

Significance
MicroRNAs (miRNAs) are small ∼22-nt RNAs that are important regulators of posttranscriptional gene expression. Since their initial discovery, they have been shown to be involved in many cellular processes, and their misexpression is associated with disease etiology. Currently, nearly 2,800 human miRNAs are annotated in public repositories. A key question in miRNA research is how many miRNAs are harbored by the human genome. To answer this question, we examined 1,323 short RNA sequence samples and identified 3,707 novel miRNAs, many of which are human-specific and tissue-specific. Our findings suggest that the human genome expresses a greater number of miRNAs than has previously been appreciated and that many more miRNA molecules may play key roles in disease etiology.
Keywords: microRNAs, isomIRs, noncoding RNA, RNA sequencing, transcriptome
Abstract
Two decades after the discovery of the first animal microRNA (miRNA), the number of miRNAs in animal genomes remains a vexing question. Here, we report findings from analyzing 1,323 short RNA sequencing samples (RNA-seq) from 13 different human tissue types. Using stringent thresholding criteria, we identified 3,707 statistically significant novel mature miRNAs at a false discovery rate of ≤0.05 arising from 3,494 novel precursors; 91.5% of these novel miRNAs were identified independently in 10 or more of the processed samples. Analysis of these novel miRNAs revealed tissue-specific dependencies and a commensurate low Jaccard similarity index in intertissue comparisons. Of these novel miRNAs, 1,657 (45%) were identified in 43 datasets that were generated by cross-linking followed by Argonaute immunoprecipitation and sequencing (Ago CLIP-seq) and represented 3 of the 13 tissues, indicating that these miRNAs are active in the RNA interference pathway. Moreover, experimental investigation through stem-loop PCR of a random collection of newly discovered miRNAs in 12 cell lines representing 5 tissues confirmed their presence and tissue dependence. Among the newly identified miRNAs are many novel miRNA clusters, new members of known miRNA clusters, previously unreported products from uncharacterized arms of miRNA precursors, and previously unrecognized paralogues of functionally important miRNA families (e.g., miR-15/107). Examination of the sequence conservation across vertebrate and invertebrate organisms showed 56.7% of the newly discovered miRNAs to be human-specific whereas the majority (94.4%) are primate lineage-specific. Our findings suggest that the repertoire of human miRNAs is far more extensive than currently represented by public repositories and that there is a significant number of lineage- and/or tissue-specific miRNAs that are uncharacterized.
MicroRNAs (miRNAs) are small, single-stranded RNAs with a length of ∼22 nt that are typically derived from endogenous hairpin transcripts. MiRNAs interact with their targeted RNA in a sequence-dependent manner (1, 2), thereby functioning as posttranscriptional regulators of gene expression. Regulation of the targeted RNAs is achieved through several mechanisms, including translational inhibition (3), disruption of cap–tail interactions (4, 5), and exonuclease-mediated mRNA degradation (6, 7).
Originally believed to regulate messenger RNAs (mRNAs) solely through interactions with the 3′ untranslated region (3′ UTR) (1), miRNAs are now known to have a very broad set of targets. These targets include loci in the protein-coding region of mRNAs (8–13), 5′ UTRs (14), intronic and intergenic transcripts (15, 16), and other non–protein-coding RNAs (ncRNAs) (17, 18), as well as embedded B retroelements (13, 19), pseudogenes (20), short interspersed elements (SINEs) (21), and circular RNAs (22, 23). As technological and research advances reveal a larger and more diverse spectrum of miRNA targets, the related question of how many miRNAs are encoded by an organism’s genome becomes one of renewed importance.
Initial attempts to characterize the miRNA repertoire of an organism assumed that miRNAs and their precursors are conserved (24–26), but, since then, it has become evident that genus-specific miRNAs also exist in the fruit fly (Drosophila melanogaster) (27), the mouse (Mus musculus) (28), and the worm (Caenorhabditis elegans) (29). This observation suggests that relying on conservation may underestimate an organism’s repertoire of miRNAs. Indeed, there is no reason to think that there will not be lineage-specific adaptive evolution in regulatory sequences and regulatory molecules. In our earlier work, we estimated that, if cross-genome conservation is not a required criterion, then the repertoire of miRNAs in the human genome likely exceeds 25,000, with an associated prediction error rate of 1% (30). A little more than 1,800 human miRNA precursors are listed in release 20 (June 2013) of miRBase (31, 32), each giving rise to one or two mature miRNA products. Recent analyses using next-generation sequencing have resulted in the identification of new human and mouse miRNAs (28, 33–37) and have suggested the existence of tissue-specific miRNAs (33). We reasoned that many more miRNAs are present and can be identified through the analysis of additional samples representing more diverse tissue types.
Here, we describe our findings from such a search of previously unrecognized miRNAs that arise from the canonical biogenesis pathway (1, 2). To this end, we examined 1,323 short RNA-seq samples representing 13 distinct human cell types, as well as sought corroborating evidence of loading onto the RNA-induced silencing complex (RISC) by examining several dozen Argonaute immunoprecipitation and sequencing (Ago CLIP-seq) samples. Our analyses revealed the presence of 3,707 new human miRNAs expressed throughout the genome. These findings suggest that the repertoire of human miRNAs is larger and more diverse than may be suggested by the publicly available repositories. Moreover, the existence of novel miRNAs that are primate- and tissue-specific indicates the existence of molecular interactions that cannot be recapitulated by mouse models and, thus, has potential implications for both disease and research endeavors.
Results
Identification of Novel miRNAs.
We deep-sequenced and generated short RNA profiles for 100 of our own samples, which we combined with an additional 1,223 public ones, to generate a collection of 1,323 samples (Dataset S1) representing 13 distinct cell types from both primary tissues and cell lines. The analyzed collection comprised over 23 billion sequenced reads from short RNA-seq: of these reads, ∼8.2 billion could be mapped unambiguously to the human genome and were subsequently used for the identification of novel miRNAs. Because the miRDeep2 algorithm (38, 39) has been shown to be a sensitive and specific method for identifying novel miRNA precursors, we used it to analyze our collection by processing each of the 1,323 samples separately (Fig. 1). Those identified precursors that received a miRDeep2 score of 1 or greater were kept for further analysis. Within the span of each retained precursor locus, we identified the most abundant miRNA isoform (isomiR) as the corresponding mature miRNA from the locus. To compensate for differences in sequence depth among the various samples and select only miRNAs with statistically significant abundance in the sample being considered, we fitted a negative binomial distribution to the data (i.e., the abundance data at each transcribed genomic locus) and used the abundance of each mature miRNA to derive the miRNA’s statistical significance within its own sample; we kept only those miRDeep2 precursor loci whose mature miRNAs had an associated false discovery rate (FDR) of ≤ 0.05 in at least one of the analyzed samples (Fig. 1) (PDF images of the predicted precursor structures can be downloaded by following each of the links contained in Dataset S2 or by visiting directly https://cm.jefferson.edu/novel-mirnas-2015/). We further postprocessed the identified mature miRNAs and precursors to remove any predicted mature miRNA that (i) is already represented in miRBase release 20 or in the mirtron catalog (28) and/or (ii) colocalized with snRNAs, tRNAs, and rRNAs even though such loci have been previously linked with miRNA production (40–43). This filtering left us with a collection of 3,494 miRNA precursors of which 213 give rise to two mature miRNA products, one from each arm of the precursor, for a grand total of 3,707 mature miRNAs that satisfied the FDR ≤ 0.05 constraint (Dataset S2). It is worth mentioning that 91.5% of the newly discovered mature miRNAs (3,392 of 3,707) were discovered independently in at least 10 of the analyzed samples.
Fig. 1.

Flow diagram depicting the steps taken in identifying novel miRNAs. Shown is a flow diagram of the process to identify candidate novel miRNAs from 1,323 deep-sequencing samples using miRDeep2. Only mature miRNA with associated FDR ≤ 0.05 were kept for further analysis. Discovered sequences that were present in release 20 of miRBase, or overlapped known tRNAs, snRNAs, or rRNAs were discarded. A total of 3,707 candidate miRNAs derived from 3,494 precursor sequences were identified. Intersection of the identified miRNAs with 43 Ago-CLIP-seq samples showed evidence of Ago loading for 1,657 newly discovered miRNAs. Sixty-six of the identified precursors produced two miRNAs, one from each arm: one product was supported by Ago CLIP-seq whereas the other was not.
The Novel miRNAs Originate Throughout the Genome.
The miRNAs in release 20 of miRBase are encoded throughout the genome, including intergenic (68.8%), exonic (4.7%), intronic (12.4%), long noncoding (5%), and repeat regions (7.9%) (Fig. 2A). We examined the genomic distribution of the newly discovered mature miRNAs in an effort to investigate any potential biases. We found that these miRNAs are distributed similarly to the existing miRBase-cataloged miRNAs, with the majority being located within the intergenic (57.6%) and intronic (17.4%) regions of the genome (Fig. 2B). Several of the novel miRNAs arise from long noncoding RNA transcripts and repeat elements in proportions that mirror those of miRBase (Fig. 2B). Taken together, these results show that the novel miRNAs that we have discovered have a proportional distribution across the genome similar to those present in miRBase.
Fig. 2.

Both known and novel miRNAs are encoded throughout the genome. Shown are the regions of the genome from which miRNAs of miRBase (A) and the novel miRNAs (B) are encoded. All annotations for genes [3′ UTR, coding DNA sequence (CDS), and 5′ UTR], long noncoding RNAs (lncRNAs), and pseudogenes are from release 72 of ENSEMBL; all repeat regions are from RepeatMasker. Intronic regions are defined to be those segments of known unspliced pre-mRNA that remain after removing all known genomic features that are sense to the pre-mRNA such as exons, miRNAs, repeat elements, etc. Intergenic regions are defined to be those segments of the genome that remain after removing all protein coding loci as well as all other already-characterized genomic features.
Many of the Newly Discovered miRNAs Are Expressed in a Tissue-Specific Manner.
Recent analyses revealed the presence of novel tissue-specific miRNAs (33). To determine whether our identified miRNAs exhibit tissue-dependent expression, we normalized the expression level of each miRNA as previously described (44, 45) and separately for each sample. From each analyzed sample, we kept only very abundant novel miRNAs [i.e., novel miRNAs with expression levels ≥1/100 the expression of the endogenous small nucleolar RNA (snoRNA) SNORD44 (44, 45)] and then formed the union for all samples across the 13 analyzed tissues. To compare the composition of the miRNA populations across the different tissues, we calculated the Jaccard similarity index between tissue i and tissue j for the novel miRNAs: This index measures what portion of the union of novel miRNAs that are in either tissue i or in tissue j are common to both tissues (Fig. 3A). The more tissue-specific the miRNAs are the smaller the Jaccard index between any two tissues. This distinction is precisely what we observed: the novel miRNAs that we discovered in a given tissue had limited presence outside that tissue, indicating their strong tissue-dependent nature. For comparison purposes, we repeated this analysis using the miRBase miRNAs that are present in the 13 tissues, instead of the newly discovered miRNAs; as can be seen from Fig. 3B, miRBase miRNAs have a markedly lower tissue specificity and thus much higher presence across multiple tissues.
Fig. 3.
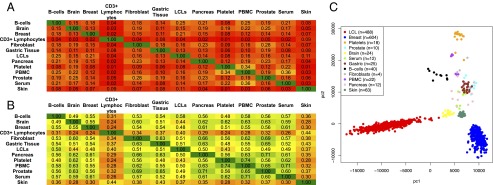
Novel miRNAs display a tissue-specific pattern of expression. Shown are the Jaccard index value for the overlap of expressed miRNAs between any two tissues for the novel miRNAs (A) and the miRBase miRNAs (B). A miRNA was considered to be expressed in each tissue if the miRNA had a normalized expression of ≥1/100 the expression of endogenous SNORD44. (C) Principal-component analysis of the sequence data can cluster the samples based upon tissue types.
Because the novel miRNA profiles are dependent upon the tissue type, we next examined whether unsupervised clustering of their expression values could cluster the samples along tissue boundaries. To this end, we performed a principal component analysis on rank-normalized (based on sequence depth) expression values for our 3,707 novel miRNAs. As can be seen from Fig. 3C, the novel miRNAs can accurately cluster lymphoblastoid cell lines (LCLs), breast, platelets, B cells, skin, and brain tissue samples. Taken together, these results suggest that the identified novel miRNAs display patterns of tissue specificity and can distinguish among tissue types.
Additional Experimental Support of the Novel miRNAs from Ago CLIP-seq Data.
MiRNAs exert their function through their association with the Ago-silencing complex. We performed Ago CLIP-seq in 10 of our own samples: two human pancreatic cell lines (the normal epithelial hTERT-HPNE and the metastatic MIA PaCa-2) and four normal and four Alzheimer’s disease human brain samples. We combined our 10 samples with an additional 33 public samples from HEK293 (46), human LCL (47), and human brain tissue (48), and sought corroborating evidence in the form of Ago loading for both known miRNAs (release 20 of miRBase) and our novel miRNAs. We stress here that the 43 Ago CLIP-seq samples we analyzed represent only 3 of the 13 tissue types that we used during the miRNA discovery phase: Ago CLIP-seq samples from HEK293 cells were also used, but these cells were not represented among the 1,232 short RNA sequence samples that were analyzed. Considering this constraint, and in conjunction with the strong tissue-specific character of the novel miRNAs (Fig. 3A), we do not expect to observe all of the newly discovered miRNAs, or all of the miRBase miRNAs for that matter, in the Ago CLIP-seq data we analyzed. Of the 2,772 miRNAs contained in miRBase release 20, 1,517 (54.7%) were found to be present in one or more of the 43 Ago CLIP-seq samples that we analyzed. Similarly to the miRBase miRNAs, 1,657 of our 3,707 newly discovered miRNAs (44.7%) were found in one or more Ago CLIP-seq samples (Fig. 1, Table 1, and Dataset S2). These results suggest that about half of our newly identified miRNAs are loaded onto the Ago-silencing complex and thus are imputed to be posttranscriptionally functional.
Table 1.
Summary of findings for known (miRBase) and novel miRNAs
| Release 20 of miRBase | Our collection of novel miRNAs | |
| No. of unique miRNAs | 2,772 | 3,707 |
| No. of unique precursors | 1,871 | 3,494 |
| No. of 3p miRNAs* | 934 | 2,130 |
| No. of 5p miRNAs* | 940 | 1,577 |
| No. of distinct seed sequences† | 1,506 | 1,761 (888 novel seeds) |
| No. of Ago-CLIP supported‡ | 1,517 (54.7%) | 1,657 (44.7%) |
There are 898 miRNAs of miRBase (release 20) that are annotated as having only one arm.
Seed sequence is defined as positions 2–7 inclusive from the 5′ end of the miRNA. A total of 1,763 unique seed sequences are identified between the two sets of miRNAs.
A miRNA was considered Ago-CLIP–supported if it was identified in at least 1 of 43 and a minimum of 5 sequence reads (Materials and Methods).
Additional Experimental Support of Novel miRNAs by a Dicer Knockdown Experiment.
For hairpin-derived miRNAs (canonical and mirtrons), the endonuclease DICER is critically important for the processing of precursors into mature miRNA products. We used published RNA-seq samples from MCF7 cells before and after siRNA knockdown of DICER (39) to determine whether the subset of our novel miRNAs that are present in the WT MCF7 cells show evidence of DICER dependence. Our analysis showed that 709 of the miRNAs in release 20 of miRBase and 278 of our novel miRNAs were endogenously expressed in MCF7 cells. After siRNA knockdown of DICER1, the miRBase miRNAs showed a median decrease in expression of ∼2×, and our novel miRNAs showed a median decrease in expression of ∼1.6× (Fig. 4). A similar result (∼1.7× decrease in expression after DICER1 knockdown) was observed for those novel miRNAs that are endogenous to MCF7 cells and show evidence of Ago loading in our Ago CLIP-seq samples. This result indicates that those of our newly discovered miRNAs that are endogenous to MCF7 cells are DICER-dependent and follow the canonical biogenesis pathway.
Fig. 4.

Dicer knockdown results in a decrease in miRNA expression. Fold change in miRNA expression levels in MCF7 cells after Dicer knockdown for release 20 miRBase miRNAs (blue), all newly discovered miRNAs (red), and the subset of Ago CLIP-seq–supported newly discovered miRNAs (green). y axis, percentage of expressed miRNAs; x axis, fold change in expression of Control vs. Dicer knockdown. A negative fold change equals decrease of the miRNA in the knockdown. inf, miRNA was absent in either the Dicer knockdown (−inf) or the Control sample (inf).
Novel miRNAs Can Be Specifically Amplified.
To further support our findings of the novel miRNAs, we set out to specifically amplify some of the newly discovered miRNAs in a panel of cell lines, representing five different tissue types (breast, pancreas, prostate, embryonic kidney, and fibroblasts). For these experiments, we selected a first group of 12 novel miRNAs that, according to our analysis, were tissue-specific and a second group of 8 novel miRNAs that our analysis indicated were present in multiple tissues, for a total of 20 tested novel miRNAs. We used a stem-loop RT-PCR system (Fig. S1) similar to what has been previously described (49), and tested 20 of our novel miRNAs in 12 cell lines representing five tissue types. As can be seen in Fig. 5, the first group of tested miRNAs indeed exhibited tissue-specific expression patterns: Each of the 12 miRNAs was present in one or more of the tested cell lines. As expected, the second group of novel miRNAs was ubiquitously expressed and present in all of the examined cell lines.
Fig. 5.

Expression of novel miRNAs in a variety of cell lines and tissue types. Stem-loop RT-PCR experiments for 20 newly discovered miRNAs (one miRNA per row). Each row represents a specific cell line.
Several Abundant Novel Mature miRNAs Arise from the “Passenger” Arms of Known miRNA Precursors.
Of the 1,871 precursors in release 20 of miRBase, 898 are annotated as giving rise to a single mature miRNA. In such instances, the corresponding precursor arm is referred to as the “driver” arm. However, recent findings (28, 37) suggest that miRNA products from the passenger arm (traditionally referred to as “miRNA*” or “miRNA-star”) may also be functionally relevant, and acting similarly to the products from the driver arm (37, 50). This observation has rekindled interest in the possibility that a double-stranded miRNA precursor can give rise to two functional miRNA products, one from each arm. Among the novel miRNAs that we have identified, 138 originated from the arms of miRBase miRNA precursors that as of release 20 (June 2013) of miRBase have remained uncharacterized (Fig. 6 and Dataset S3). Importantly, 99 of these 138 miRNAs (71.7%) also received corroborating evidence of Ago loading from our Ago-CLIP samples.
Fig. 6.

Examples of novel mature miRNAs from previously uncharacterized arms of precursors linked to important cell processes. (A) Novel miRNA TJU_CMC.MD2.ID00400.5p-miR arises from the 5′ arm of miR-107’s precursor (MI0000114; chr10:91,352,549-91,352,572). (B) Novel miRNA TJU_CMC.MD2.ID02736.5p-miR arises from the 5′ end of the miR-103-a- precursor (MI0000109; chr5:2167,987,901-167,987,978). The y axis is logarithmic (base 2).
Many Novel miRNAs Are Seed-Paralogues of Known miRBase miRNAs.
The “seed” sequence of a miRNA (positions 2–7 inclusive from the 5′ end) has been shown to play a pivotal role in determining a miRNA’s set of targets (2, 10). Consequently, similarities in the seed sequences have been taken to imply commonality among the targeted mRNAs. With this framework in mind, we set out to determine the composition of seed sequences of our 3,707 novel miRNAs in relation to those annotated in miRBase and known mirtrons. We used a strict definition for the seed as the sequence spanning positions 2–7 inclusive from the 5′ end of the mature miRNA and clustered all miRNAs into groups based upon this sequence. The current release of miRBase has 2,772 annotated miRNAs comprising 1,506 distinct seed sequences. Our set of 3,707 novel miRNAs comprised 1,761 unique seed sequences, of which 873 are common with the seeds of miRBase miRNAs (Dataset S4). The 873 common seeds captured 2,146 of our novel miRNAs; the remaining 1,561 novel miRNAs had 888 distinct seed sequences not present in an annotated miRBase miRNA (Dataset S4). Fig. 7 shows multiple sequence alignments for the sequences of several seed families that comprise well-characterized miRNAs, such as miR-107/103a and miR-21, as well as instances of novel seed clusters consisting of multiple newly discovered miRNAs. Table S1 shows some characteristic examples of seed-paralogues for miRNAs that are frequently cited in the literature. As can be seen, in several instances, each of the listed known miRNAs has multiple, currently uncharacterized, seed-paralogues among the newly discovered miRNAs.
Fig. 7.
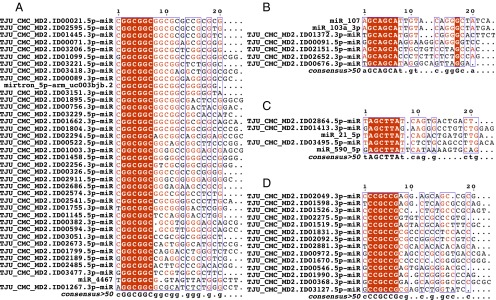
Multiple sequence alignments of seed-based paralogues. The alignments shown in each panel comprise novel miRNAs and miRBase miRNAs that have been clustered based on their shared seed sequences (red highlight). (A–C) Novel miRNAs that are previously uncharacterized seed-paralogues of known miRNAs. (D) A new seed family consisting of 14 newly discovered miRNAs.
Several of the Newly Discovered miRNAs Are Arranged in Genomic Clusters.
The term “miRNA cluster” has been used in multiple ways in the literature. In some instances, it is used to refer to multiple precursors of a polycistronic transcript: e.g., the two-miRNA cluster miR-29a/29b, the six-miRNA cluster miR-17/18/19a/19b/20/92 (51), etc. In other instances, it is used to refer to precursors that are genomically proximal: e.g., a few hundred nucleotides from one another, but which could arise from different transcripts (e.g., miR-371/372/373). In yet other instances, it is used to refer to a collection of precursors that are far from one another but which, as a collection, appear as dense aggregates when viewed from the standpoint of the genome (e.g., the cluster of 49 miRNAs in 19q13.42 that spans more than 120 kb).
With these variable definitions in mind, we sought clusters that contained two or more miRNAs, comprised either novel miRNAs exclusively or a mix of novel and known miRNAs, were transcribed from the same strand, and any two consecutive of the miRNAs forming the cluster were separated by no more than 1,500 nt. We identified 31 such clusters, 21 of which were comprised exclusively of novel miRNAs (Dataset S5). When we expanded the definition of a cluster to include larger regions of the genome [such as the DLK1-D103 locus (52) on chromosome 14], we discovered that one of our newly discovered miRNAs resides within this locus between positions 101,506,189 and 101,506,245 (Dataset S5).
Some of the Novel miRNAs Are Antisense to Known miRNAs or to Other Novel miRNAs.
We expanded our cluster analyses to both genomic strands, in search of potential instances of miRNAs that were transcribed from the same genomic locus but from opposite strands. In particular, we investigated whether any of our novel miRNAs were antisense to either a known miRNA or to another novel miRNA. We limited our searches to only those miRNAs whose respective precursors directly overlapped with each other on opposite strands and identified 13 such instances, 9 of which comprise only novel miRNA pairs. A complete list of sense/antisense pairs with the respective genomic coordinates is given in Dataset S5.
Many of the Novel miRNAs Are Specific to the Hominidae Family of Primates.
Many of the mature miRNA products that are contained in release 20 of miRBase are conserved among different genera: e.g., the well-conserved miRNA let-7. Several exceptions have also been reported, with the corresponding miRNAs being genus-specific (27, 53–56). To determine the degree of conservation of our novel miRNAs, we performed a search where we sought instances not only of the mature miRNA but also of the full-length precursor (at two different thresholds) in several model organisms.
For this purpose, we used GLSEARCH (57) to look for the 3,494 newly identified human miRNA precursors and their respective 3,707 mature miRNAs in the genome assemblies of chimpanzee, gorilla, orangutan, macaque, mouse, Drosophila, and worm. During these searches, we imposed two requirements: (i) At least 85% of the miRNA precursor positions should be identically present in the genome being searched, and (ii) at least 85% of the human mature miRNA sequence should be identically present in the identified orthologous precursor, including an identically present seed. We found that 2,140 (58.1%) precursor/mature miRNA combinations were specific to humans: i.e., they were absent from the other primates, rodents, and invertebrates that we examined (Table 2 and Table S2). As the phylogenetic distance from the human genome increased, we found that progressively fewer of our novel miRNA precursors were conserved: only 476 (12.8%) precursor/mature miRNA combinations were shared by all of the members of the Hominidae family of primates that we examined (Table 2). Beyond primates, the extent of conservation of precursors and their respective mature miRNAs dropped abruptly and substantially: 109 (2.9%) of them were present in mouse whereas none were present in the Drosophila or worm genomes. On the other hand, using similar criteria, we found that only 10% of the precursor/mature miRNA combinations from miRBase were human-specific (Table 2), suggesting that the identified novel miRNAs have a more recent evolutionary origin. We also reran the analysis after relaxing the conservation requirement for the precursor to a more permissive 50% (from 85%) in step i above: The results remained largely unchanged, with the large majority of the newly identified sequences continuing to be primate-specific (Table S2).
Table 2.
Conservation of novel miRNA precursors
| Genome where present | No. of precursor:mature combinations for all novel miRNAs (n = 3,707) | No. of precursor:mature combinations for Ago CLIP-seq (n = 1,657) | No. of precursor:mature combinations from miRBase (n = 2,772) |
| Human | 3,707 (100.0%) | 1,657 (100.0%) | 2,772 (100.0%) |
| Chimpanzee | 1,275 (34.3%) | 543 (32.7%) | 2,136 (77.1%) |
| Gorilla | 1,321 (35.6%) | 582 (35.1%) | 2,303 (83.1%) |
| Orangutan | 1,071 (28.9%) | 442 (26.6%) | 1,938 (69.9%) |
| Macaque | 749 (20.2%) | 327 (19.7%) | 1,811 (65.3%) |
| Mouse | 161 (4.3%) | 86 (5.2%) | 659 (23.7%) |
| Drosophila | 6 (0.25%) | 5 (0.3%) | 4 (0.14%) |
| Worm | 2 (0.05%) | 1 (0.06%) | 1 (0.03%) |
Our search of other model organisms showed that both the miRBase release 20 entries and our novel miRNAs are prevalent among primates. After a more tolerant search (Table S2), the results remain largely unchanged from what is shown here.
These results highlight the limitations that can result from imposing the requirement that miRNAs be conserved across organisms. Such requirements will in turn result in our missing bona fide organism-specific miRNAs and could perhaps explain why many of these novel miRNAs have not been previously identified.
Evaluation of the Novel miRNA Targetome.
It is reasonable to assume that these novel miRNAs will affect many pathways and exert their effects on a wide range of targets. To further characterize the potential targetome for this collection of novel miRNAs, we computationally predicted their mRNA targets using RNA22 (30, 58) to generate predictions of the mRNA targets for each of the 3,707 newly discovered miRNAs. We opted to use RNA22 (https://cm.jefferson.edu/rna22v2/) because of its demonstrated ability to correctly predict targets in amino acid coding regions and in 5′ UTRs, as well as targets that do not contain contiguous Watson–Crick base pairs in their seed region. The precomputed collection of targets can be downloaded from https://cm.jefferson.edu/novel-mirnas-2015/. In Dataset S2, and in addition to the genomic and cross-genome information, we provide separate links for each miRNA to tab-separated text files that contain the miRNA’s collection of predicted targets. We anticipate that this compilation will enable investigators across multiple laboratories and help them embark on such studies. At the same time, these predictions will also facilitate analyses across several levels of the targetome hierarchy: from the targets of an individual miRNA and the mRNAs collectively targeted by miRNAs with the same seed, to the miRNAs targeting a specific pathway and to previously unsuspected tissue-centered networks of interactions. We conclude with a small example that highlights the possibilities. In particular, we analyzed the targets of one set of four miRNAs that share an identical seed sequence; the miRNAs were miR-miR-19a, miR-19b, TJU_CMC_MD2.ID00745.5p, and TJU_CMC_MD2.ID03086.5p. We processed our predicted targets using DAVID (59, 60) and found a broad range of significantly enriched GO terms among them (P ≤ 0.05 and FDR ≤ 0.05). Notably, and despite the shared seed sequence of these four miRNAs, we found limited overlap between the predicted targets (Table S3).
Discussion
In this study, we report on the discovery of 3,707 novel miRNAs in the human genome. By comparison, there are 2,772 miRNAs in release 20 of miRBase (61). The increasing importance of miRNAs and their multifaceted involvement in various cellular processes make it pressing to obtain an accurate estimate of their numbers, to profile their patterns of expression across cell types, and to determine their targets. To address these questions, we examined 1,323 samples, representing 13 human cell types (Dataset S1) for the presence of novel miRNAs using the miRDeep2 algorithm (39). By analyzing in excess of 23 billion sequenced reads, we identified 3,707 novel human miRNAs, each with an associated FDR ≤ 0.05 (see Fig. 1 and Dataset S2 for specific information on genomic location and cross-genome conservation).
Just as protein-coding genes display tissue-specific patterns of expression, it is reasonable to assume that miRNAs would exhibit a similar behavior. For example, the C. elegans cel-lsy-6 miRNA is specifically expressed in the brain and controls left/right neuronal asymmetry (62). In analogy to cel-lsy-6, the novel miRNAs that we identified characteristically display a tissue-specific pattern of expression, as evidenced by the low Jaccard similarity index of the miRNAs that are expressed in pairs of tissues (Fig. 3). This specificity was further demonstrated by including, in the 20 miRNAs that we experimentally tested, 12 novel miRNAs that, according to our analysis, exhibited strong tissue-specific expression patterns (Fig. 5). Additionally, (unsupervised) principal component analysis (PCA) clustering of our novel miRNAs showed that they are able to correctly cluster the analyzed samples into distinct groups based upon their tissue of origin (Fig. 3A). We note that, of our 3,707 novel miRNAs, only 292 are in common with the miRNAs discovered by another recently reported effort (33) that also used miRDeep2. There are two reasons for this small overlap: first, the 1,323 samples that we analyzed and the 94 samples analyzed in ref. 33 have only one dataset in common (NIH GEO accession no. GSE15229; 30 samples). And second, our novel miRNAs (as evidenced by Fig. 3A) and those discovered in ref. 33 are tissue-specific. Taken together, these results suggest that the complete human miRNA-ome is just beginning to be elucidated: as more studies are performed with more varied tissue types, we expect that many additional miRNAs will be discovered.
Both the novel miRNAs identified in our work and the miRBase miRNAs span a wide range of expression levels. Naturally, the expression levels of both the newly discovered and known miRNAs change from tissue to tissue. Before determining a novel miRNA’s expression, we made sure to compensate for the sequence depth of the sample at hand: in particular, we used an adaptive thresholding strategy that required a miRNA to be supported by a higher number of sequence reads in samples that were more deeply sequenced. Generally, we found their average expression level to be somewhat lower than that of miRBase miRNAs, in agreement with previous reports (39, 54–56). We nonetheless stress that all of the novel miRNAs that we discovered are statistically significant (FDR ≤ 0.05) in at least one and typically in many of the analyzed samples: as a matter of fact, 91.5% of the newly discovered miRNAs are statistically significant in 10 or more of the processed samples. Additionally, it is important to note that many of the novel miRNAs are loaded onto the Ago-silencing complex, which suggests that they are biologically active.
In the early days of the miRNA field, there was an emphasis on identifying miRNAs that are conserved across organisms: e.g., let-7 first described in 2000 (63, 64). Nonetheless, species-specific miRNAs (e.g., cel-lsy-6 in C. elegans) (62) have also been described and characterized as have been miRNAs that are present only in one or a few species of the same genus. Therefore, enforcing an organism-conservation requirement during miRNA searches is bound to limit the number of potential miRNAs that can be discovered, leaving organism- and lineage-specific miRNAs undiscovered (53–56). In our effort to further characterize the human miRNA repertoire, we liberated ourselves from the conservation requirement: not surprisingly then, 56.7% of our newly discovered miRNAs are human-specific whereas 94.4% are primate-specific (Table 2). Considering that many miRNA studies to date have focused on seeking and analyzing conserved miRNAs, it is not surprising that, of the human miRNAs in miRBase, we found a larger fraction to be conserved in rodents and invertebrates (Table 2). These findings strongly suggest the possibility of a wide-ranging species-specific miRNA-ome that has yet to be characterized. Indeed, it is reasonable to expect that at least some of these novel primate-specific miRNAs participate in unexplored aspects of regulatory processes that cannot be captured by the currently available mouse disease models. Thus, not only could these newly discovered miRNAs provide new molecular insights but they could also help us define novel biomarkers for tissue or disease states.
MiRNAs exert their function through their association with the Argonaute complex, and, thus, those miRNAs loaded onto Ago are expected to be capable of interacting with target RNAs. Using 43 Ago CLIP-seq samples, 10 of which we generated ourselves, we found evidence of Ago loading for 1,657 of the 3,707 novel miRNAs (44.7%) (Table 1). In complete analogy, of all of the miRNAs in miRBase20, we find evidence of Ago loading for 1,517 (54.7%): i.e., a comparable fraction, among the 43 Ago CLIP-seq samples. These seemingly moderate percentages (44.7% and 54.7%, respectively) of Ago-loading support are in fact expected, considering that the Ago CLIP-seq samples we analyzed represent only 3 of the 13 distinct tissue types in which we carried out the novel miRNA discovery. Consequently, in the 43 Ago CLIP-seq samples, we expect to find support for only a portion of the novel miRNAs that we discovered and of those miRNAs currently in miRBase. Moreover, the lower fraction of Ago support for the newly discovered miRNAs (44.7% vs. 54.7%) is also expected, considering the markedly higher degree of tissue dependence exhibited by the novel miRNAs compared with those in miRBase (Fig. 3).
In addition to being loaded onto the Ago complex, we found that more than 50% of our novel miRNAs were bona fide seed-paralogues of miRNAs that are already present in miRBase (Fig. 7, Table S1, and Dataset S4). This similarity in their seed regions generally suggests overlapping sets of targets and shared or related functions (65–67). Nonetheless, the remainder of the miRNA sequence can also assist in defining the eventual targetome. We examined our results of rna22-predicted targets for two seed-paralogues of miR-19a/b. As shown in Table S3, the predictions indicate little overlap between the GO pathways that are enriched among the predicted targets. Although uncommon, miRNAs with the same seed can in principle have different targeting preferences that are dictated by variations outside of the seed sequence (68). For example, the members of the miR-29 family share the same seed sequence but display differences in the predicted targetomes: miR-29b localizes predominantly in the nucleus whereas miR-29a and miR-29c do not (69). Furthermore, we note that, in addition to discovering previously unsuspected paralogues of known miRNAs, we established 888 new seeds and their associated miRNA-seed families.
A key question of this collection of miRNAs will be to unravel the specifics of their functional impact. The sheer magnitude of the identified miRNAs and the very high number of predicted targets for each such miRNA make the experimental validation of the targetome by a single laboratory an intractable proposition. To facilitate such investigations and to assist other laboratories as well as our own in embarking on such studies, we have computationally generated candidate mRNA targets for all 3,707 novel miRNAs using RNA22 (30, 58) (for each miRNA, we provided links in Dataset S2 and https://cm.jefferson.edu/novel-mirnas-2015/ that give access to the RNA22-predicted targets). One important property of RNA22 is that it is not confined on the 3′ UTR alone but can generate predictions across the entire length of an mRNA. Although additional experimental work will be required to functionally validate these putative interactions, it is reasonable to assume that a portion of these novel miRNAs will be found to be involved in critical pathways and thus can serve as biomarkers for disease research.
Based on the results presented here and taking into account previous studies (28, 33), we conclude that the miRNAs currently listed in the public repositories represent only a small fraction of the total human miRNA repertoire and that many more human miRNAs await discovery. Our study focused on identifying only canonical miRNAs that arise in a Dicer-dependent fashion, and we need to keep in mind that bona fide miRNAs can arise through other mechanisms as well (28, 70), in turn suggesting an even larger miRNA-ome. Nonetheless, our work makes a concrete and very tangible contribution toward a comprehensive understanding of the human miRNA-ome, its functional relevance, and its potential roles in disease etiology.
Materials and Methods
Makeup of the Analyzed Collection of Samples.
The 1,323 samples we analyzed represent 13 distinct cell types derived from human primary tissues [platelets, prostate tissue, breast tissue, pancreas, B cells, serum, peripheral blood mononuclear cells (PBMCs), gastric tissue, CD3+ lymphocytes, brain tissue, skin tissue] and human cell lines (lymphoblastoid cell lines, the breast cell line MCF7, and the pancreatic cell lines HPNE and MIA PaCa-2). One hundred of these samples were collected, deep-sequenced, and analyzed by the Computational Medicine Center of Thomas Jefferson University in the context of studies approved by the Institutional Review Boards of the universities whose teams participated in this project. The remaining samples were obtained from various public sequence data repositories. Dataset S1 provides information on all of the used samples.
RNA Sequencing.
The 100 short RNA-seq samples were generated at Thomas Jefferson University using LifeTech’s SOLiD 4 and SOLiD 5500xl sequencing platforms. Short RNA sequence library construction, emulsion PCR, and subsequent sequencing runs were performed following the manufacturer’s protocols. Sequencing was performed by fragment-end sequencing of 50-nt fragments at the Cancer Genomics Laboratory of the Kimmel Cancer Center of Thomas Jefferson University.
Reference miRNA Database.
All of the identified miRNAs were compared with those represented in release 20 of miRBase (June 2013) (31, 32). This release of miRBase comprises 1,871 precursor sequences and 2,772 mature miRNAs. Additional comparisons were made to a recently identified collection of 458 mirtrons (28). It should be noted that, while we were nearing completion of our data analyses, a new miRBase release (release 21) became publicly available. This new version of miRBase includes only a very small (1.48%) increase in the number of mature miRNAs (2,813 mature miRNAs). Analysis of our collection of identified miRNAs revealed that 10 of the novel miRNAs that we discovered were included in release 21 of miRBase. Considering the very small increase in the number of human miRNAs that is represented by release 21 of miRBase, we maintained our focus on release 20 of miRBase.
Mapping of the Sequenced Reads.
The short RNA sequence reads were mapped onto the human genome assembly GRCH37 (hg19) using the Short Read Mapping Package (SHRiMP2) (71). Before mapping, the sequence adaptors were trimmed using cutadapt (72). All reads were quality-trimmed using the reads’ associated quality values. During mapping, we allowed only mismatches (replacements) that comprised no more than 4% of a given read’s length; no insertions or deletions were permitted. Also, trimmed reads that were shorter than 16 nt were discarded and not considered further. Only reads that mapped unambiguously on the human genome under these settings were kept and considered in the analysis.
Argonaute CLIP Sequencing.
The hTERT-HPNE and MIA PaCa-2 cell lines (obtained from the American Type Culture Collection or kindly provided by Jonathan Brody, Thomas Jefferson University, Philadelphia) were propagated in Dulbecco’s modified Eagle medium supplemented with 10% FBS and 1% penicillin/streptomycin (Cellgro). Total RNA was extracted with TRIzol reagent as per the manufacturer’s protocol (Life Technologies) and depleted of ribosomal RNA using a RiboZero Kit (Epicentre Biotechnologies). Ago HITS-CLIP was performed as described previously (18) with the recently described modifications (73) to increase stringency. Briefly, cells were grown to 70% confluency, washed once with PBS, and UV-irradiated at 254 nm for a total energy deposition of 600 mJ/cm2 (Spectroline). RNA digestion was carried out as per Hafner et al. (74) whereby cell lysates were treated initially with RNase T1 at a concentration of 1 U/μL for 15 min at room temperature in PXL buffer before coimmunoprecipitation of RNA–protein complexes on protein A Dynabeads (Life Technologies) using the pan-Ago antibody 2A8 for 4 h at 4 °C (Millipore). Beads were then washed twice with PXL buffer and subjected to a secondary, complete RNA digestion with 100 U/μL RNase T1 for 15 min at room temperature. After complete digestion, CLIP-RNAs were released from their on-bead protein complexes by treatment with 4 mg/mL proteinase K and subsequent phenol/chloroform extraction as described (18).
Identification of Significantly Expressed Sample-Dependent Novel miRNAs.
Novel miRNA discovery was performed using the miRDeep2 algorithm (38) using default settings. Each sample was processed independently (there was no sample pooling). Only those identified hairpins with a miRDeep2 score of 1 or greater were kept for further analysis. For each sample, we identified the end points of the mature miRNA by looking at the most prominently expressed isomiR located within the miRDeep2 predicted mature loci. If the identified hairpin was not at least 50 nt in length or the most prominent isomiR was not 20–24 nt in length inclusive, we discarded the prediction. Additionally, to eliminate noisy lowly expressed miRs, we kept a discovered miRNA only if it had an associated FDR ≤ 0.05. To derive FDR values for each sample, we fitted a negative binomial distribution to the available data (i.e., the abundance data at every transcribed genomic locus), followed by a correction for multiple testing using the Benjamini–Hochberg procedure. These steps allow us to compensate for differences in sequence depth among the various samples and to select only miRNAs with statistically significant abundance in the sample being considered each time. Because we used the abundance of each mature miRNA to derive the miRNA’s statistical significance within its own sample, it follows that any miRNA that satisfies this FDR level is comparatively very abundant within the samples in which it is discovered. At this stage, any and all of miRDeep2’s predictions that intersected known miRNAs, mirtrons, tRNAs, snRNAs, scRNAs, or rRNAs were excluded from further consideration.
Because each sample was run independently, we identified hairpins and mature coordinates with end points typically differing by 1–2 base pairs across different samples. To compensate for this effect, overlapping hairpins were merged into a single larger “island”, and the mature isomiR with the highest read support across samples was determined to be the mature miRNA. The genomic coordinates of the mature and hairpin sequences are listed in Dataset S2. The hairpins of all discovered miRNA precursors were inspected manually (through the PDF output generated by miRDeep2) to ensure quality and can be downloaded from links located in Dataset S2 and at https://cm.jefferson.edu/novel-mirnas-2015/.
Finally, we used 10 internally generated Ago-CLIP-seq samples (HPNE and MIA PaCa-2 cells and human brain tissue) and combined them with an additional 33 public datasets from HEK293 cells (46), LCLs (47), and human brain (48) (Dataset S1). Each mature miRNA candidate had to completely coincide with an Ago CLIP-seq site, be observed in at least 1 of the 43 CLIP-seq samples, and be supported by five or more unambiguously mapped CLIP-seq reads.
Seed-Based Clustering of Known and Novel Mature miRNAs.
We collected the unique seed sequences (positions 2–7 inclusive) by examining the mature miRNAs in release 20 of miRBase (June 2013), the mature mirtrons from the mirtron catalog (28), and our collection of novel mature miRNAs. Known and novel miRNAs that shared the same seed formed a cluster identifiable by the shared-seed 6-mer. All of the cluster’s members contain the same 6-mer in positions 2–7. A novel miRNA that is in the same cluster as a known miRNA is thus classified as a seed-paralogue of the known miRNA. Novel mature miRNAs whose seeds are not among those of known miRNAs or mirtrons form their own clusters and are, by definition, seed-paralogues of one another.
Genome Conservation of Hairpin and Mature miRNAs.
To determine which of the newly discovered miRNAs were conserved, we used GLSEARCH (57) and sought the miRDeep2-identified novel miRNA hairpins and their mature miRNA(s) in the chimpanzee, gorilla, orangutan, macaque, mouse, Drosophila, and worm genome assemblies. During these searches, we required that (i) at least 85% of the miRNA precursor positions be identically conserved in the searched genome and (ii) at least 85% of the human mature miRNA positions be identically present in the identified orthologous precursor, including an identically present seed. Those hairpin/mature miRNA combinations that did not meet these criteria were considered to be not conserved. We also repeated the search of each model genome, this time imposing a 50% identical match for the precursor (instead of 85%).
Experimental Amplification of Novel miRNAs.
To experimentally validate the presence of the miRNAs, we specifically amplified miRNA by stem-loop RT-PCR followed by PCR amplification of the miRNAs (49). The method is schematically represented in Fig. S1: briefly, a stem-loop RT-PCR specific to the last 6 nt of the 3′ end of the miRNA that is used to reverse transcribe the miRNA along with the miRNA. The RT-PCR product is then used as a template for PCR with a forward primer specific to the miRNA and a reverse primer specific to the hairpin region. All reactions were performed with 50 ng of RNA and performed using standard protocols. PCR products were designed to be 50–60 nt in length and were run on 2% agarose gels. In total, 20 novel miRNAs were tested. As a negative control, a primer to a scrambled miRNA sequence was designed. U6 RT-PCR was performed on all samples to control for the RNA concentrations. All primer sequences are listed in Table S4. All PCRs were performed to 35 PCR cycles.
PCRs were performed on a panel of RNAs from 12 cell lines representing five different tissues: breast cancer cell lines MCF-7, MCF-10A, and DCIS; pancreas cell lines HPNE, MIA PaCa-2, and PANC-1; prostate cancer cell lines DU145, LnCaPs, and C42; the human embryonic kidney cell line HEK293; and two fibroblast cell lines N1 and N5 (created at Thomas Jefferson University).
Supplementary Material
Acknowledgments
We thank Eric Lai and his team for providing the genomic coordinates of human mirtrons. This research was supported by the William M. Keck Foundation (I.R.), the Hirshberg Foundation for Pancreatic Cancer Research (I.R.), the Tolz Foundation Weizmann Institute of Science-Thomas Jefferson University Collaboration Program (I.R. and J.R.B.), a Pilot Project Award by the NIH Autoimmune Centers of Excellence (2U19-AI056363-06/20309840 to I.R., S.A.J., and P.F.), NIH-National Cancer Institute Core Grant P30CA56036 (to K.D. and P.F.), and Thomas Jefferson University Institutional funds. J.J.Y. is supported by NIH Grant CA140424. M.L. and B.R. are supported by Lifespan/Tufts/Brown Center for AIDS Research Grant P30 AI042853. P.T.N. is supported by NIH Grants AG042419, NS085830, and AG028383. S.A.J. is supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant R01 AR 19616. G.A.C. is supported in part by the CLL Global Research Foundation, a Sister Institution Network Foundation MD Anderson Cancer Center-German Cancer Research Center grant on Chronic Lymphocytic Leukemia, the Laura and John Arnold Foundation, the RGK Foundation, and the Estate of C. G. Johnson, Jr. C.Y. is supported by the Jefferson Pancreas, Biliary and Related Cancer Center. P.B. is supported by Grant HL102482 from the National Heart, Lung, and Blood Institute of the National Institutes of Health. K.E.K. is supported by a Pennsylvania Commonwealth Universal Research Enhancement grant and Grant R01 CA099996. Y.K. is supported by NIH grant GM106047. C.E.S.C. was supported by Department of Defense Grant PC094507.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: All sequence data generated at Thomas Jefferson University have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo. For a list of accession numbers, see Dataset S1.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420955112/-/DCSupplemental.
References
- 1.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012;336(6078):237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eulalio A, et al. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev. 2007;21(20):2558–2570. doi: 10.1101/gad.443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui Q, Yu Z, Purisima EO, Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol Syst Biol. 2006;2:46. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramachandran V, Chen X. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science. 2008;321(5895):1490–1492. doi: 10.1126/science.1163728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009;461(7263):546–549. doi: 10.1038/nature08349. [DOI] [PubMed] [Google Scholar]
- 8.Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci USA. 2008;105(39):14879–14884. doi: 10.1073/pnas.0803230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson PT, et al. Specific sequence determinants of miR-15/107 microRNA gene group targets. Nucleic Acids Res. 2011;39(18):8163–8172. doi: 10.1093/nar/gkr532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rigoutsos I. New tricks for animal microRNAS: Targeting of amino acid coding regions at conserved and nonconserved sites. Cancer Res. 2009;69(8):3245–3248. doi: 10.1158/0008-5472.CAN-09-0352. [DOI] [PubMed] [Google Scholar]
- 11.Schnall-Levin M, et al. Unusually effective microRNA targeting within repeat-rich coding regions of mammalian mRNAs. Genome Res. 2011;21(9):1395–1403. doi: 10.1101/gr.121210.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen WF, Hu YL, Uttarwar L, Passegue E, Largman C. MicroRNA-126 regulates HOXA9 by binding to the homeobox. Mol Cell Biol. 2008;28(14):4609–4619. doi: 10.1128/MCB.01652-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455(7216):1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H, Rigoutsos I. MiR-103a-3p targets the 5′ UTR of GPRC5A in pancreatic cells. RNA. 2014;20(9):1431–1439. doi: 10.1261/rna.045757.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zisoulis DG, et al. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol. 2010;17(2):173–179. doi: 10.1038/nsmb.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung AK, et al. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol. 2011;18(2):237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cesana M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460(7254):479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tay YM, et al. MicroRNA-134 modulates the differentiation of mouse embryonic stem cells, where it causes post-transcriptional attenuation of Nanog and LRH1. Stem Cells. 2008;26(1):17–29. doi: 10.1634/stemcells.2007-0295. [DOI] [PubMed] [Google Scholar]
- 20.Tay Y, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147(2):344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu TJ, Yi X, Zhao XW, Zhao Y, Yin JQ. Alu-directed transcriptional regulation of some novel miRNAs. BMC Genomics. 2009;10:563. doi: 10.1186/1471-2164-10-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen TB, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 23.Memczak S, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 24.Ambros V, et al. A uniform system for microRNA annotation. RNA. 2003;9(3):277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai EC, Tomancak P, Williams RW, Rubin GM. Computational identification of Drosophila microRNA genes. Genome Biol. 2003;4(7):R42. doi: 10.1186/gb-2003-4-7-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299(5612):1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- 27.Stark A, et al. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res. 2007;17(12):1865–1879. doi: 10.1101/gr.6593807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012;22(9):1634–1645. doi: 10.1101/gr.133553.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruby JG, et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127(6):1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 30.Miranda KC, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32(Database issue):D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedländer MR, et al. Evidence for the biogenesis of more than 1,000 novel human microRNAs. Genome Biol. 2014;15(4):R57. doi: 10.1186/gb-2014-15-4-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jima DD, et al. Hematologic Malignancies Research Consortium Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood. 2010;116(23):e118–e127. doi: 10.1182/blood-2010-05-285403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joyce CE, et al. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum Mol Genet. 2011;20(20):4025–4040. doi: 10.1093/hmg/ddr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meiri E, et al. Discovery of microRNAs and other small RNAs in solid tumors. Nucleic Acids Res. 2010;38(18):6234–6246. doi: 10.1093/nar/gkq376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plé H, et al. The repertoire and features of human platelet microRNAs. PLoS ONE. 2012;7(12):e50746. doi: 10.1371/journal.pone.0050746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedländer MR, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26(4):407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 39.Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cole C, et al. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA. 2009;15(12):2147–2160. doi: 10.1261/rna.1738409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falaleeva M, Stamm S. Processing of snoRNAs as a new source of regulatory non-coding RNAs: snoRNA fragments form a new class of functional RNAs. BioEssays. 2013;35(1):46–54. doi: 10.1002/bies.201200117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes Dev. 2009;23(22):2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maute RL, et al. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci USA. 2013;110(4):1404–1409. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bray PF, et al. The complex transcriptional landscape of the anucleate human platelet. BMC Genomics. 2013;14:1. doi: 10.1186/1471-2164-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Londin ER, et al. The human platelet: Strong transcriptome correlations among individuals associate weakly with the platelet proteome. Biol Direct. 2014;9(1):3. doi: 10.1186/1745-6150-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kishore S, et al. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011;8(7):559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 47.Skalsky RL, et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012;8(1):e1002484. doi: 10.1371/journal.ppat.1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boudreau RL, et al. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron. 2014;81(2):294–305. doi: 10.1016/j.neuron.2013.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen C, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Almeida MI, et al. Strand-specific miR-28-5p and miR-28-3p have distinct effects in colorectal cancer cells. Gastroenterology. 2012;142(4):886–896, e9. doi: 10.1053/j.gastro.2011.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20(12):1603–1614. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benetatos L, et al. The microRNAs within the DLK1-DIO3 genomic region: Involvement in disease pathogenesis. Cell Mol Life Sci. 2013;70(5):795–814. doi: 10.1007/s00018-012-1080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman EJ, Carrington JC. Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet. 2007;8(11):884–896. doi: 10.1038/nrg2179. [DOI] [PubMed] [Google Scholar]
- 54.Meunier J, et al. Birth and expression evolution of mammalian microRNA genes. Genome Res. 2013;23(1):34–45. doi: 10.1101/gr.140269.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23(2):431–442. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 2011;12(12):846–860. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 57.Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- 58.Loher P, Rigoutsos I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics. 2012;28(24):3322–3323. doi: 10.1093/bioinformatics/bts615. [DOI] [PubMed] [Google Scholar]
- 59.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 60.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kozomara A, Griffiths-Jones S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database issue):D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426(6968):845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- 63.Reinhart BJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 64.Slack FJ, et al. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5(4):659–669. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- 65.Grad Y, et al. Computational and experimental identification of C. elegans microRNAs. Mol Cell. 2003;11(5):1253–1263. doi: 10.1016/s1097-2765(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 66.Lim LP, et al. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17(8):991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18(10):505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 68.Martin HC, et al. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol. 2014;15(3):R51. doi: 10.1186/gb-2014-15-3-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315(5808):97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 70.Chak LL, Okamura K. Argonaute-dependent small RNAs derived from single-stranded, non-structured precursors. Front Genet. 2014;5:172. doi: 10.3389/fgene.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rumble SM, et al. SHRiMP: Accurate mapping of short color-space reads. PLOS Comput Biol. 2009;5(5):e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17(1):10–12.
- 73.Vourekas A, et al. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat Struct Mol Biol. 2012;19(8):773–781. doi: 10.1038/nsmb.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hafner M, et al. PAR-CliP: A method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp. 2010;41:2034. doi: 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.