

Abstract
MicroRNAs (miRNAs) are small RNAs that regulate target gene expression. Using microarray-based miRNA expression profiling, we compared the miRNA expression in granulocytes from four patients with acute myeloid leukemia and four healthy controls. Thirty-four miRNAs were found to be differentially expressed, including 20 miRNAs that were up-regulated and 14 miRNAs that were down-regulated. The expression of selected miRNAs (miR-26a-5p and miR-23b-3p) was independently validated in 20 patients and 12 healthy controls. Notably, we demonstrated that peroxiredoxin III (PrxIII) is a common direct target of both miR-26a-5p and miR-23b-3p. Furthermore, these results indicate that the two decreased miRNAs could scavenge cellular reactive oxygen species (ROS) by targeting the PrxIII gene. These findings are discussed with regard to the known function of PrxIII as a ROS scavenger and the high endogenous ROS levels required for hematopoietic stem cell differentiation. These findings may potentially offer insights into the pathological relationships between miR-26a-5p, miR-23b-3p and leukemogenesis.
Keywords: AML, miR-26a-5p, miR-23b-3p, PrxIII
Introduction
Acute myeloid leukemia (AML) is a malignant disease characterized by proliferation and maturation arrest of myeloid blasts in bone marrow and blood [1,2]. Genetic or epigenetic abnormalities confer the leukemic clones with unlimited self-renewal and blockage of differentiation at the specific stage of development [3]. In addition to recurrent structural and numerical chromosomal aberrations [e.g. t(15;17), − 5q, − 7q, + 8], the discovery of mutant genes (e.g. FLT3-ITD, CEBPA and NPM1) and dysregulated oncogenes (ERG and BAALC overexpression) has improved the molecular and prognostic classification of AML [2,4].
Over the past few years, a new class of small, non-coding RNAs, named microRNAs (miRNAs), has been demonstrated to be altered in AML [5–7]. Evolutionarily conserved miRNAs are 18–24 nucleotides (nt) in length and regulate the expression of target mRNAs post-transcriptionally, either through translational inhibition or destabilization of target mRNAs [8]. Several studies have shown that miR-155 expression is elevated in many solid tumors, lymphomas and acute leukemias [9–12]. miR-196b has been reported to be up-regulated in patients with AML with t(11q23)/MLL [13–15]. The up-regulation of miR-15a/16-1 has been observed in patients with AML with retinoic acid treatment [16]. Correspondingly, lower levels of miR-223 and miR-29 have been detected in AML samples [17–19]. These data support tumor suppressor functions of the down-regulated miRNAs, and provide a rationale for the use of synthetic miRNAs as novel therapeutic options in AML [20–22]. Despite these advances, additional miRNAs and their functional roles in AML remain essentially unclear and are still a matter of interest.
Peroxiredoxins (Prxs) belong to a family of thiol-specific antioxidant proteins that also participate in mammalian cell signal transduction [23–25]. The human Prxs include six isoforms (PrxI to PrxVI), and are classified into three subgroups (2-Cys, atypical 2-Cys and 1-Cys) based on the number and positions of the Cys residues that participate in catalysis [26]. PrxIII belongs to the 2-Cys subgroup, and contains both N- and C-terminal Cys residues [27]. During PrxIII catalysis, there are two active sites where Cys residues are oxidized by peroxide substrates to form disulfide bonds [28]. Notably, alterations in the protein levels of Prxs have been observed in several types of cancer [3,29]. PrxIII has been reported to be elevated in the formation and development of hepatocellular carcinomas [30].
The present study aimed to identify the specific miRNAs associated with AML. To accomplish this, we applied microarray-based miRNA expression profiling to characterize the miRNAs that are differentially expressed in granulocyte cells of peripheral blood from untreated patients with AML and healthy controls. Several differentially expressed miRNAs were selected and tested. Among all the validated miRNAs, we report down-regulated miR-26a-5p and miR-23b-3p expression for patients with AML and demonstrate that the two miRNAs modulate the common target PrxIII gene. Moreover, the down-regulated miR-26a-5p and miR-23b-3p exhibited elevation of PrxIII in AML granulocyte samples and transfected cells. Taken together, our findings demonstrate that down-regulation of miR-26a-5p and miR-23b-3p by targeting PrxIII may reveal important insights into the pathogenesis of AML.
Materials and methods
Study population
The present study included 24 patients with AML (13 females, 11 males) and 16 age- and gender-matched healthy subjects (eight females, eight males). The 24 patients with AML included three with M1 (acute myeloblastic leukemia with minimal maturation), eight with M2 (acute myeloblastic with maturation) and 13 with M4 disease (acute myelomonocytic leukemia). The mean age of the patients and control subjects was 41 ± 8 years and 34 ± 6 years, respectively. The diagnosis of leukemia was made by morphologic and cytochemical studies of bone marrow smears. All patients were not taking any anti-leukemic therapy at the moment of blood sampling and were newly diagnosed with AML. The study was approved by the Ethics Review Committee for Human Studies of the Shandong University School of Medicine. All participants provided written informed consent before any blood sampling.
Cell preparation and RNA extraction
Five-milliliter K2EDTA (dipotassium ethylenediaminetetraacetic acid) peripheral blood (PB) samples were collected from patients with AML and healthy individuals in Lymphoprep separation medium (Solarbio, Beijing, China). Blood cells were separated by centrifugation at 2000 rpm for 20 min at 4°C. The blood samples were then separated into four parts, namely plasma, mononuclear, Lymphoprep and sediment layer (including granulocytes and erythrocytes). For enough cell amounts for miRNA microarray, quantitative polymerase chain reaction (PCR) and Western blotting analysis in this study, we used granulocytes isolated from 5 mL PB to detect miRNA expression and verify the cellular results. The sediment layer was treated with red blood cell lysis buffer and was then separated by centrifugation at 2000 rpm for 10 min. The isolated leukocytes were washed three times in phosphate buffered saline (PBS). As shown in Supplementary Figure 1 to be found online at http://informahealthcare.com/doi/abs/10.3109/10428194.2014.924115, the percentage of granulocytes (CD11b+ CD15+) after the isolation procedures was assessed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and was higher than 80%. Total RNA was isolated from granulocytes using TRIzol reagent (Invitrogen, Carlsbad, CA) and was treated with RNase-Free DNase (Promega, Fitchburg, WI), according to the manufacturer's instructions.
Figure 1.
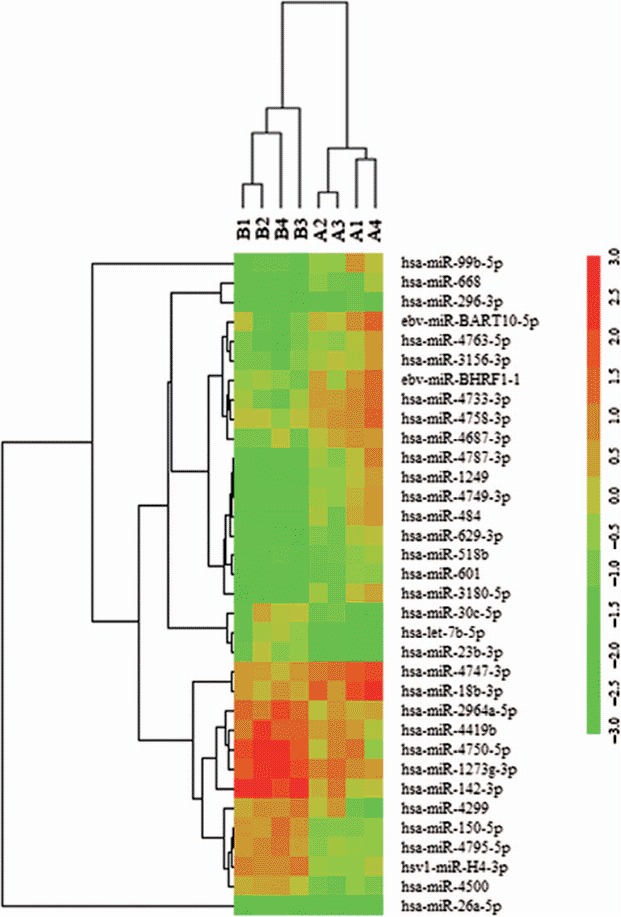
Heat map of miRNA microarray expression data in granulocytes of peripheral blood from patients with AML (n = 4) and healthy controls (n = 4). The expression of miRNA is hierarchically clustered on the y-axis, and AML granulocyte samples or healthy control granulocyte samples are hierarchically clustered on the x-axis. Legend on the right indicates miRNA represented in the corresponding row. Relative miRNA expression is depicted according to the color scale shown on the right. Red indicates up-regulation, green down-regulation. Numbers A1, A2, A3, A4 indicate AML patient samples; numbers B1, B2, B3, B4 indicate healthy control samples.
miRNA profile
The miRNA microarray profiling was conducted by KangChen Bio-tech (Shanghai, China). miRNA expression profiling of the granulocyte samples was performed using the miRCURY LNA Array (version 11.0) system (Exiqon Inc., Woburn, MA). RNA samples were labeled using a miRCURY Hy3/Hy5 Power labeling kit (Exiqon Inc.) and hybridized on the miRCURY LNA Array station (version 11.0; Exiqon Inc.). Scanning was performed with an Axon GenePix 4000B microarray scanner (Molecular Devices, LLC, Sunnyvale, CA). GenePix Pro version 6.0 was used to read the raw data of the images. The intensity of the green signal was calculated after background subtraction, and replicated spots in the same image were averaged to obtain the median intensity. The median normalization method was used to obtain normalized data (foreground minus background divided by median). The median represents the 50th quartile of miRNA intensity, which is > 50% in all samples after background correction. The significance of the results was determined using fold change and t-tests. The threshold value for significance used to define up-regulation or down-regulation of miRNAs was a fold change > 2 and p-value < 0.05. The miRNAs selected for investigation in this study were further filtered on the basis of expression levels and previously published data.
Detection and quantification of miRNAs by quantitative PCR
To confirm the findings obtained by miRNA profiling, real-time quantitative reverse transcription PCR (qRT-PCR) was performed using an ABI 7500 system (Applied Biosystems, Foster City, CA). The relative expression level of miRNAs was normalized to that of the internal control human U6 by using the 2−ΔΔCt cycle threshold method. The qPCR mixture contained the following components: 12.5 μL SYBR Premix Ex TaqII (Takara Bio, Otsu, Japan), 2.0 μL cDNA, 8.5 μL H2O and 1.0 μL specific miRNA primer (Takara Bio). The qPCR reaction cycles were: one cycle of 30 s at 95°C, 40 cycles of 5 s at 95°C, 34 s at 60°C, 30 s at 72°C and final extension 10 min at 72°C.
3′-UTR luciferase reporter assays
Fluorescent reporter experiments were conducted to confirm that the PrxIII gene, which was predicted to contain miR-26a-5p and miR-23b-3p binding sites, was indeed the target of these two miRNAs. The PrxIII 3′-untranslated region (UTR) luciferase reporter construct was made by amplifying the human PrxIII mRNA 3′-UTR sequence and cloning it to the 3′ end of a firefly luciferase reporter gene in the pmirGLO dual luciferase vector (Promega). Site-directed mutagenesis of a miR-26a-5p or miR-23b-3p target site of PrxIII mRNA was performed using a site-directed gene mutagenesis kit (Beyotime, Beijing, China) using the pmirGLO-PrxIII 3′-UTR plasmid as a template. Mutagenesis primers for a miR-26a-5p target site of PrxIII were 5′-GTG CGG ATC GTA TTA TTT TAA AAA GTG G-3′ (forward) and 5′-GAT TGA CGC AAG GCT AAG AAA GAA G-3′ (reverse). Mutagenesis primers for a miR-23b-3p target site of PrxIII were 5′-GCC TTG CGT CAA TCT TTT CAT CTT G-3′ (forward) and 5′-GAT CCG CAC TTG CTT CAA TCA CAT AAA C-3′ (reverse). HEK293 cells were co-transfected with 10 ng of luciferase reporter plasmid, and miR-26a-5p and miR-23b-3p (final concentration, 5 pM) using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Luciferase activity was measured using the Dual-Luciferase Reporter Assay system (Promega).
Cell culture, transfection and Western blotting analysis
HEK293 and K562 cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) and Iscove's modified Dulbecco's medium with 10% fetal bovine serum (FBS) plus penicillin and streptomycin in a humidified incubator at 37°C with 5% CO2. HEK293 or K562 cells were transfected the following day using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For overexpression of miRNA, 120 pmol of miR-26a-5p mimic, miR-23b-3p mimic or negative control mock-miRNA (GenePharma, Beijing, China) was used. For knockdown of miRNA, 100 pmol of miR-26a-5p inhibitor, miR-23b-3p inhibitor or negative control miRNA inhibitor (GenePharma) was used. Cells were harvested 72 h after transfection. For siRNA-mediated knockdown of PrxIII, cells were transfected with 100 pmol of small interfering RNA (siRNA) duplexes (5′-GUG GCA GAG UGA CUU AAC UUT T-3′) or negative control siRNA duplexes (5′-UUC UCC GAA CGU GUC ACG UTT-3′) (GenePharma). Analysis was performed after an additional 48 h. Plasmid methods for generating expression constructs of human PrxIII (pcDNA3.1/Myc-HisA-PrxIII) were conducted as previously described [25].
Antibodies against anti-PrxIII (Abcam, Cambridge, UK), anti-actin (Santa Cruz Biotechnology, Dallas, TX) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology) were purchased. The protein samples prepared from cell cultures were quantified using the Bradford assay, subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto polyvinylidene fluoride (PVDF) membranes (GE Healthcare, Fairfield, CT) for 1 h at 100 V using a standard transfer solution. The membranes were then incubated overnight at 4°C with the appropriate primary antibody at a 1:1000 dilution, followed by incubation with anti-goat or anti-mouse immunoglobulin G (IgG)-conjugated horseradish peroxidase at a 1:1000 dilution. The proteins were visualized by chemiluminescence using an enhanced chemiluminescence (ECL) kit (Thermo Fisher Scientific, Waltham, MA).
Cell differentiation assay and determination of cellular ROS by flow cytometry
Human K562 is a human leukemia cell line used as model of hematopoietic differentiation. K562 cells induced by phorbol myristate acetate (PMA) (Sigma-Aldrich, St. Louis, MO) were stained with anti-human CD11b phycoerythrin (PE) (eBioscience, San Diego, CA) in accordance with the manufacturer's instructions. Differentiation was analyzed by quantifying the CD11b-positive cell population by flow cytometry using the FACSCalibur flow cytometer (BD Biosciences). To measure cellular reactive oxygen species (ROS) levels, the cells were incubated with 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) (Beyotime) in the dark for 15 min at 37°C. After washing, the cells were analyzed by flow cytometry, which was performed using the FACSCalibur flow cytometer (BD Biosciences). Data were analyzed using the FCSExpress V3 program (DeNovo Software, Los Angeles, CA).
Statistical analysis
All continuous variables are expressed as mean ± standard deviation (SD), unless stated otherwise. Categorical variables were compared using the χ2 test, and continuous variables were compared using Student's t-test. All tests were performed using a two-sided significance level, and p < 0.05 was considered statistically significant (∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001). GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA) was used for all statistical analyses. All images of Western blotting are representative of at least three independent experiments. qRT-PCR was performed in triplicate and repeated three times.
Results
miRNA profiles in patients with AML versus healthy subjects
To identify aberrantly expressed miRNAs in AML, a miRNA expression assay was performed in granulocytes from four untreated patients with AML and four healthy donors using the miRCURY LNA Array (version 11.0) system. The levels of miRNAs differed significantly between patients and healthy controls (Figure 1). Of 1700 miRNAs detected on the microarray, 34 miRNAs were found to be differentially expressed in patients with AML compared with controls. Quantification revealed that multiple miRNAs were significantly differentially expressed in patients compared with healthy controls. According to their known and postulated functions, five miRNAs were selected and further tested. Two of these miRNAs were up-regulated in patients with AML: miR-296-3p and miR-99b-5p. Three miRNAs were down-regulated: miR-150-5p, miR-26a-5p and miR-23b-3p. The miRNA names, fold changes and p-values are presented in Table I.
Table I.
Properties of microRNAs differentially expressed in patients with AML compared with healthy control subjects.
| miRNA | Fold-change | p-Value | Chromosome location | Potential targets |
|---|---|---|---|---|
| Up-regulated miRNAs | ||||
| miR-296-3p | 2.33 | < 0.05 | 20q13.32 | ZNF732, ZNF138 |
| miR-99b-5p | 3.68 | < 0.05 | 19q13.41 | HES7, AP1AR |
| Down-regulated miRNAs | ||||
| miR-26a-5p | 0.39 | < 0.05 | 3p22.2 | PrxIII, B3GNT5, NEK6 |
| miR-23b-3p | 0.28 | < 0.01 | 9q22.32 | PrxIII, WBP2, AUH |
| miR-150-5p | 0.32 | < 0.01 | 19q13.33 | SV2B, ADIPOR2 |
AML, acute myeloid leukemia.
Quantitative reverse-transcription polymerase chain reaction validation of profiling data
To confirm the findings of the miRNA profile, the expression of these five differentially regulated miRNAs was measured using qRT-PCR. The relative expression of each miRNA in patients with AML compared with healthy controls is shown in Figure 2. The results demonstrated a 0.21-fold decrease of miR-26a-5p (p < 0.01) and a 0.53-fold decrease of miR-23b-3p (p < 0.05) in patients with AML. miR-26a-5p and miR-23b-3p are of particular interest among all the validated miRNAs. miR-26a-5p has been reported to inhibit monocytic differentiation of AML cells [31]. In the present study, we chose to use granulocytes isolated from 5 mL PB for sufficient amounts of cells, and miR-26a-5p/miR-23b-3p were significantly decreased in AML as shown in the distinct granulocyte-based miRNA expression pattern. Most importantly, computational analysis from several widely used mammalian target prediction programs, including miRBase, TargetScan, miRanda, and PicTar, indicated that both miR-26a-5p and miR-23b-3p regulate the same target gene: PrxIII, potentially contributing to impaired self-renewal and differentiation of hematopoietic stem cells (HSCs) in leukemogenesis.
Figure 2.
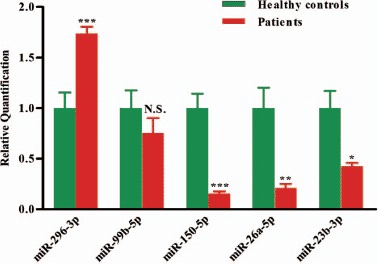
Validation of miRNA microarray data by quantitative reverse transcription-polymerase chain reaction. The microarray cohort included four patients with AML and four healthy controls. Relative expression of five miRNAs was normalized to expression of the internal control (U6). p-Values were calculated by two-sided Student's t-test. ∗p < 0.05; ∗∗p < 0.01 and ∗∗∗p < 0.001.
Independent validation of miRNA expression
To further verify the decreased miR-26a-5p and miR-23b-3p in patients with AML, we studied a second set of granulocyte samples from 20 patients with AML and 12 healthy controls. As shown in Figure 3, the two miRNAs were significantly decreased in patients compared with healthy controls. Our results showed a 0.14-fold decrease of miR-26a-5p and a 0.18-fold decrease of miR-23b-3p in patients with AML compared with healthy control subjects (both p < 0.0001).
Figure 3.
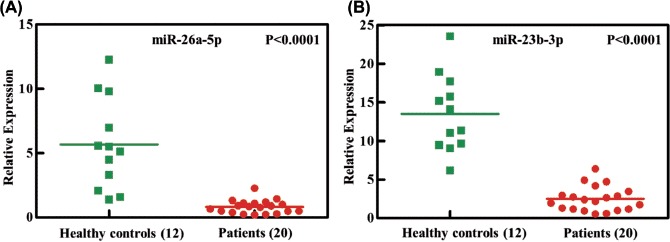
Independent validation of differential expression of miRNA in patients with AML. Quantitative reverse-transcription polymerase chain reaction for two miRNAs, (A) miR-26a-5p and (B) miR-23b-3p, in an independent validation set of 20 patients with AML and 12 healthy controls. Relative expression of each miRNA in patients with AML compared with healthy controls was normalized to human U6 expression. Horizontal lines indicate medians. p-Values (p < 0.0001) were calculated by Student's t-test.
PrxIII is a direct target of both miR-26a-5p and miR-23b-3p
To identify the direct target genes of miR-26a-5p or miR-23b-3p, we combined several widely used mammalian target prediction programs, including miRBase, miRanda, TargetScan and PicTar [32–34]. Among all the putative genes, PrxIII is of particular interest because it is present in the lists of candidate genes targeted by both miR-26b-5p and miR-23b-3p. Thus, we investigated whether miR-26a-5p and miR-23b-3p target PrxIII for expression regulation. To this end, Western blot and qRT-PCR were performed to demonstrate the effects of miR-26a-5p and miR-23b-3p on the regulation of PrxIII expression in vitro. HEK293 cells were treated with inhibitors specific for miR-26a-5p, miR-23b-3p or miR-26a-5p + miR-23b-3p or with control microRNAs [Figure 4(A)]. Quantitative summary of three replicated experiments indicated that 3.1-fold of PrxIII protein expression was simultaneously increased following transfection with miR-26a-5p + miR-23b-3p compared with 2.3-fold in cells transfected with miR-26a-5p or 2.2-fold in cells transfected with miR-23b-3p [Figures 4(A) and 4(C)]. When inhibitors of miR-26a-5p or miR-23b-3p were transfected into cells, PrxIII mRNA levels increased compared with the negative controls [Figure 4(D)]. Accordingly, miR-26a-5p mimic or miR-23b-3p mimic could significantly decrease the mRNA and protein levels of PrxIII gene [Figures 4(B), 4(C) and 4(D)].
Figure 4.
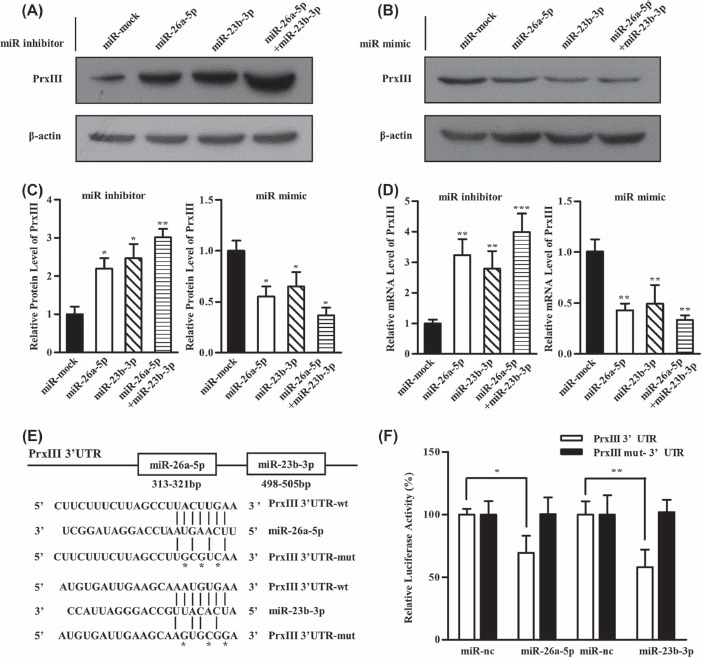
PrxIII is a direct target of both miR-26a-5p and miR-23b-3p. (A) HEK293 cells were transfected with miRNA inhibitors specific for miR-26a-5p, miR-23b-3p and miR-26a-5p + miR-23b-3p. Equivalent amounts (30 μg) of whole-cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins. (B) HEK293 cells were transfected with miRNA mimics specific for miR-26a-5p, miR-23b-3p and miR-26a-5p + miR-23b-3p. Equivalent amounts (30 μg) of whole-cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins. (C) Compiled data of PrxIII protein analysis from three independent experiments after transfection of miRNAs is shown. Columns, mean; bars, ± SD; ∗p < 0.05, ∗∗p < 0.01. (D) Compiled data of PrxIII mRNA analysis from three independent experiments after transfection of miRNAs is shown. Columns, mean; bars, ± SD; ∗∗p < 0.01, ∗∗∗p < 0.001. (E) Predicted human PrxIII-encoded mRNA contains a 3′-UTR element that is partially complementary to the indicated miRNAs. Above: predicted target binding sites for miR-26a-5p and miR-23b-3p, respectively. Below: predicted duplex combination between human PrxIII 3′-UTR-wt/mut and miR-26a-5p, PrxIII 3′-UTR-wt/mut and miR-23b-3p. (F) Luciferase activity of pmirGLO3-PrxIII-wt or pmirGLO-PrxIII-mut in HEK293 cells after miR-26a-5p and miR-23b-3p transfections. Data are shown as mean ± SD based on three independent experiments, ∗p < 0.05, ∗∗p < 0.01.
We further determined whether miR-26a-5p and miR-23b-3p could directly alter the expression of PrxIII. The PrxIII-encoded mRNA contains a 3′-UTR element that is partially complementary to miR-26a-5p and miR-23b-3p [Figure 4(E)], indicating that the miRNAs might directly target the corresponding sites. A fragment of the 3′-UTR of PrxIII mRNA containing the wild-type or mutated putative miR-26a-5p or miR-23b-3p corresponding binding sequence was cloned into a luciferase reporter construct, respectively [Figure 4(E)]. The luciferase reporter assay in HEK293 cells showed that the luciferase activity was decreased by 31% when co-transfected with miR-26a-5p and its corresponding pmirGLO-PrxIII-wt [Figure 4(F), open bars] and 43% when co-transfected with miR-23b-3p and its corresponding pmirGLO-PrxIII-wt [Figure 4(F), open bars], whereas no significant reduction in luciferase activity was observed when the cells were transfected with miR-mock. The construct pmirGLO-PrxIII-mut was then used to repeat the luciferase assay experiments in HEK293 cells, which showed that mutating the seed region for miR-26a-5p or miR-23b-3p in the pmirGLO-PrxIII-wt plasmid completely abrogated its regulatory activity [Figure 4(F), filled bars]. The results demonstrated that miR-26a-5p or miR-23b-3p might suppress PrxIII expression through corresponding binding sites at the 3′-UTR of PrxIII.
Detection of PrxIII in K562 cells and granulocytes from patients with AML
To study the significance of the miR-26a-5p- and miR-23b-3p-dependent suppression of PrxIII expression, a human myeloid leukemia cell line (K562) was treated with inhibitors and mimics specific for miR-26a-5p and miR-23b-3p. As shown in Figures 5(A) and 5(B), the mRNA and protein levels of the PrxIII gene increased significantly compared with the negative controls when inhibitors of miR-26a-5p or miR-23b-3p were transfected into K562 cells. Correspondingly, mimics of miR-26a-5p or miR-23b-3p decreased PrxIII mRNA and protein levels [Figures 5(A) and 5(B)]. In further analysis of PrxIII in granulocytes from untreated patients with AML, we found that the protein levels of PrxIII were significantly higher in 11 patients with AML compared with six control subjects [Figures 5(C) and 5(D)]. Our findings suggest that elevated PrxIII protein levels were regulated by decreased miR-26a-5p and miR-23b-3p in patients with AML compared with control subjects. In addition, the increase in PrxIII expression caused by inhibitor specific for miR-26a-5p or miR-23b-3p was offset in siPrxIII + miR-26a-5p/miR-23b-3p inhibitor cells, demonstrating that the elevated PrxIII was indeed mediated by the down-regulation of miR-26a-5p or miR-23b-3p [Figure 5(E)].
Figure 5.
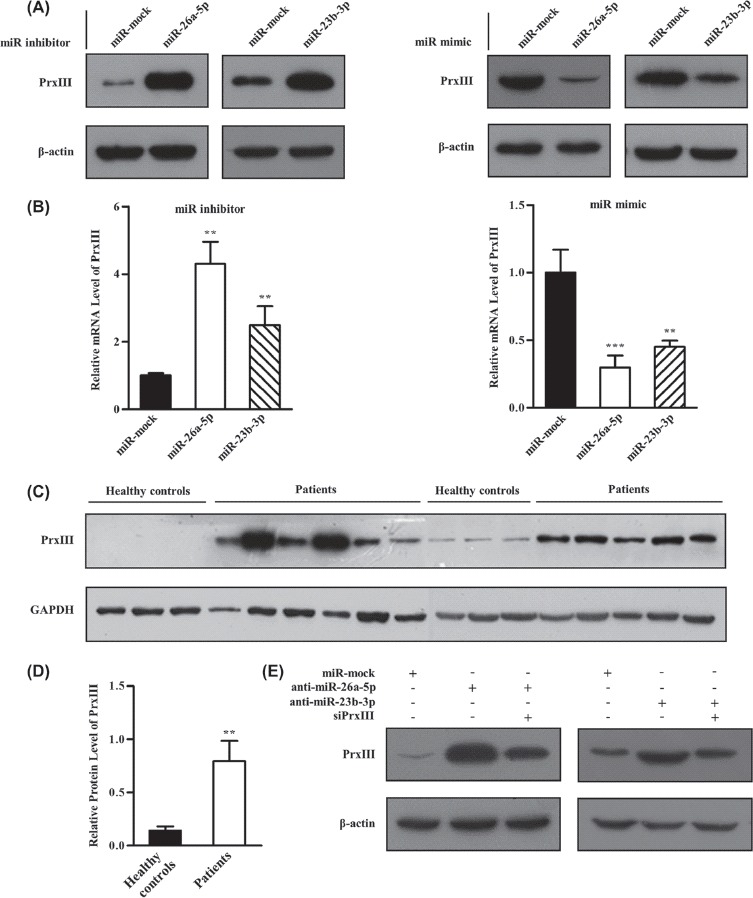
miR-26a-5p and miR-23b-3p repressed PrxIII expression in K562 cells and granulocytes from patients with AML. (A) K562 cells were transfected with miRNA inhibitors (left) and mimics (right) specific for miR-26a-5p or miR-23b-3p. Equivalent amounts (30 μg) of whole-cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins. (B) PrxIII mRNA analysis from three independent experiments after transfection of miRNAs is shown. Columns, mean; bars, ± SD; ∗∗p < 0.01, ∗∗∗p < 0.001. (C) Equivalent amounts (35 μg) of whole-granulocyte lysates from 11 untreated patients with AML and six healthy controls were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins. (D) Relative expression of PrxIII in patients with AML compared with healthy controls was normalized to GAPDH expression. Columns, mean; bars, ± SD; ∗∗p < 0.01. (E) Transfection with indicated siRNA and miRNA inhibitors. Equivalent amounts (30 μg) of whole-cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins.
Determination of cellular ROS by flow cytometry
Next, we investigated the contribution of the miR-26a-5p/miR-23b-3p-PrxIII regulatory pathway to the ROS status in HEK293 and K562 cell lines. ROS are involved in regulating the balance between self-renewal and differentiation of HSCs. The increased ROS levels might act as an intracellular trigger for HSC differentiation to myeloid lineage fates in hematopoietic systems [35,36]. Therefore, we predicted that overexpression of PrxIII caused by down-regulation of miR-26a-5p and miR-23b-3p would have similar effects. To this end, HEK293 and K562 cells were first treated with control miRNA and miR-26a-5p/miR-23b-3p inhibitor. Next, cells were pre-labeled with DCFH-DA (a free radical-recognizing dye) and then analyzed using flow cytometry. Importantly, the transfection of miR-26a-5p inhibitor or miR-23b-3p inhibitor led to a significant decrease in ROS production in both HEK293 cells and K562 cells [Figure 6(A), red and blue lines]. Quantitative summary of three replicated experiments indicated that ROS production was reduced significantly in miR-26a-5p/miR-23b-3p down-regulated cells compared with negative control cells [Figure 6(B)]. ROS induction by miR-26a-5p or miR-23b-3p mimic was also measured. As expected, the results showed that ROS levels were significantly higher in miR-26a-5p/miR-23b-3p up-regulated cells compared with control cells [Figures 6(C) and 6(D)]. To determine whether the difference was due to the PrxIII expression, ROS production was measured in miR-26a-5p/miR-23b-3p inhibitor + siPrxIII cells. As shown in Figure 6(E), the decrease in ROS production caused by miR-26a-5p/miR-23b-3p inhibitor (red line) was offset in miR-26a-5p/miR-23b-3p inhibitor + siPrxIII (blue line), demonstrating that the reduction in ROS production of the transfection of miR-26a-5p/miR-23b-3p inhibitor was indeed mediated by the up-regulation of PrxIII. Quantitative summary of three replicated experiments indicated that ROS production was reduced significantly in anti-miR-26a-5p (anti-miR-23b-3p) cells compared with negative control cells, whereas in miR-26a-5p/miR-23b-3p inhibitor + siPrxIII cells, ROS production was restored to the level in negative control cells [Figure 6(F)].
Figure 6.
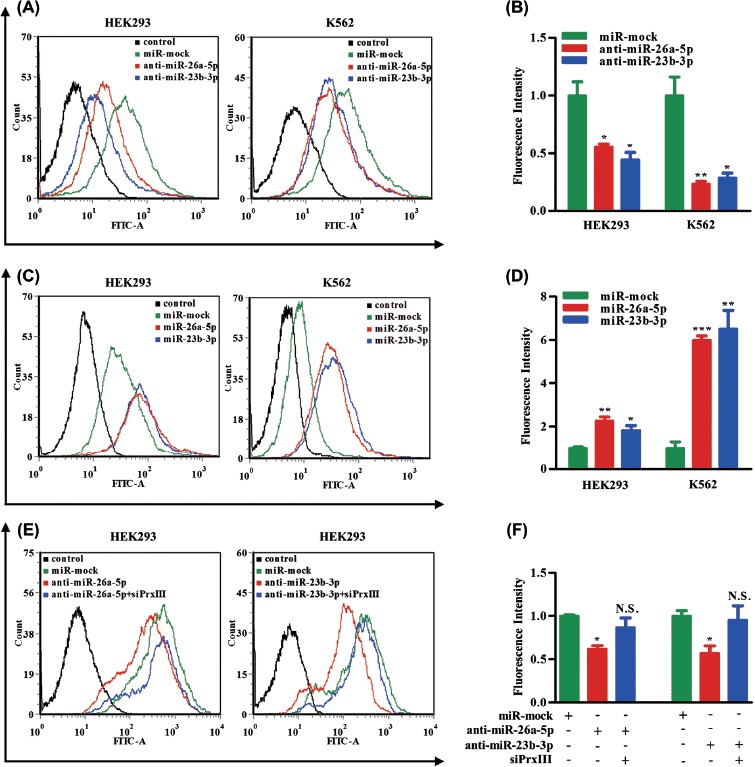
miR-26a-5p and miR-23b-3p decrease levels of ROS. (A) HEK293 and K562 cells transfected with control, miR-mock, miR-26a-5p inhibitor and miR-23b-3p inhibitor were labeled with DCFH-DA and analyzed by flow cytometry. A representative histogram (green, red and blue lines indicate ROS production in transfection of miR-mock, miR-26a-5p inhibitor and miR-23b-3p inhibitor, respectively) is shown. (B) DCFH-DA fluorescence measurements of cellular ROS levels in transfection of miR-mock, miR-26a-5p inhibitor and miR-23b-3p inhibitor in HEK293 or K562 cells. Columns, mean; bars, ± SD; ∗p < 0.05, ∗∗p < 0.01. Compiled data were produced from three independent experiments. (C) HEK293 and K562 cells transfected with control, miR-mock, miR-26a-5p mimic and miR-23b-3p mimic were labeled with DCFH-DA and analyzed by flow cytometry. A representative histogram (green, red and blue lines indicate ROS production in transfection of miR-mock, miR-26a-5p mimic and miR-23b-3p mimic, respectively) is shown. (D) DCFH-DA fluorescence measurements of cellular ROS levels in transfection of miR-mock, miR-26a-5p mimic and miR-23b-3p mimic in HEK293 or K562 cells. Columns, mean; bars, ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.01. Compiled data were produced from three independent experiments. (E) HEK293 and K562 cells transfected with control, miR-mock, miR-26a-5p inhibitor + siPrxIII and miR-23b-3p inhibitor + siPrxIII were labeled with DCFH-DA and analyzed by flow cytometry. A representative histogram (green, red and blue lines indicate ROS production in transfection of miR-mock, miR-26a-5p/miR-23b-3p inhibitor and miR-26a-5p/miR-23b-3p inhibitor + siPrxIII, respectively) is shown. (F) DCFH-DA fluorescence measurements of cellular ROS levels in transfection of miR-mock, miR-26a-5p/miR-23b-3p inhibitor and miR-26a-5p/miR-23b-3p inhibitor + siPrxIII in HEK293 cells. Columns, mean; bars, ± SD; ∗p < 0.05. Compiled data were produced from three independent experiments.
PrxIII responsible for ROS production and K562 cell differentiation
Last, we investigated the contribution made by the PrxIII/ROS regulatory pathway to AML cell differentiation. The K562 cell line has been used as a model for the study of AML cell differentiation, which could be achieved by exposure to PMA [37]. As shown in Figure 7(A), Western blotting analysis revealed that the expression of PrxIII was significantly repressed during differentiation of K562 cells induced by PMA. ROS has been reported as a key factor for PMA-induced differentiation [38]. PMA-differentiated K562 cells were conducted with silencing/overexpression of PrxIII, to clarify its function during differentiation by detection of the CD11b differentiation marker. To this end, K562 cells were treated with siRNA specific for PrxIII and with the pcDNA3.1A-PrxIII plasmid [Figures 7(B) and 7(C)]. Next, cells were pre-labeled with DCFH-DA and anti-human CD11b and then analyzed using flow cytometry, respectively. Importantly, silencing of PrxIII led to a significant increase in ROS production and vice versa [Figure 7(D), red and blue lines]. Quantitative summary of three replicated experiments indicated that ROS production was increased 2.6-fold in PrxIII-silenced cells compared with negative control cells, whereas in pcDNA3.1A-PrxIII transfected cells, ROS production was reduced 0.7-fold compared with negative control cells [Figure 7(E)]. As shown in Figures 7(F) and 7(G), our data showed that the percentage of CD11b+ was 27.6 ± 3.6% induced by PMA, 78.2 ± 9.9% with PMA induction and silencing of PrxIII, and 1.5 ± 0.2% with PMA induction and transfection with pcDNA3.1A-PrxIII plasmid. Collectively, these results support the conclusion that the level of PrxIII was responsible for ROS production and AML cell differentiation.
Figure 7.
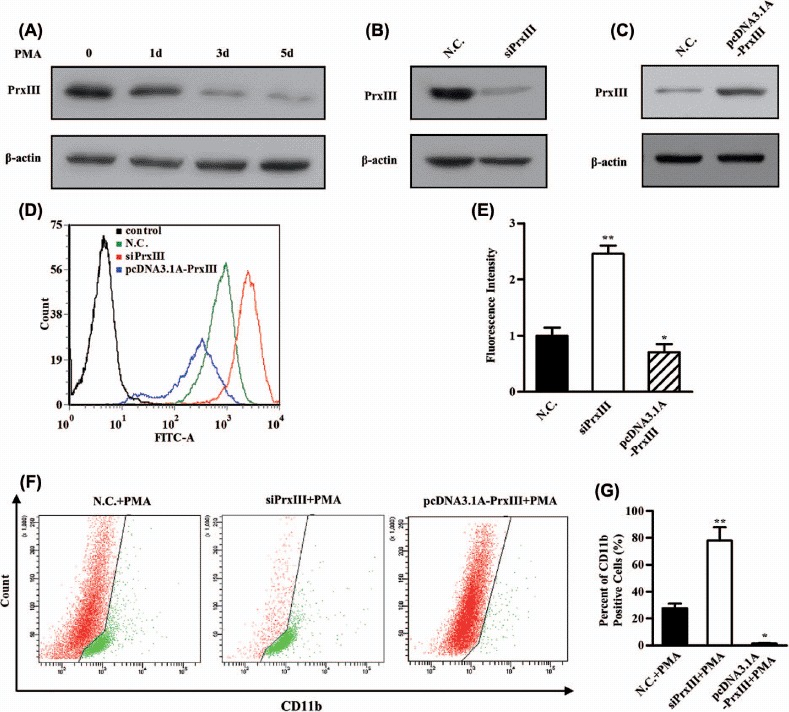
PrxIII responsible for ROS production and K562 cell differentiation. (A) K562 cells were induced by PMA. Equivalent amounts (30 μg) of whole-cell lysates were extracted at several time points (0 day, 1 day, 3 days and 5 days), separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins. (B, C) Transfection with the indicated siRNA and plasmid. Equivalent amounts (30 μg) of whole-cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for the indicated proteins. (D) K562 cells transfected with siPrxIIII, pcDNA3.1A-PrxIII and control were labeled with DCFH-DA and analyzed by flow cytometry. A representative histogram (green, red and blue lines indicate ROS production in transfection of normal control (N.C.), siPrxIII and pcDNA3.1A-PrxIII, respectively) is shown. (E) DCFDA fluorescence measurements of cellular ROS levels in transfection of N.C., siPrxIII and pcDNA3.1A-PrxIII in K562 cells, respectively. Columns, mean; bars, ± SD; ∗p < 0.05, ∗∗p < 0.01. Compiled data were produced from three independent experiments. (F) Representative CD11b fluorescence measurements of differentiation percentage in transfection of N.C., siPrxIII and pcDNA3.1A-PrxIII in K562 cells, respectively. (G) CD11b positive percentage of differentiation in transfection of N.C., siPrxIII and pcDNA3.1A-PrxIII in K562 cells, respectively. Columns, mean; bars, ± SD; ∗∗p < 0.01. Compiled data were produced from three independent experiments.
Discussion
Leukemogenesis is an evolutionary process that involves multiple independent genetic and epigenetic events. Many different causes may alter normal leukemogenesis, including aberrant behavior of chromosomal abnormalities, dysregulation of certain oncogenes and altered intracellular pathways. Over the past few years, emerging evidence suggests that aberrantly expressed miRNAs play a significant role in leukemogenesis [5,39]. miRNAs involved in leukemogenesis are divided into two types: oncogenic miRNA (oncomir), such as miR-155, and tumor-suppressive miRNA, such as miR-29b [2,5,40].
For enough cell amounts for miRNA microarray, quantitative PCR and Western blotting analysis in the present study, we used granulocytes isolated from 5 mL PB to detect miRNA expression and verify the cellular results. We demonstrated a distinct granulocyte-based miRNA expression pattern in patients with AML compared with healthy controls, including 20 miRNAs that were up-regulated and 14 miRNAs that were down-regulated. Particularly, we observed that two miRNAs, miR-26a-5p and miR-23b-3p, were significantly decreased in patients with AML. Both miR-26 and miR-23 are functional miRNAs that have been previously studied. miR-26 has been reported in several types of tumor (such as bladder and breast cancers), and may exhibit tumor-suppressive activities during tumorigenesis [40–42]. miR-23 has also been reported to be dysregulated in renal, breast and prostate cancers [43,44]. Our results indicate that both miR-26a-5p and miR-23b-3p are dysregulated in AML and that they directly target the PrxIII gene.
PrxIII is an important regulator of cellular ROS. Elevated PrxIII induced decreased levels of cellular ROS. Although PrxIII is widely believed to function as a scavenger of H2O2 and to protect against ROS-induced damage, other functions have been proposed, such as the regulation of cellular signal transduction [45,46]. We have shown that miR-26a-5p/miR-23b-3p-inhibited cells are able to scavenge excess ROS. The K562 cell line has been used as a model for the study of differentiation, which could be achieved by exposure to PMA [37]. PMA-differentiated K562 cells were conducted with silencing/overexpression of PrxIII to clarify its function during differentiation. Our data indicated that PrxIII is responsible for ROS production and AML cell differentiation. Although the majority of studies have focused on the damaging effects of ROS, emerging evidence now suggests that the ROS signal plays a critical role in regulating the balance between self-renewal and differentiation of HSCs. High ROS levels could actually act as an intracellular trigger for HSC differentiation to myeloid lineage fate [35]. For example, a high ROS state enhances proliferation and self-renewal of HSCs [47]. It has been found that Drosophila multipotent hematopoietic progenitors maintain a high ROS status and are highly responsive to ROS stimulation [36]. Correspondingly, a decrease in normal cellular ROS levels had negative impacts on the differentiation potential into mature blood cells [36]. In addition, iron deprivation was shown to promote leukemic cell differentiation by resulting in dose- and time-dependent ROS formation [48].
In the present study, we observed that the accumulation of PrxIII caused by decreased miR-26a-5p and miR-23b-3p leads to a considerable decrease in ROS production. In support of these findings, alternations in the protein levels of Prxs were recently found in various types of cancer. PrxI and PrxII have been found to be elevated in several types of cancer cells and tissues such as oral, esophageal, pancreatic, follicular thyroid, breast and lung cancers [29]. PrxII was found to be significantly increased in AML cells by using two-dimensional electrophoresis coupled with mass spectrometry [49]. Notably, PrxII was also demonstrated as an inhibitor of myeloid cell growth by reducing ROS levels in AML [50]. Also PrxIII has been reported to be elevated in the formation and development of hepatocellular carcinomas [30]. In light of these data, the down-regulation of miR-26a-5p and miR-23b-3p is expected to cause the accumulation of PrxIII, and thus enhance its ROS scavenging capacity. It is possible that decreased ROS levels as a consequence of miR-26a-5p/miR-23b-3p deficiency might not be high enough to sustain normal hematopoietic stem cell proliferation and impair normal hematopoiesis.
In conclusion, the present study demonstrated a miRNA expression profile in granulocytes for AML and that both miR-26a-5p and miR-23b-3p directly target the PrxIII gene. It would be interesting to further investigate the effects of miR-26a-5p and miR-23b-3p and the resulting stability of PrxIII in stem cells and/or animal models. However, a large number of miRNAs might be involved in AML and leukemogenesis. In addition, our findings based on Chinese patients with AML may not be generalized to other populations and a large number of specific miRNAs might be involved in the progression and development of AML in different populations. Thus, investigation of additional miRNAs and their roles in AML are necessary to offer insights into the pathology of AML and their potential role in targeted therapies for AML.
Acknowledgments
Potential conflict of interest
Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
This work was supported by the National Science Foundation of China (81070223, 81271198), State Program of National Natural Science Foundation of China for Innovative Research Group (81021001), the Science Foundation of Shandong Province (ZR2010HM038, BS2010YY034) and Independent Innovation Foundation of Shandong University, IIFSDU.
Supplementary material available online
Supplementary Figure 1 showing CD11b+ and CD15 + percentages in peripheral blood samples from healthy controls after granulocyte isolation.
References
- [1].Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- [2].Garzon R, Heaphy CE, Havelange V, et al. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009;114:5331–5341. doi: 10.1182/blood-2009-03-211938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu CX, Zhou HC, Yin QQ, et al. Targeting peroxiredoxins against leukemia. Exp Cell Res. 2013;319:170–176. doi: 10.1016/j.yexcr.2012.06.013. [DOI] [PubMed] [Google Scholar]
- [4].Mrozek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Croce CM. MicroRNA dysregulation in acute myeloid leukemia. J Clin Oncol. 2013;31:2065–2066. doi: 10.1200/JCO.2013.48.8239. [DOI] [PubMed] [Google Scholar]
- [6].Marcucci G, Mrozek K, Radmacher MD, et al. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011;117:1121–1129. doi: 10.1182/blood-2010-09-191312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Erdogan B, Bosompem A, Peng D, et al. Methylation of promoters of microRNAs and their host genes in myelodysplastic syndromes. Leuk Lymphoma. 2013;54:2720–2727. doi: 10.3109/10428194.2013.790542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wu L, Belasco JG. Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell. 2008;29:1–7. doi: 10.1016/j.molcel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- [9].Garzon R, Croce CM. MicroRNAs in normal and malignant hematopoiesis. Curr Opin Hematol. 2008;15:352–358. doi: 10.1097/MOH.0b013e328303e15d. [DOI] [PubMed] [Google Scholar]
- [10].Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- [11].Jongen-Lavrencic M, Sun SM, Dijkstra MK, et al. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood. 2008;111:5078–5085. doi: 10.1182/blood-2008-01-133355. [DOI] [PubMed] [Google Scholar]
- [12].Garzon R, Volinia S, Liu CG, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111:3183–3189. doi: 10.1182/blood-2007-07-098749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li Z, Lu J, Sun M, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dixon-McIver A, East P, Mein CA, et al. Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS One. 2008;3:e2141. doi: 10.1371/journal.pone.0002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Popovic R, Riesbeck LE, Velu CS, et al. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood. 2009;113:3314–3322. doi: 10.1182/blood-2008-04-154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gao SM, Yang J, Chen C, et al. miR-15a/16-1 enhances retinoic acid-mediated differentiation of leukemic cells and is up-regulated by retinoic acid. Leuk Lymphoma. 2011;52:2365–2371. doi: 10.3109/10428194.2011.601476. [DOI] [PubMed] [Google Scholar]
- [17].Fazi F, Racanicchi S, Zardo G, et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell. 2007;12:457–466. doi: 10.1016/j.ccr.2007.09.020. [DOI] [PubMed] [Google Scholar]
- [18].Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- [19].Mott JL, Kobayashi S, Bronk SF, et al. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Garzon R, Liu S, Fabbri M, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pulikkan JA, Dengler V, Peramangalam PS, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood. 2010;115:1768–1778. doi: 10.1182/blood-2009-08-240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu S, Wu LC, Pang J, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010;17:333–347. doi: 10.1016/j.ccr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chang TS, Cho CS, Park S, et al. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- [24].Wood ZA, Schroder E, Robin Harris J, et al. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- [25].Li X, Lu D, He F, et al. Cullin 4B protein ubiquitin ligase targets peroxiredoxin III for degradation. J Biol Chem. 2011;286:32344–32354. doi: 10.1074/jbc.M111.249003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Monteiro G, Horta BB, Pimenta DC, et al. Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc Natl Acad Sci USA. 2007;104:4886–4891. doi: 10.1073/pnas.0700481104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- [28].Lehtonen ST, Markkanen PM, Peltoniemi M, et al. Variable overoxidation of peroxiredoxins in human lung cells in severe oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2005;288:L997–L1001. doi: 10.1152/ajplung.00432.2004. [DOI] [PubMed] [Google Scholar]
- [29].Kang SW, Rhee SG, Chang TS, et al. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med. 2005;11:571–578. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Choi JH, Kim TN, Kim S, et al. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 2002;22:3331–3335. [PubMed] [Google Scholar]
- [31].Salvatori B, Iosue I, Mangiavacchi A, et al. The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis. 2012;3:e413. doi: 10.1038/cddis.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Betel D, Wilson M, Gabow A, et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- [34].Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- [35].Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007;1:140–152. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- [36].Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sutherland JA, Turner AR, Mannoni P, et al. Differentiation of K562 leukemia cells along erythroid, macrophage, and megakaryocyte lineages. J Biol Response Mod. 1986;5:250–262. [PubMed] [Google Scholar]
- [38].Ojima Y, Duncan MT, Nurhayati RW, et al. Synergistic effect of hydrogen peroxide on polyploidization during the megakaryocytic differentiation of K562 leukemia cells by PMA. Exp Cell Res. 2013;319:2205–2215. doi: 10.1016/j.yexcr.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Garzon R, Garofalo M, Martelli MP, et al. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA. 2008;105:3945–3950. doi: 10.1073/pnas.0800135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gao J, Liu QG. The role of miR-26 in tumors and normal tissues (Review) Oncol Lett. 2011;2:1019–1023. doi: 10.3892/ol.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maillot G, Lacroix-Triki M, Pierredon S, et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009;69:8332–8340. doi: 10.1158/0008-5472.CAN-09-2206. [DOI] [PubMed] [Google Scholar]
- [42].Wang G, Zhang H, He H, et al. Up-regulation of microRNA in bladder tumor tissue is not common. Int Urol Nephrol. 2004;42:95–102. doi: 10.1007/s11255-009-9584-3. [DOI] [PubMed] [Google Scholar]
- [43].Zaman MS, Thamminana S, Shahryari V, et al. Inhibition of PTEN gene expression by oncogenic miR-23b-3p in renal cancer. PLoS One. 2012;7:e50203. doi: 10.1371/journal.pone.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu Q, Wang C, Lu Z, et al. Analysis of serum genome-wide microRNAs for breast cancer detection. Clin Chim Acta. 2012;413:1058–1065. doi: 10.1016/j.cca.2012.02.016. [DOI] [PubMed] [Google Scholar]
- [45].Baier M, Dietz KJ. The plant 2-Cys peroxiredoxin BAS1 is a nuclear-encoded chloroplast protein: its expressional regulation, phylogenetic origin, and implications for its specific physiological function in plants. Plant J. 1997;12:179–190. doi: 10.1046/j.1365-313x.1997.12010179.x. [DOI] [PubMed] [Google Scholar]
- [46].Muller FL, Lustgarten MS, Jang Y, et al. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- [47].Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Callens C, Coulon S, Naudin J, et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med. 2010;207:731–750. doi: 10.1084/jem.20091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lopez-Pedrera C, Villalba JM, Siendones E, et al. Proteomic analysis of acute myeloid leukemia: Identification of potential early biomarkers and therapeutic targets. Proteomics. 2006;6(Suppl. 1):S293–S299. doi: 10.1002/pmic.200500384. [DOI] [PubMed] [Google Scholar]
- [50].Agrawal-Singh S, Isken F, Agelopoulos K, et al. Genome-wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood. 2012;119:2346–2357. doi: 10.1182/blood-2011-06-358705. [DOI] [PubMed] [Google Scholar]