K mining identifies the top K scoring itemsets. Nevertheless, these theoretically elegant heuristics do not necessarily reflect biological foundations.
Another aspect that is relevant for the bioinformatics community, but has not yet been fully explored, is the visualization of patterns. In general, table-based, matrix-based and graph-based visualization methods exist. Commonly known examples are arulesviz [84], FPViz [85] and WiFIsViz [86], which are all available as R packages. Although these visualizations allow deeper understanding of the data, there is room for future work.
Last but not least, pattern mining and association rule discovery is vulnerable to false discoveries, as it searches the entire sample space for frequent co-occurrences. Owing to the massive scale, it is prone to find relations that are true in the sample set, but do not necessarily hold any relation to the actual underlying process in the dataset and may identify uninteresting rules, with many type I errors [82]. Several solutions to these problems have been proposed, of which the most popular can be attributed to two families: family-wise error rate, such as the Bonferroni correction, and false discovery rate [87]. Owing to the difficulties inherent to family-wise error rate, control of the false discovery rate has become increasingly popular. Some examples of false discovery rate methods are the Benjamini–Liu [88] and Benjamini–Hochberg [89] procedures. Various permutation-based and holdout approaches also exist [90]. Shrinkage estimates and Bayesian smoothing have also been proposed to limit overestimation of measures, such as support, and to reduce type I errors [82].
Key Points.
Frequent itemset mining (and derived association rule mining) is a group of pattern mining techniques designed to identify elements that frequently co-occur, like sets of products that often end up together in the supermarket basket.
Owing to the straightforward interpretability of the resulting patterns, frequent itemset mining techniques are powerful tools to extract relevant knowledge from complex biological data.
The flexibility of frequent itemset mining techniques is demonstrated by the diverse range of bioinformatics problems they have been applied to, including annotation mining, structural motif discovery, subgraph detection, SNP analysis and biclustering of expression profiles. Furthermore, they can be used as input to construct classifiers.
FUNDING
This work was supported by the Research Foundation-Flanders (FWO) [grant number: G.0903.13N]; the agency for Innovation by Science and Technology (IWT) [grant number 120025]; and the University of Antwerp [BOF docpro to S.N.; BOF ID to T.N.V.].
Biographies
Stefan Naulaerts is a PhD student in bioinformatics at the University of Antwerp. He works on the integration and mining of ‘omics’ data, often using frequent itemset mining.
Pieter Meysman is a post-doctoral researcher in bioinformatics at the University of Antwerp. His research focuses on the application of data mining algorithms for pattern discovery in transcriptomics and evolutionary genomics.
Wout Bittremieux is a PhD student in bioinformatics at the University of Antwerp. His research focuses on using data mining techniques in mass spectrometry-based proteomics.
Trung Nghia Vu is a final year PhD student at the University of Antwerp. His research interests include data mining on mass spectrometry-based proteomics and metabolomics data.
Wim Vanden Berghe is Professor in Epigenetics at the University of Antwerp. His research interest focuses on nutritional and phytomedicinal epigenetic mechanisms in cancer, neuroinflammation and cardiovascular diseases.
Bart Goethals is a professor in data mining at the University of Antwerp. His research focuses on data mining, data science and big data analytics. He has won several awards in the data mining field.
Kris Laukens is coordinator of the biomedical informatics research center and professor at the University of Antwerp. His research lies in the application of data mining to complex life science data.
References
- 1.Agrawal R, Imieliński T, Swami A. Proceedings of the 1993 ACM SIGMOD International Conference on Management of Data, Washington, DC, USA, 1993. Vol. 22. New York: ACM; Mining association rules between sets of items in large databases; pp. 207–16. [Google Scholar]
- 2.Carmona-Saez P, Chagoyen M, Rodriguez A, et al. Integrated analysis of gene expression by association rules discovery. BMC Bioinformatics. 2006;7:54. doi: 10.1186/1471-2105-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manda P, Ozkan S, Wang H, et al. Cross-ontology multi-level association rule mining in the gene ontology. PLoS One. 2012;7:e47411. doi: 10.1371/journal.pone.0047411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koyutürk M, Kim Y, Subramaniam S, et al. Detecting conserved interaction patterns in biological networks. J Comput Biol. 2006;13:1299–322. doi: 10.1089/cmb.2006.13.1299. [DOI] [PubMed] [Google Scholar]
- 5.Yoon Y, Lee GG. Subcellular localization prediction through boosting association rules. IEEE/ACM Trans Comput Biol Bioinform. 2012;9:609–18. doi: 10.1109/TCBB.2011.131. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal R, Srikant R. Fast algorithms for mining association rules. In: Bocca JB, Jarke M, Zaniolo C, editors. Proceedings of the 20th VLDB Conference. Santiago: Morgan Kaufman; 1994. pp. 487–99. [Google Scholar]
- 7.Goethals B. Frequent Set Mining. In: Maimon O, Rokach L, editors. The Data Mining and Knowledge Discovery Handbook. Heidelberg: Springer Berlin; 2010. pp. 321–38. [Google Scholar]
- 8.Tan P-N, Steinbach M, Kumar V. Introduction to Data Mining. Addison-Wesley: Boston; 2005. Chapter 6. Association analysis: basic concepts and algorithms; p. 769. [Google Scholar]
- 9.Antonie ML, Zaïane OR. Mining positive and negative association rules: an approach for confined rules. In: Boulicaut JF, Esposito F, Giannotti F, Pedreschi D, editors. Knowledge Discovery in Databases: PKDD 2004. Heidelberg: Springer Berlin; 2004. pp. 27–38. [Google Scholar]
- 10.Besson J, Boulicaut JF, Guns T, Nijssen S. Generalizing itemset mining in a constraint programming setting. In: Džeroski S, Goethals B, Panov P, editors. Inductive Databases and Constraint-Based Data Mining. Heidelberg: Springer Berlin; 2010. pp. 107–26. [Google Scholar]
- 11.Tan P-N, Kumar V, Srivastava J. Proceedings of the 8th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Edmonton, Alberta, Canada, 2002. New York: ACM; Selecting the right interestingness measure for association patterns; pp. 32–41. [Google Scholar]
- 12.Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012;41:D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaki M, Parthasarathy S, Ogihara M, Li W. Proceedings of the 3rd International Conference on Knowledge Discovery and Data Mining (KDD). Newport Beach, CA, USA, 1997. Palo Alto, CA: AAAI Press; New algorithms for fast discovery of association rules; pp. 283–6. [Google Scholar]
- 14.Han J, Pei J, Yin Y, et al. Mining frequent patterns without candidate generation: a frequent-pattern tree approach. Data Min Knowl Discov. 2004;8:53–87. [Google Scholar]
- 15.Artamonova II, Frishman G, Gelfand MS, et al. Mining sequence annotation databanks for association patterns. Bioinformatics. 2005;21:iii49–57. doi: 10.1093/bioinformatics/bti1206. [DOI] [PubMed] [Google Scholar]
- 16.Karpinets TV, Park BH, Uberbacher EC. Analyzing large biological datasets with association networks. Nucleic Acids Res. 2012;40:e131. doi: 10.1093/nar/gks403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung K-S, Wong K-C, Chan T-M, et al. Discovering protein–DNA binding sequence patterns using association rule mining. Nucleic Acids Res. 2010;38:6324–37. doi: 10.1093/nar/gkq500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ha N, Polychronidou M, Lohmann I. COPS: detecting co-occurrence and spatial arrangement of transcription factor binding motifs in genome-wide datasets. PLoS One. 2012;7:e52055. doi: 10.1371/journal.pone.0052055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J, Zhao S, Heber S. Proceedings of the 45th Annual Southeast Regional Conference, Winstom-Salem, NC, USA, 2007. New York: ACM; Finding association rules of cis-regulatory elements involved in alternative splicing; pp. 232–37. [Google Scholar]
- 20.Tweedie-Cullen RY, Brunner AM, Grossmann J, et al. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS One. 2012;7:e36980. doi: 10.1371/journal.pone.0036980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaki MJ, Jin S, Bystroff C. Mining residue contacts in proteins using local structure predictions. Trans Sys Man Cyber Part B. 2003;33:789–801. doi: 10.1109/TSMCB.2003.816916. [DOI] [PubMed] [Google Scholar]
- 22.Aung Z, Tan KL. Proceedings of the 4th IEEE symposium on Bioinformatics and Bioengeering (BIBE 2004), Taichung, Taiwan, 2004. Washington, DC: IEEE Computer Society; Automatic protein structure classification through structural fingerprinting; pp. 508–15. [Google Scholar]
- 23.Icev A, Ruiz C, Ryder EF. Proceedings of the 3rd ACM SIGKDD Workshop on Data Mining in Bioinformatics (BIOKDD 2003), Washington, DC, USA, 2003. New York: ACM; Distance-Enhanced Association Rules for Gene Expression. [Google Scholar]
- 24.Zhou C, Meysman P, Cule B, et al. Proceedings of the 12th International Workshop on Data Mining in Bioinformatics (BIOKDD 2013), Chicago, IL, 2013. Mining spatially cohesive itemsets in protein molecular structures. accepted. New York: ACM. [Google Scholar]
- 25.Meydan C, Out HH, Sezerman OU. Prediction of peptides binding to MHC class I and II alleles by temporal motif mining. BMC Bioinformatics. 2013;14(Suppl 2):S13. doi: 10.1186/1471-2105-14-S2-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siu WY, Mamoulis N, Yiu SM, Chan HL. A data-mining approach for multiple structural alignment of proteins. Bioinformation. 2011;4:366–70. doi: 10.6026/97320630004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez FJ, Blanco A, Garcia F, et al. Fuzzy association rules for biological data analysis: a case study on yeast. BMC Bioinformatics. 2008;9:107. doi: 10.1186/1471-2105-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alves R, Rodriguez-Baena DS, Aguilar-Ruiz JS. Gene association analysis: a survey of frequent pattern mining from gene expression data. Brief Bioinform. 2010;11:210–24. doi: 10.1093/bib/bbp042. [DOI] [PubMed] [Google Scholar]
- 29.Serin A, Vingron M. DeBi: Discovering differentially expressed biclusters using a frequent itemset approach. Algorithms Mol Biol. 2011;6:18. doi: 10.1186/1748-7188-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay A, Maulik U, Bandyopadhyay S. A novel biclustering approach to association rule mining for predicting HIV-1–human protein interactions. PLoS One. 2013;7:e32289. doi: 10.1371/journal.pone.0032289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Remmerie N, de Vijlder T, Valkenborg D, et al. Unraveling tobacco BY-2 protein complexes with BN PAGE/LCMS/MS and clustering methods. J Proteomics. 2011;74:1201–17. doi: 10.1016/j.jprot.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Becquet C, Blachon S, Jeudy B, et al. Strong-association-rule mining for large-scale gene-expression data analysis: a case study on human SAGE data. Genome Biol. 2002;3:research0067. doi: 10.1186/gb-2002-3-12-research0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Creighton C, Hanash S. Mining gene expression databases for association rules. Bioinformatics. 2003;19:79–86. doi: 10.1093/bioinformatics/19.1.79. [DOI] [PubMed] [Google Scholar]
- 34.Tuzhilin A. Proceedings of the 8th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Edmonton, Alberta, Canada, 2002. New York: ACM; Handling very large numbers of association rules in the analysis of microarray data. proc; pp. 23–6. [Google Scholar]
- 35.Bayardo RJ. Proceedings of the 1998 ACM SIGMOD International Conference on Management of Data, Seattle, WA, USA, 1998. Vol. 27. New York: ACM; Efficiently mining long patterns from databases; pp. 85–93. [Google Scholar]
- 36.Gouda K, Zaki MJ. GenMax: an efficient algorithm for mining maximal frequent itemsets. Data Min Knowl Discov. 2005;11:223–42. [Google Scholar]
- 37.Cai R, Hao Z, Wen W, et al. Kernel based gene expression pattern discovery and its application on cancer classification. Neurocomputing. 2010;73:2562–70. [Google Scholar]
- 38.Georgii E, Richter L, Rückert U, et al. Analyzing microarray data using quantitative association rules. Bioinformatics. 2005;11:123–9. doi: 10.1093/bioinformatics/bti1121. [DOI] [PubMed] [Google Scholar]
- 39.Pan F, Cong G, Tung AK, et al. Proceedings of the 9th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Washington, DC, USA, 2003. New York: ACM; Carpenter: finding closed patterns in long biological datasets; pp. 637–42. [Google Scholar]
- 40.Pan F, Tung A, Cong G, et al. Proceedings of the 16th International Conference on Scientific and Statistical Database Management SSDBM, Santorini Island, Greece, 2004. Washington, DC: IEEE Computer Society; COBBLER: combining column and row enumeration for closed pattern discovery; pp. 21–30. [Google Scholar]
- 41.Cong G, Tung AK, Xu X, et al. Proceedings of the ACM SIGMOD International Conference on Management of Data, Paris, France, 2004. New York: ACM; FARMER: finding interesting rule groups in microarray datasets; pp. 143–54. [Google Scholar]
- 42.Liu H, Han J, Xin D, et al. Proceedings of the 22nd international conference on Data Engineering, Long Beach, California, USA, 2006. Washington, DC: IEEE Computer Society; Top-Down Mining of Interesting Patterns from Very High Dimensional Data; p. 114. [Google Scholar]
- 43.Cong G, Tan K, Tung AK, et al. Proceedings of the ACM SIGMOD International Conference on Management of Data, Baltimore, MD, USA, 2005. New York: ACM; Mining top-K covering rule groups for gene expression data; pp. 670–81. [Google Scholar]
- 44.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez R, Pasquier N, Pasquier C. Mining association rule bases from integrated genomic data and annotations. In: Masulli F, Tagliaferri R, Verkhivker GM, editors. Computational Intelligence Methods for Bioinformatics and Biostatistics. Heidelberg: Springer Berlin; 2009. pp. 78–90. [Google Scholar]
- 47.Tseng VS, Yu H-H, Yang S-C. Efficient mining of multilevel gene association rules from microarray and gene ontology. Inf Syst Front. 2009;11:433–47. [Google Scholar]
- 48.Liu Y-C, Cheng C-P, Tseng VS. Discovering relational-based association rules with multiple minimum supports on microarray datasets. Bioinformatics. 2011;27:3142–8. doi: 10.1093/bioinformatics/btr526. [DOI] [PubMed] [Google Scholar]
- 49.Lemmens K, De Bie T, Dhollander T, et al. The condition-dependent transcriptional network in Escherichia coli. Ann N Y Acad Sci. 2009;1158:29–35. doi: 10.1111/j.1749-6632.2008.03746.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang M, Wu S, Cai R. Two novel interestingness measures for gene association rule mining. Neural Comput Appl. 2013;23:835–41. [Google Scholar]
- 51.Ma L, Assimes T, Asadi N, et al. An almost exhaustive search-based sequential permutation method for detecting epistasis in disease association studies. Genet Epidemiol. 2010;34:434–43. doi: 10.1002/gepi.20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang G, Haznadar M, Wang W, et al. High-order SNP combinations associated with complex diseases: efficient discovery, statistical power and functional interactions. PLoS One. 2012;7:e33531. doi: 10.1371/journal.pone.0033531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shoemaker CA, Ruiz C. Association rule mining algorithms for set-valued data. In: Liu J, Cheung Y, Yin H, editors. Intelligent Data Engineering and Automated Learning. Heidelberg: Springer Berlin; 2003. pp. 669–76. [Google Scholar]
- 54.Liu G, Wang Y, Wong L. FastTagger: an efficient algorithm for genome-wide tag SNP selection using multi-marker linkage disequilibrium. BMC Bioinformatics. 2010;11:66. doi: 10.1186/1471-2105-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Leemput K, Verschoren A. Modeling networks as probabilistic sequences of frequent subgraphs. http://win.ua.ac.be/∼adrem/bibrem/pubs/MLSB08.pdf (16 October 2013, date last accessed) [Google Scholar]
- 56.Bringmann B, Nijssen S. What Is Frequent in a Single Graph? In: Washio T, Suzuki E, Ting KM, Inokuchi A, editors. Advances in Knowledge Discovery and Data Mining. Heidelberg: Springer Berlin; 2008. pp. 858–63. [Google Scholar]
- 57.Inokuchi A, Washio T, Motoda H. An Apriori-based algorithm for mining frequent substructures from graph data. Princ Data Min Knowl Discov. 2000;1910:13–23. [Google Scholar]
- 58.Kuramochi M, Karypis G. Proceedings of the 2001 IEEE International Conference on Data Mining, San Jose, California, USA, 2001. Washington, DC: IEEE Computer Society; Frequent Subgraph Discovery; pp. 313–20. [Google Scholar]
- 59.Yan X, Han J. Proceedings of the 2002 IEEE International Conference on Data Mining, Maebashi City, Japan, 2002. Washington, DC: IEEE Computer Society; gSpan: graph-based substructure pattern mining; pp. 721–4. [Google Scholar]
- 60.Yan X, Han J. Proceedings of the 9th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Washington, DC, USA, 2003. New York: ACM; CloseGraph: mining closed frequent graph patterns; pp. 286–95. [Google Scholar]
- 61.Nijssen S, Kok JN. The gaston tool for frequent subgraph mining. Electron Notes Theor Comput Sci. 2005;127:77–87. [Google Scholar]
- 62.Hu H, Yan X, Huang Y, et al. Mining coherent dense subgraphs across massive biological networks for functional discovery. Bioinformatics. 2005;21:213–21. doi: 10.1093/bioinformatics/bti1049. [DOI] [PubMed] [Google Scholar]
- 63.Pandey G, Garg T, Steinbach M, et al. Proceedings of the 13th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Jose, CA, USA, 2007. New York: ACM; Association analysis-based transformations for protein interaction networks: a function prediction case study; pp. 540–9. [Google Scholar]
- 64.Bebek G, Yang J. PathFinder: mining signal transduction pathway segments from protein-protein interaction networks. BMC Bioinformatics. 2007;8:335. doi: 10.1186/1471-2105-8-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Besemann C, Denton A, Yekkirala A, et al. Proceedings of the 4th Workshop on Data Mining in Bioinformatics (SigKDD 2004) Toronto, ON: ACM; 2004. Differential association rule mining for the study of protein-protein interaction networks; pp. 72–80. [Google Scholar]
- 66.Oyama T, Kitano K, Satou K, et al. Extraction of knowledge on protein–protein interaction by association rule discovery. Bioinformatics. 2002;18:705–14. doi: 10.1093/bioinformatics/18.5.705. [DOI] [PubMed] [Google Scholar]
- 67.Liu B, Hsu W, Ma Y. Proceedings of the 4th International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 1998. Palo Alto, CA: AAAI Press; Integrating classification and association rule mining; pp. 335–40. [Google Scholar]
- 68.Li W, Han J, Pei J. Proceedings of the IEEE international conference on Data Mining (ICDM 2001). San Jose, CA, USA, 2001. Washington, DC: IEEE Computer Society; CMAR: accurate and efficient classification based on multiple class-association rules; pp. 369–76. [Google Scholar]
- 69.Yin X, Han J. Proceedings of the SIAM International Conference on Data Mining (SDM'03). San Francisco, CA, USA, 2003. Philadelphia: SIAM; CPAR: classification based on predictive association rules; pp. 331–5. [Google Scholar]
- 70.Vapnik VN. The nature of statistical learning theory. In: Jordan M, Lawless JF, Lauritzen SL, editors. Statistics for Engineering and Information Science. New York: Springer New York; 1995. [Google Scholar]
- 71.He J, Hu H, Chen B, et al. Rule extraction from SVM for protein structure prediction. Rule Extr Support Vector Mach. 2008;80:227–52. [Google Scholar]
- 72.Giugno R, Pulvirenti A, Cascione L, et al. MIDClass: Microarray Data Classification by association rules and gene expression intervals. PLoS One. 2013;8:e69873. doi: 10.1371/journal.pone.0069873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Antonie L, Bessonov K. Proceedings of the 2011 ACM Symposium on Applied Computing. Taichung, Taiwan, 2011. New York: ACM; Classifying microarray data with association rules; pp. 94–9. [Google Scholar]
- 74.Karabatak M, Ince MC. An expert system for detection of breast cancer based on association rules and neural network. Expert Syst Appl. 2009;36:3465–9. [Google Scholar]
- 75.Park SH, Reyes JA, Gilbert DR, et al. Prediction of protein-protein interaction types using association rule based classification. BMC Bioinformatics. 2009;10:36. doi: 10.1186/1471-2105-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tang Y, Jin B, Zhang Y-Q. Granular support vector machines with association rules mining for protein homology prediction. Artif Intell Med. 2005;35:121–34. doi: 10.1016/j.artmed.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 77.Tamura M, D’haeseleer P. Microbial genotype–phenotype mapping by class association rule mining. Bioinformatics. 2008;24:1523–29. doi: 10.1093/bioinformatics/btn210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He Y, Hui SC. Exploring ant-based algorithms for gene expression data analysis. Artif Intell Med. 2009;47:105–19. doi: 10.1016/j.artmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 79.Tang L, Zhang L, Luo P, et al. Proceedings of the 21st ACM international conference on Information and Knowledge Management. Maui, Hawaii, 2012. New York: ACM; Incorporating occupancy into frequent pattern mining for high quality pattern recommendation (CIKM’12) pp. 75–84. [Google Scholar]
- 80.Tatti N, Mampaey M. Using background knowledge to rank itemsets. Data Min Knowl Discov. 2010;21:293–309. [Google Scholar]
- 81.Bayardo JR, Agrawal R, Gunopulos D. Constraint-based rule mining in large, dense databases. Data Min Knowl Discov. 2000;4:217–40. [Google Scholar]
- 82.Webb GI. Discovering significant patterns. Mach Learn. 2007;68:1–33. [Google Scholar]
- 83.Vreeken J, van Leeuwen M, Siebes A. Krimp: mining itemsets that compress. Data Min Knowl Discov. 2011;23:169–214. [Google Scholar]
- 84.Hahsler M, Chelluboina S, Hornik K, et al. The arules R-package ecosystem: analyzing interesting patterns from large transaction data sets. J Mach Learn Res. 2011;12:2021–5. [Google Scholar]
- 85.Leung CK-S, Carmichael CL. Proceedings of the ACM SIGKDD Workshop on Visual Analytics and Knowledge Discovery: Integrating Automated Analysis with Interactive Exploration (VAK'09), Paris, France. New York: ACM; 2009. FpViz: a visualizer for frequent pattern mining; pp. 30–9. [Google Scholar]
- 86.Leung CK-S, Irani PP, Carmichael CL. Proceedings of the eighth IEEE international conference on Data Mining (ICDM’08), Pisa, Italy, 2008. Washington, DC: IEEE Computer Society; WiFIsViz: Effective Visualization of Frequent Itemsets; pp. 875–80. [Google Scholar]
- 87.Lallich S, Teytaud O, Prudhomme E. Association rule interestingness: measure and statistical validation. In: Guillet F, Hamilton HJ, editors. Quality Measures in Data Mining. Vol. 43. Heidelberg: Springer Berlin; 2007. pp. 251–75. [Google Scholar]
- 88.Benjamini Y, Liu W. A step-down multiple hypotheses testing procedure that controls the false discovery rate under independence. J Stat Planning Inf. 1999;82:163–70. [Google Scholar]
- 89.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 90.Liu G, Zhang H, Wong L. Controlling false positives in association rule mining. Proc Vldb Endow. 2011;5:145–56. [Google Scholar]
- 91.Dong G, Li J. Proceedings of the fifth ACM SIGKDD international conference on Knowledge discovery and data mining (KDD'99), San Diego, CA, USA. New York: ACM; 1999. Efficient Mining of Emerging Patterns: Discovering Trends and Differences; pp. 43–52. [Google Scholar]
- 92.Bailey J, Manoukian T, Ramamohanarao K. Fast Algorithms for Mining Emerging Patterns. Princ Data Min Knowl Discov. 2002;2431:39–50. [Google Scholar]
- 93.Chen X, Chen J. Emerging patterns and classification algorithms for DNA sequence. J Softw. 2011;6:6. [Google Scholar]
- 94.Borgelt C. Frequent item set mining. Wiley Interdisciplinary Reviews. Data Min Knowl Discov. 2012;2:437–56. [Google Scholar]
- 95.Burdick D, Calimlim M, Gehrke J. Proceedings of the 17th International Conference on Data Engineering, Heidelberg, Germany, 2001. Washington, DC: IEEE Computer Society; MAFIA: A Maximal Frequent Itemset Algorithm for Transactional Databases; pp. 443–52. [Google Scholar]
- 96.McIntosh T, Chawla S. High confidence rule mining for microarray analysis. IEEE/ACM Trans Comput Biol Bioinformatics. 2007;4:611–23. doi: 10.1109/tcbb.2007.1050. [DOI] [PubMed] [Google Scholar]
- 97.Cristofor L, Simovici D. Proceedings of the 2002 IEEE International Conference on Data Mining, Maebashi City, Japan, 2002. Washington, DC: IEEE Computer Society; Generating an informative cover for association rules; pp. 597–600. [Google Scholar]
- 98.Berthold MR, Cebron N, Dill F, et al. KNIME—the Konstanz information miner: version 2.0 and beyond. SIGKDD Explor Newslett. 2009;11:26–31. [Google Scholar]
- 99.Goethals B, Moens S, Vreeken J. Proceedings of the 2011 European Conference on Machine Learning and Knowledge Discovery in Databases - Volume Part III, Athens, Greece, 2011. Heidelberg: Springer Berlin; MIME: a framework for interactive visual pattern mining; pp. 634–7. [Google Scholar]
- 100.Demšar J, Zupan B, Leban G, Curk T. Orange: from experimental machine learning to interactive data mining. In: Boulicaut JF, et al., editors. Knowledge Discovery in Databases (PKDD’04) Heidelberg: Springer Berlin; 2004. pp. 537–9. [Google Scholar]
- 101.Mierswa I, Wurst M, Klinkenberg R, et al. YALE: rapid prototyping for complex data mining tasks. In: Ungar L, Craven M, Gunopulos D, Eliassi-Rad T, editors. Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD’06), Philadelphia, PA, USA. New York: ACM; 2006. pp. 935–40. [Google Scholar]
- 102.Hall M, Frank E, Holmes G, et al. The WEKA data mining software: an update. SIGKDD Explor Newsl. 2009;11:10–8. [Google Scholar]