

Summary
Assembly of the divisome in E. coli occurs in two temporally distinct steps. First, FtsZ filaments attached to the membrane through interaction with FtsA and ZipA coalesce into a Z ring at midcell. After a delay, additional proteins are recruited to the Z ring in a hierarchical manner to form a complete divisome, activated by the arrival of FtsN. Recently, we proposed the interaction of FtsA with itself competes with its ability to recruit downstream division proteins (both require the same IC domain of FtsA) and that ZipA’s essential function is to promote the formation of FtsA monomers. Here, we tested whether overexpression of a downstream division protein could make ZipA dispensable, presumably by shifting the FtsA equilibrium to monomers. Only overexpression of FtsN bypassed ZipA and we identified a motif in the cytoplasmic domain of FtsN required for both the bypass of ZipA and interaction with FtsA. In addition, this cytoplasmic motif has to be linked to the periplasmic E domain of FtsN in order to bypass ZipA, suggesting that FtsN was linking FtsA to periplasmic components of the divisome. These results are used to further elaborate our model for the role of FtsA in recruiting downstream division proteins.
Keywords: FtsA, FtsN, ZipA, FtsZ, divisome, cell division
Introduction
Cytokinesis in bacteria relies on a relatively conserved trans-envelope protein complex called the divisome. In E. coli this complex is organized in a ring-shaped structure composed of 12 essential core proteins, which are recruited to the division site in a sequential manner in two temporally distinct stages (Lutkenhaus et al., 2012, de Boer, 2010, Aarsman et al., 2005). In the first stage, FtsZ is recruited to the cytoplasmic side of the inner membrane through interaction with FtsA and ZipA to form the Z ring (Pichoff & Lutkenhaus, 2002, Hale & de Boer, 1999, Hale & de Boer, 1997, Bi & Lutkenhaus, 1991). The Z ring is restricted to midcell by the combined action of the Min and the Nucleoid Occlusion systems (Bernhardt & de Boer, 2005, Pichoff & Lutkenhaus, 2001, de Boer et al., 1989, Bi & Lutkenhaus, 1993, Lutkenhaus, 2007). Although a Z ring can form with either FtsA or ZipA, both are necessary for a Z ring to mature into a divisome (Pichoff & Lutkenhaus, 2005, Hale & de Boer, 2002). In the second stage, occurring after a lag equivalent to about half a division cycle in rapidly growing cells (Aarsman et al., 2005), the late proteins are recruited. These include the transmembrane proteins, FtsE/FtsX, FtsK, FtsQ/ FtsL/ FtsB, FtsW/ FtsI and finally FtsN. This recruitment occurs in a linear hierarchy to form a complete divisome that initiates formation of the septum leading to constriction (for reviews of this process see Lutkenhaus et al. (2012), de Boer (2010)). It is not currently clear why there is a delay between the two stages and what regulates the second wave of protein recruitment. In addition to the above 12 core proteins, about two dozen additional proteins are recruited to the divisome, but are non-essential, in part, because they have redundant functions (de Boer, 2010, Durand-Heredia et al., 2012, Hale et al., 2011, Schwille, 2014). These accessory proteins improve the efficiency or regulate the septation process.
Many proteins interact with the conserved C-terminal peptide sequence of FtsZ (“CCTP”) in E. coli, including ZipA, FtsA, MinCD, SlmA, ZapD and ClpX (Du & Lutkenhaus, 2014, Shen & Lutkenhaus, 2009, Durand-Heredia et al., 2012, Camberg et al., 2009, Haney et al., 2001, Pichoff & Lutkenhaus, 2002). Modifying the ratio of these proteins usually results in inhibition of cell division (Begg et al., 1998, Dewar et al., 1992, Rueda et al., 2003, Hale & de Boer, 1997, Pichoff & Lutkenhaus, 2001, Dai & Lutkenhaus, 1992, Shen & Lutkenhaus, 2010) suggesting that the delay in recruitment of the late cell division proteins could be due to competition for the “CCTP”. The facts that Z rings can form with either ZipA or FtsA alone (in conditions of FtsA or ZipA depletions respectively) but in these cases none of the late cell division proteins are recruited (Pichoff & Lutkenhaus, 2002, Hale & de Boer, 2002), and also that slight overexpressions of ZipA or FtsA block cell division, suggests that the second phase of recruitment of cell division proteins might be delayed until the correct ratio of FtsZ/ZipA/FtsA at the Z ring is reached. In support of this, our previous published results indicate that when FtsA is impaired for its ability to self-interact, the normally essential ZipA protein can be bypassed (Pichoff et al., 2012). It appears that lowering FtsA’s ability to self-interact allows it to recruit the late cell division proteins to the Z ring in the absence of ZipA. Based upon the above results, we suggested a model in which the more multimeric FtsA is, the less efficient FtsA is in the recruitment of late cell division proteins because the domain (FtsA IC) involved in the interaction with these late cell division proteins is sequestered by FtsA self-interaction (Pichoff et al., 2012, Lutkenhaus et al., 2012). In this model the essential role of ZipA is to modulate the ability of FtsA to self-interact at the Z ring, causing FtsA IC domain to become accessible for other proteins and as a consequence, ZipA regulates the ability of FtsA to recruit the late cell division proteins to the Z ring.
FtsN is the last known essential protein recruited to the divisome in E. coli and its arrival is thought to be the trigger to initiate constriction. Its recruitment requires that FtsA, FtsQ and FtsI be at the divisome (Addinall et al., 1997, Chen & Beckwith, 2001, Wang et al., 1998, Wissel & Weiss, 2004). Once FtsN starts to arrive at the divisome, it accumulates strongly in a self-enhancing manner following the onset of constriction (Gerding et al., 2009). In the current model, the short N-terminal cytoplasmic domain of FtsN (NcytoFtsN or FtsN1–31) interacts with domain IC of FtsA (Busiek & Margolin, 2014, Busiek et al., 2012, Corbin et al., 2004, Rico et al., 2004). This localization with FtsA which is weak and probably reinforced by the interaction of FtsN’s essential domain (EFtsN [FtsN71–105]) to other proteins of the divisome in the periplasm, is sufficient to promote the start of constriction by “activating” FtsI (PBP3), leading to the synthesis of new septal peptidoglycan (Gerding et al., 2009). The essential domain of FtsN (EFtsN) is contained within a 34 residue segment close to the periplasmic face of the inner membrane (FtsN transmembrane domain is FtsN32–54). The subsequent action of amidases produces a transient form of septal peptidoglycan which interacts with the C-terminal SPOR domain of FtsN (SPORFtsN or FtsN243–319), a strong septal localization determinant. Interestingly, neither NcytoFtsN nor SPORFtsN is absolutely essential for the function of FtsN in cell division (Ursinus et al., 2004, Dai et al., 1996, Gerding et al., 2009). When EFtsN is overexpressed and exported to the periplasm, it can rescue the depletion of FtsN (Gerding et al., 2009).
FtsN was originally isolated as a multicopy suppressor of a thermosensitive mutant of FtsA (Dai et al., 1993). Overexpression of FtsN also suppresses defects in FtsQ and FtsI and even suppresses the depletion of FtsK or FtsE/FtsX (Goehring et al., 2007, Reddy, 2007, Geissler & Margolin, 2005, Draper et al., 1998). These and other results indicate that FtsN makes multiple contacts (both in the cytoplasm and the periplasm) with early and late recruits to the divisome and may act as a control switch for the start of constriction (Corbin et al., 2004, Goehring & Beckwith, 2005, Lutkenhaus, 2009, Gerding et al., 2009). Recent work from Margolin’s lab (Busiek & Margolin, 2014, Busiek et al., 2012) confirmed previous genetic and protein–protein interaction assays showing that FtsN, despite being a late recruit to the division site, interacts with the early recruit FtsA via FtsA’s 1C domain (FtsA87–164) (Corbin et al., 2004, Rico et al., 2004, Di Lallo et al., 2003) .
In this report we find that overexpression of FtsN, but not any of the other known late cell division proteins, allows cells to grow without the essential cell division protein ZipA and determine that the cytoplasmic domain of FtsN is essential for this property. We also identify a conserved motif in the cytoplasmic domain of FtsN, which is important for its interaction with FtsA and is probably involved in the initial recruitment of FtsN to the divisome. We also show that this interaction with FtsA is essential for FtsN to function at a physiological level and for overexpressed FtsN to suppress division defects. Based on these new results and our earlier work showing that FtsA mutants impaired for self-interaction are able to grow without ZipA (Pichoff et al., 2012), we argue that this interaction between FtsN and FtsA increases the monomeric state of FtsA at the Z ring by antogonizing FtsA self-interaction and allows the recruitment of the late cell division proteins in the absence of the essential cell division protein ZipA, leading to cell division.
Results
Overexpression of FtsN suppresses ZipATs and bypasses the need for ZipA
Previously, we proposed that the late cell division proteins are recruited by monomeric FtsA and that ZipA’s essential role is to modulate FtsA’s self-interaction at the Z ring, thereby regulating the recruitment of the late cell division proteins (Pichoff et al., 2012, Lutkenhaus et al., 2012). In this model domain IC of FtsA is involved in two mutually exclusive interactions: FtsA’s self-interaction and FtsA’s interaction with the late cell division proteins. One prediction of this model is that overexpression of a late cell division protein interacting directly with FtsA might promote FtsA’s monomeric state and bypass ZipA because it is no longer needed to regulate FtsA’s self-interaction at the Z ring.
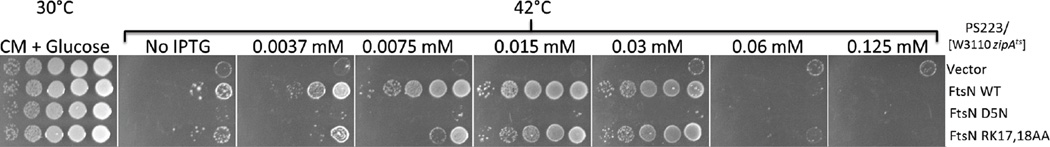
In order to test this prediction, we overexpressed each of the known late cell division proteins in PS223 [W3110 zipA1Ts], a strain expressing a thermosensitive mutant of ZipA (ZipA1Ts) (Pichoff & Lutkenhaus, 2002). We wanted to know which, if any, of these division proteins when overexpressed would allow growth at the non-permissive temperature of 42°C. The controls confirmed that PS223 transformed with the vector pDSW208 did not grow at 42°C whereas the construct expressing ZipA complemented the ZipA1Ts defect at low induction levels (Fig. 1). At higher induction levels overexpression becomes toxic as previously reported (Hale & de Boer, 1997). Of the cell division proteins tested, only overexpression of FtsN suppressed the lethal phenotype of ZipA1Ts at the non-permissive temperature. FtsN is already known to be a multicopy suppressor of other cell division defects such as those caused by ftsA12(Ts), ftsQ1(Ts), ftsI23(Ts), ftsK44(Ts), ΔftsK and ΔftsEX (Dai et al., 1993, Geissler & Margolin, 2005, Draper et al., 1998, Reddy, 2007).
Figure 1.

Among the late cell division proteins only overexpression of FtsN suppressed zipA1 (Ts). The plasmids used for overexpression all contain inserts in the vector pDSW208 (or pDSW210 for ZipA) and were transformed into PS223 [W3110 zipA1(Ts)] and selected on ampicillin plates containing 0.2% glucose at 30°C. Colonies of each strain were re-suspended in LB, serially diluted 10 fold, spotted onto plates containing increasing IPTG concentrations and incubated at 42°C. The protein expressed by each plasmid is indicated.
Two other proteins have been proposed to have a role in stabilizing and protecting divisome assembly under various conditions when they are overexpressed, SufI (FtsP) and DapE (Samaluru et al., 2007). DapE (MsgB) and FtsN (which is also annotated as MsgA in protein databanks), were isolated as multicopy suppressors of grpE280 (Wu et al., 1992), a thermosensitive allele of the chaperone protein GrpE. It is thought that the mutated GrpE280 is deficient for growth at nonpermissive temperature because of defects in cell wall synthesis and cell division (Wu et al., 1992). dapE has also been isolated as a multicopy suppressor of ΔftsEX and ΔsufI (ftsP) (Samaluru et al., 2007). Overexpression of FtsP (SufI) is also known to suppress multiple cell division defects such as ftsA12(Ts), ftsQ1(Ts), ftsI23(Ts), ftsK44(Ts) or ΔftsEX deletion (Samaluru et al., 2007, Reddy, 2007). For these reasons, we tested both DapE and SufI (FtsP) overexpression to see if they would suppress ZipA1Ts. Neither of these, however, was able to suppress ZipA1Ts (Fig. 1) despite the fact that the same expression vectors could suppress the other defects indicated above (Fig. S1). It is interesting to note that overexpression of DapE somewhat helps the growth of the zipA1(Ts) strain at the non-permissive condition (especially in the higher cell density spots) but it does not allow formation of strong growing individual colonies at the lowest dilutions even when the IPTG concentration keep increasing above 60 µM. These results indicates that the suppression of ZipA temperature sensitivity does not respond to general suppressors of cell division defects and appears to be specific to overexpression of FtsN.
Having determined that FtsN can suppress ZipA1Ts when overexpressed we wanted to know if the overexpression of FtsN alone was also sufficient to allow the complete bypass of ZipA. To do this we P1 transduced zipA::kan into W3110 expressing different FtsN constructs on a plasmid (pDSW208) under lac promoter control (Table S1). Only recipient cells expressing full length FtsN or a version of FtsN deleted for the C-terminal SPOR domain (FtsNΔSPOR) were able to acquire zipA::kan and form colonies on plates containing kanamycin, ampicillin and 1 mM IPTG. A spot test of these transductants confirmed that the growth was IPTG dependent demonstrating that the bypass of ZipA was dependent on the expression of FtsN or FtsNΔSPOR (Fig. 2). Interestingly, both constructs required the same level of IPTG to bypass ZipA (0.125–0.25 mM) and Western analysis revealed that FtsN had to be overexpressed at about 10–12 times the physiological level (Fig. S2).
Figure 2.

FtsN overexpression suppresses depletion of ZipA independently of the SPOR domain. Plasmids expressing FtsN (pSEB417 [pDSW208-FtsN]) or FtsN lacking the SPOR domain (pSEB418 [pDSW208-FtsN1-140]) were transformed into W3110. ZipA::Kan was then P1 transduced into these cells in the presence of 1 mM IPTG and individual colonies were re-suspended in LB and tested for IPTG-dependent survival at 37°C by spotting serial dilutions on plates containing ampicilin and increasing IPTG concentrations as described Fig. 1.
In an unbiased approach to identify suppressors of ZipA deficiency, we searched for multicopy suppressors of a ZipA depletion strain W3110ΩPbad-zipA, which expresses ZipA at its native chromosomal location under the control of the araBAD promoter (Liu et al., 1999). The suppressor plasmids were selected from a library that contains chromosomal DNA partially digested by TaqI and Sau3A ligated to the pBR322 double digested with BamHI and ClaI (Dai et al., 1993). Plasmids obtained from colonies growing in the absence of arabinose were re-tested for suppression in two ways: 1) by re-transformation into a naïve ZipA depletion strain, and 2) by checking if they allowed a ZipA1Ts strain to grow at 42°C (non-permissive temperature) (See Fig. S3). All plasmids that allowed the depletion strain to grow also allowed the ZipA1Ts strain to grow at the non-permissive condition. Sequence analysis of the inserts from the confirmed suppressor plasmids revealed 6 different plasmids (pMCSΔZipA A to F). Three of these had the sdiA gene in common while the other three had only the ftsN gene in common (Fig. S3A). SdiA, a transcriptional regulator, has been isolated as a multicopy suppressor of cell division inhibition due to ftsZ84 (Ts), a temperature sensitive mutant of FtsZ, and the overexpression of MinCD (Wang et al., 1991). Finding sdiA in our screen was not that surprising since multicopy sdiA has been shown to increase the expression of the ftsQAZ genes (Wang et al., 1991) and it is known that pZAQ (a pBR322 derivative carrying the ftsQAZ genes) allows the bypass of zipA (Geissler et al., 2003). We did not isolate a plasmid carrying the ftsQAZ genes in our screen, but we independently confirmed that pZAQ allows the growth of both the W3110ΩPbad-zipA strain and the ZipA1Ts strain under non-permissive conditions (Fig. S3B).
Taken together, these results indicate that the bypass of ZipA by the overexpression of FtsN is quite specific and different from the general suppression of cell division defects observed with overexpression of DapE or FtsP (SufI). Also, because FtsN is known to interact directly with FtsA, our result is also in accordance with our hypothesis in which we proposed that overexpression of a late cell division protein that interacts directly with FtsA should bypass the essential role of ZipA.
A newly identified conserved sequence in the cytoplasmic region of FtsN is required for the bypass of ZipA
If our hypothesis about the bypass of ZipA is correct, FtsN must interact with FtsA in the cytoplasm and the cytoplasmic domain of FtsN has to be essential for this even though it is not otherwise absolutely essential for growth (Dai et al., 1996). From the results presented in Fig. 2 it is clear that the periplasmic SPOR domain of FtsN is dispensable for the suppression of ZipA defects. In order to test the essentiality of the N-terminus of FtsN for this we overexpressed a functional MalG1–33-FtsN46–319 fusion by introducing plasmid pKD157 (Dai et al., 1996) into PS223 [W3110 zipA1Ts]. In the MalG1–33-FtsN46–319 fusion the cytoplasmic domain and most of the transmembrane domain of FtsN are replaced by an N-terminal segment of the unrelated MalG protein, including MalG’s first membrane spanning domain. This construct has been shown before to have a bitopic membrane topology and complement an ftsN-null strain (Dai et al., 1996). Even though this fusion is able to complement the ΔftsN strain and cause some toxicity when highly overexpressed, similar to wild type FtsN (Fig. S4A), it is unable to suppress ZipA1Ts (Fig. S4B). This experiment supports the idea the N-terminal cytoplasmic region of FtsN is necessary for overexpressed FtsN to suppress the ZipA defect.
Recent reports from Margolin’s lab (Busiek & Margolin, 2014, Busiek et al., 2012) clearly demonstrate in vitro, but also in vivo, that the cytoplasmic region of FtsN is essential for its interaction with FtsA. This cytoplasmic region is short (33 residues) and positively charged overall (see Fig. 3). It was proposed that a stretch of basic residues within this region (RRKK at amino acid positions 16 to 19) was responsible for the direct interaction with FtsA (Busiek et al., 2012). This conclusion stemmed from the observation that changing these residues to negatively charged residues (RRKK to DDEE mutation) abolished the interaction observed by Far Western between FtsA and cytoFtsN. However, no phenotypic effects of these substitutions were observed in vivo and the mutated protein behaved like the wild type FtsN in complementation tests (Busiek et al., 2012). When compared to wild type FtsN, besides a slight stability issue as described below, we also failed to observe any impairment of FtsN in various assays when we mutated this region of charged residues to give the RK17,18AA mutant (R17 K18 to AA mutations). These results suggested that the sequence of FtsN that directly interacts with FtsA has not yet been clearly identified and led us to revisit sequence alignments of FtsN.
Figure 3.

Alignment of FtsN sequences and domain organization of FtsN. A) Comparison of FtsN’s N-terminal region from various bacteria [Escherichia coli str. K12 substr.W3110 (GI:388479326); Klebsiella pneumonia 342 (GI:206578823); Haemophilus influenza (GI:491956398); Legionella pneumophila subsp. pneumophila ATCC 43290 (GI:378777884); Shewanella amazonensis (GI:500082549); Methylotenera versatilis (GI:502914455); Geobacter metallireducens RCH3 (GI:373563253)] as indicated. The conserved charged residues in these sequences are highlighted in magenta. The conserved sequence we identified in this study is highlighted in yellow. Residue changes made to the E. coli sequence for this report are indicated above the blue arrows. B) Schematic representation of FtsN and its different domains; cyto - cytoplasmic domain, TM - transmembrane region, E domain - the essential region of FtsN, and SPOR - the conserved domain that is shared with other proteins and is thought to bind peptidoglycan.
FtsN has clear homologues in α, β, δ and γ Proteobacteria indicating that it is a core component of the division apparatus among these Gram-negative bacteria (Moll & Thanbichler, 2009). Although published work suggests that the only conserved region of FtsN is the SPOR domain (Ursinus et al., 2004, Yang et al., 2004), our sequence alignments of FtsN allowed us to identify a conserved sequence in the cytoplasmic N-terminal part of the protein (shaded yellow in Fig. 3). The sequence is R/KDY at position 4–6 and was probably not detected earlier since the databases contain N-terminal truncations (30% of the sequences designated FtsN use the wrong start codon [E. coli FtsN uses GTG] and are missing the N-terminal cytoplasmic domain due to early errors in annotating FtsN). Due to the conservation, this sequence was a candidate for interacting with FtsA to drive the initial localization of FtsN to the septum. Because residues D5 and Y6 are the most conserved in the sequence we tested different substitutions of these residues. After assessing the effect of these changes on protein stability, we decided to use the D5N mutation since it is as stable as the WT (Fig. S5) but displays a mutant phenotype as described below.
The effect of the D5N and RK17,18AA substitutions on the ability of FtsN to complement an ftsN –null strain was examined (Fig. 4A). To do this we used pDSW210 to express the different FtsN constructs (with or without the SPOR domain) at levels low enough to assess differences between the mutant proteins. pDSW210 has a mutated Trc promoter that allows lower expression than the pDSW208 expression vector we used above (Weiss et al., 1999). The plasmids expressing the various ftsN alleles were introduced into JKD4-2 [ftsN::kan recA56]/pKD123 [contains a repTs allele on a compatible CmR derivative that expresses FtsN constitutively]. The transformants were tested for their ability to complement the depletion of FtsN by spotting serial dilutions of the different strains onto selective plates containing increasing concentrations of IPTG at 42°C, the non-permissive temperature for pKD123 replication.
Figure 4.

Complementation of the FtsN depletion strain by various FtsN constructs and the level of expression of FtsN mutant proteins. A) Spot test to examine the efficiency of various FtsN constructs to complement an FtsN depletion strain. pDSW210 derivatives pSEB413 (ftsN) and pSEB414 (ftsNΔSPOR) expressing the different ftsN mutants (as indicated) under the control of the lac promoter were transformed into JKD4-2 [W3110 ftsN::kan recA56]/ pKD123 [ftsN repTs CmR] and selected on plates containing kanamycin, chloramphenicol, ampicillin and 0.2% glucose at 30°C. Colonies were then re-suspended in LB and tested for growth under ftsN-null conditions (pKD123 is unable to replicate at 42°C) by spotting serial dilutions onto plates containing kanamycin, ampicillin and increasing IPTG concentrations as described in Fig 1. B) Western blot analysis of the minimal level of each FtsN mutant sufficient to complement the ftsN-null strain (based on colony formation and cell morphology) and comparison to the chromosomal level of FtsN. An equal amount of total protein extract of JKD4-2 containing pSEB413, pSEB413-D5N or pSEB413-RK grown to OD540 of 0.4 at 37°C with the minimal IPTG concentration required to allow the strain to survive on plates along with the control strain W3110/pDSW210 in the presence of 1 mM IPTG (indicative of FtsN chromosomal level of expression) were loaded onto a SDS page gel and transferred to a nitrocellulose membrane. FtsN was detected using a polyclonal rabbit anti-FtsN. Note the band above FtsN, is MalE [the antiserum was raised against a MalE-FtsN fusion as described earlier (Dai et al., 1996)] and serves here as an internal loading control. The lane marked M contains size markers.
At 0.06 mM IPTG all of the FtsN constructs tested complemented the ftsN-null strain. This is consistent with previously published results (Gerding et al., 2009), showing that as long as the FtsN’s E-domain (FtsN71–105) is present and expressed at a high enough level in the periplasm, it is sufficient to complement the FtsN depletion strain. For IPTG concentrations below 0.06 mM the constructs behave differently. As a general rule constructs in which the SPOR domain is not present require higher IPTG levels than the corresponding one with the intact SPOR domain indicating FtsN is more efficient when the SPOR domain is present. This is in contrast with the observation that the presence or absence of the SPOR domain did not affect the suppression of ZipA1Ts (Fig. 2), but is consistent with the previous observation that FtsN is more effective than FtsN lacking the SPOR domain in complementing ΔftsN (Gerding et al., 2009, Ursinus et al., 2004). In the presence or absence of the SPOR domain, mutations D5N and RK17,18AA affected FtsN function as higher IPTG levels were required to complement the ftsN-null strain. For RK17,18AA the spot assay is a little misleading because Western analysis (Fig. 4B) indicates that the mutation affects the protein expression/stability and a slightly higher level of IPTG is required to reach the same level of expression as wild type FtsN. Thus, the minimal level of FtsNRK17,18AA to complement the ftsN-null strain is about the same level as for wild type FtsN (compare lanes 3 and 5 in Fig. 4B). Also, FtsN and FtsNRK17,18AA can complement the ftsN-null strain at less than the physiological level of FtsN (compare lanes 2 with 3 and 5 in Fig. 4B) suggesting that the level provided by the chromosomal copy of FtsN is in excess in rich medium. This is not too surprising as it was reported that in Caulobacter the level of FtsN can be reduced by 90% without producing an apparent phenotype (Moll & Thanbichler, 2009). Importantly, the Western analysis (Fig. 4B) clearly shows that FtsND5N has to be expressed at a higher level than FtsN to allow comparable cell growth, suggesting that its function is impaired (compare lanes 3 and 4, no IPTG). We confirmed by additional Western analysis, using different expression vectors that FtsND5N is expressed at the same level as FtsN, whereas FtsNRK17,18AA is always about 30% less (Fig. S6).
High overexpression of FtsN is known to be toxic (Aarsman et al., 2005, Dai et al., 1993), which offered an alternative way to examine the differences in our mutant proteins’ stability/expression. This toxicity is largely due to the periplasmic domain since the MalG1–33-FtsN46–319 fusion is toxic (Fig. S4A). In order to reach a high enough level of FtsN to become toxic we cloned our FtsN constructs into the pSEB436 vector (pACYC184 derivative expressing ftsN with an ATG start codon [instead of its normal GTG]). We then introduced these plasmids into the ftsN-null strain and expressed our FtsN mutant proteins by increasing the IPTG concentration. As shown in Fig. 5, the mutant proteins complement at low induction levels but then become toxic when overexpressed. In accordance with the Western blot results that show FtsN and FtsND5N have comparable stability, both of these proteins become toxic at the same level of IPTG induction and kill cells at 0.06 mM. FtsNRK17,18AA, which is slightly less stable than FtsN, requires approximately 2-fold more IPTG to become toxic.
Figure 5.
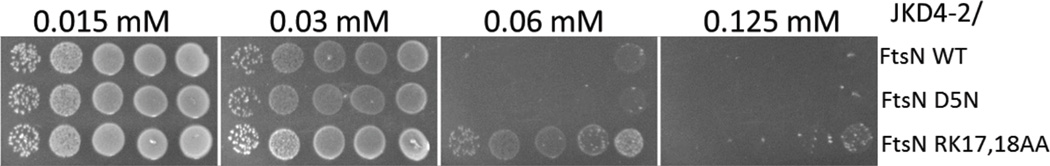
Overexpression toxicity of different FtsN mutants. JKD4-2 expressing different ftsN alleles in pDSW208 (derivatives pSEB417, pSEB417-D5N and pSEB417-RK) were spotted as described in Fig. 1 on plates with increasing IPTG concentrations. The plates were incubated at 42°C in order to compare the quantity of IPTG necessary to induce toxicity.
Because we think that the R/KDY sequence at position 4–6 of FtsN is important for direct interaction with FtsA and that this interaction with FtsA is essential for the bypass of ZipA when FtsN is overexpressed, we proceeded to test if the FtsND5N mutant had lost the ability to suppress ZipA1Ts (Fig. 6). In order to do this we introduced the pSEB436 derivatives into PS223 [W3110 zipA1Ts] and plated serial dilutions of the different strains on plates with increasing IPTG concentrations. Overexpression of FtsN suppresses ZipA1Ts (as shown in Fig. 1 with a lower expression vector), but because of the higher expression vector used here, it becomes toxic at concentrations around 0.03 mM IPTG (Fig. 5). FtsNRK17,18AA requires a little more IPTG to overcome its stability issue, but it also allows the cells to grow at low IPTG and causes toxicity around 0.06 mM IPTG, similar to what was observed in Fig. 5. Western analysis (Fig. S6) confirms that the level of overexpression of FtsNRK17,18AA necessary to suppress ZipA1Ts is about the same as for FtsN. In contrast, overexpression of FtsND5N never suppresses ZipA1Ts even though it is overexpressed to the same level as wild type FtsN. These results indicate that the D5 residue is very important for overexpressed FtsN to bypass ZipA, which is in accordance with our hypothesis that this residue is in the core of the sequence important for the direct interaction with FtsA.
Figure 6.
Suppression of ZipATs by FtsN mutants. PS233 [W3110 zipA1(Ts)] was transformed with pDSW208 or its derivatives pSEB417, pSEB417-D5N and pSEB417-RK, expressing the different ftsN mutants as indicated in the figure, and selected at 30°C on plates containing ampicilin and 0.2% glucose. Colonies were picked, re-suspended in LB and serial dilutions spotted on plates containing ampicillin with increasing IPTG concentrations as described in Fig. 1. The plates were incubated at 42°C.
FtsND5N has a reduced interaction with FtsA
Recent publications from Margolin’s laboratory (Busiek & Margolin, 2014, Busiek et al., 2012) confirmed earlier results suggesting that the cytoplasmic N-terminus of FtsN interacts directly with FtsA’s IC domain (Corbin et al., 2004, Rico et al., 2004, Karimova et al., 2005). In these recent reports, the authors use a battery of in vitro and in vivo assays such as co-affinity purification, Far Western analysis, BACTH assay and in vivo localization of FtsNΔSPOR derivatives to demonstrate the interaction. The authors also reported that a stretch of “more” conserved basic residues (RRKK at position 16 to 19) of FtsN are important for the interaction with FtsA. This is based on a mutant in which these four residues were changed to negatively charged residues (DDEE) that no longer interacts with FtsA in Far Western analysis (Busiek et al., 2012). However, that mutant complemented the ftsN-null strain and was not tested in other in vivo assays for interaction with FtsA. So, it is not clear if the loss of interaction in the Far Western was a nonspecific consequence of the extensive change in charge of the N-terminus of FtsN. In addition, as described above, we did not observe any obvious phenotype when we mutated the two central residues of that sequence to alanines (RK17,18AA) whereas we found that a change of the aspartic acid residue to asparagine at position 5 (D5N) clearly caused a demonstrable change in the ability of FtsN protein to bypass ZipA and complement the ftsN-null strain. The behavior of the D5N mutant is in accordance with our proposition that the conserved sequence R/KDY at position 4 to 6 is responsible for the direct interaction with FtsA. We decided to look at the effect of the FtsN mutants on the interaction with FtsA in some of the same tests used in Margolin’s studies (Busiek & Margolin, 2014, Busiek et al., 2012). The plasmids and strains necessary to address these questions were generously provided to us by Margolin’s lab.
First, we examined the FtsA-FtsN interaction in the BACTH assay as described in Busiek et al. (2012) but instead of doing streaks of cells on reporter plates, and in order to better appreciate the differences in color after incubation, we re-suspended cells in LB and spotted them on the test plates (Fig. 7). In this assay we used the following plasmids pWM3014 (pKT25-ftsA) (Shiomi & Margolin, 2007), pWM3021 (pUT18c–ftsA), pWM3861 (pKT25F–ftsN1–128), pWM3451 (pKT25F–ftsN1–54’phoA) (Busiek et al., 2012). The mutations to be tested were introduced into the latter two plasmids by site directed mutagenesis. In addition, to make sure we were measuring the direct interaction between FtsA and FtsN and not some bridging effect from recruitment to the divisome, we mutated the ftsA carried by pWM3021 and pWM3014 to ftsAR300E, a well characterized mutant of FtsA that is not recruited to the Z ring (Pichoff & Lutkenhaus, 2007). We also used versions of FtsN without the SPOR domain (FtsN1–54 is truncated after the transmembrane domain and FtsN1–128 is truncated after the FtsN essential domain) to minimize the recruitment to the septum. Our reasoning behind avoiding the use of proteins that are efficiently recruited to the divisome is based on the following observations. Previous published studies agree that the interaction of FtsA* [FtsAR286W] with FtsZ is stronger than the FtsA-FtsZ interaction when measured by BACTH assay or by Yeast 2 Hybrid (Y2H), but they disagree on the ability of these same FtsA proteins to self-interact (Pichoff et al., 2012, Geissler et al., 2007, Shiomi & Margolin, 2007). By Y2H, and consistent with the results obtained with the other mutants of FtsA that allow the bypass of ZipA (Pichoff et al., 2012), FtsA* self-interaction is impaired when compared to FtsA, while, by the BACTH assay FtsA* self-interaction appears to be increased compared to FtsA (Shiomi & Margolin, 2007). We found that introduction of the R300E mutation, which abolishes the interaction with FtsZ, can reconcile these apparently different results for the FtsA* self-interaction (Figs. 7A and 7B). The addition of the R300E mutation to the FtsA* allele resulted in a decrease in the FtsA* self-interaction in the BACTH assay, suggesting that the previously reported increased self-interaction for FtsA* was probably indirect due to the better recruitment of FtsA* to the Z ring.
Figure 7.

Interaction of FtsA with itself and with FtsN by BACTH assay. A-C) Variants of PKT25-ftsA, pUT18c–ftsA, pKT25F–ftsN1-128 and pKT25F–ftsN1-54 were transformed in various combinations as indicated in the figure into the BACTH assay strain DHM1. For each strain, 3 independent colonies were re-suspended in 1 ml of LB and spotted onto selective plates containing 50 µg/ml X-Gal and 0.5 mM IPTG. A) Comparison of FtsA* [FtsAR286W] self-interaction when the protein can interact with FtsZ and localize to the septum or when this localization is impaired by adding the R300E mutation. B) Comparison of FtsA and FtsA* self-interaction when proteins are impaired for their interaction with FtsZ by the R300E mutation. C) Interaction of FtsAR300E in combination with different FtsN constructs and mutants as indicated. Here, the qualitative plate assay is supported by a quantitative measurement of the β-galactosidase assay for each combination. To assess β-galactosidase activity at least 3 different colonies for each combination were taken from fresh transformation plates (different colonies than tested on the plate assay) and grown to mid-logarithmic phase in the appropriate media before the assay. Assays were performed as described in materials and methods.
Results from the BACTH assay (Fig. 7C) indicate that both ftsN mutations (D5N or RK17,18AA) reduce the interaction between FtsA and FtsN1–54 or FtsN1–128. The qualitative results (Blue/White colors) are supported by quantitative β-galactosidase assays and demonstrate that the D5N mutation causes a larger decrease in the FtsA-FtsN interaction than RK17,18AA. Although the D5N mutation causes a significant reduction in the FtsN-FtsA interaction, yielding only a third of the β-galactosidase activity observed between FtsA with FtsN, it does not result in a complete loss of interaction. The decrease in the FtsA-FtsN interaction seen with the RK17,18AA mutation is likely due to the reduction in the stability of FtsN (about 30% lower level than the wild type FtsN or FtsND5N) (Fig. 3) but we can’t exclude that this mutation also has an effect on the interaction between FtsN with FtsA.
Another way to test the effect of the D5N mutation on the interaction of FtsN with FtsA is to examine the localization of FtsN2–55. Busiek and Margolin (2014) reported that a GFP-FtsN2–55 fusion protein expressed from pWM4528 [pDSW207-gfp-ftsNCyto-Tm] localized efficiently at the Z ring. By using thermosensitive alleles of ftsA (ftsA12), ftsQ (ftsQ1) and ftsI (ftsI23) they were able to show that this localization of GFP-FtsN2–55 is dependent on FtsA but not on FtsQ or FtsI. We found that introduction of the D5N mutation into the pWM4528 plasmid resulted in GFP-FtsN2–55+D5N no longer being localized at the septum (Fig. 8). This result is in accordance with what is expected if the D5N mutation impairs FtsN’s interaction with FtsA.
Figure 8.

Localization of GFP-FtsN2-55 and its D5N mutant version. Fluorescence images of W3110 cells expressing different constructs from pWM4528 [pDSW207-ftsN2-55] or pWM4528-D5N [pDSW207-ftsN2-55(D5N)] after strains were grown in LB plus ampicillin without IPTG to the same low OD600 as described in Busiek and Margolin (2014).
In earlier work we observed that overexpression of FtsAR300E (which does not interact with FtsZ) is toxic (Pichoff & Lutkenhaus, 2007). However, unlike overexpression of FtsA, which causes the formation of smooth filaments, it leads to filamenting cells with a “sausage” phenotype, cells connected by the presence of an elongated septal region of a smaller diameter that we call “grey necks” (Pichoff & Lutkenhaus, 2007). In that earlier work, we proposed that FtsAR300E, which is distributed throughout the membrane because it does not localize to the Z ring, recruits one or more late cell division proteins away from the septum, resulting in the aberrant constrictions. A likely late cell division candidate is FtsN since it interacts directly with FtsA. If so, then increasing the level of FtsN should suppress the cell division inhibition phenotype caused by overexpression of FtsAR300E for potentially two reasons. One is that the excess FtsN should overcome its titration away from the septum and, two, the interaction of FtsN’s SPOR domain with septal peptidoglycan, could actually “back recruit” FtsAR300E to the septum. This would help counter the titration away from the septum due to any unlocalized FtsAR300E. In contrast, increasing FtsND5N should be less helpful since its interaction with FtsA is impaired. This hypothesis was tested in the spot assay shown Fig. 9. As predicted, because it efficiently interacts with FtsA, FtsN was able to counteract the toxic effects of overexpression of FtsAR300E, whereas, FtsND5N, which is impaired for interaction with FtsA, and FtsNRK17,18AA which is less stable, were less able to suppress the toxicity. The inability of FtsND5N to suppress FtsAR300E overexpression is in accordance with it being impaired for its interaction with FtsA as observed in the BACTH.
Figure 9.

Suppression of FtsAR300E toxicity by increasing the level of different FtsN constructs. The FtsAR300E protein was expressed under IPTG control from plasmid pSEB440-R300 contained in JS238 [MC1061 lacIQ recA]. FtsN, FtsND5N or FtsNRK17,18AA levels were increased in JS238/pSEB440-R300E by transforming the strain with pKD140, a pBR322 derivative carrying ftsN under its own promoter, or a derivative containing an ftsN mutation as indicated in the figure. A spot test assay was performed at 37°C on selective plates containing ampicillin, chloramphenicol and increasing concentrations of IPTG.
The interaction of FtsN with FtsA is essential for the suppression of other cell division defects by FtsN overexpression
As mentioned above, FtsN overexpression, in addition to bypassing ZipA, can also compensate for the complete lack of FtsK or FtsE/FtsX and suppress temperature sensitive mutations in ftsA, ftsK, ftsQ and ftsI (Dai et al., 1993, Draper et al., 1998, Reddy, 2007, Geissler & Margolin, 2005). Because Cyto+TmFtsN is essential for the suppression of ftsQ1(Ts) and the depletion of FtsK (Goehring et al., 2007) we wanted to know if these suppression effects, like the suppression of zipA-null, are directly dependent on the interaction with FtsA. The various constructs expressing FtsN or FtsN mutants D5N or RK17,18AA were transformed into MCQ1 [MC4100 ftsQ1(Ts) leu::Tn10] (Fig. 10, top panel) and EC1779 [BW25113 ftsEX::CmR] (Fig. 10, bottom panel). Single colonies were picked into 300 µl of media and serial dilutions spotted onto selective plates incubated under non-permissive conditions. As shown before, overexpression of FtsN suppresses ftsQ1(Ts) and ftsEX-null and FtsN’s SPOR domain is dispensable for this effect ((Goehring et al., 2007), (Samaluru et al., 2007) and Fig. 10). FtsNRK17,18AA is also able to suppress, and in accordance with the fact that it is a little less stable than FtsN, the suppression requires slightly more IPTG. In contrast, FtsND5N, which is impaired for its interaction with FtsA, is unable to suppress the temperature sensitivity of FtsQ1 or the growth defect of the ftsEX deletion grown on LB plates in the absence of an osmoprotectant. These results strongly indicate that the suppression of these phenotypes requires the ability of FtsN to interact efficiently with FtsA.
Figure 10.

Suppression of ftsQ1 and ftsEX-null phenotypes by overexpression of different FtsN variants. MCQ1 [MC4100 ftsQ1] (top panel) or EC1779 [ftsEX-null] (bottom panel) strains were transformed with pSEB417 [pDSW208 ftsN] or pSEB418 [pDSW208 ftsN1-140] derivatives expressing ftsN or ftsN mutants as indicated in the figure. Suppression of the cell division phenotypes was assessed by a spot assay on selective plates with increasing IPTG under non permissive conditions; 42°C for MCQ1 and no sucrose for EC1779.
As we have shown, the suppression of cell division defects by overexpression of FtsN is independent of FtsN’s SPOR domain but absolutely requires FtsN’s interaction with FtsA. Even though the interaction between FtsN with FtsA is essential for the suppression of the cell division defect, additional tests show it is not sufficient (Fig. S7). PS223 [W3110 zipA1Ts] was transformed with pDSW208 derivatives expressing FtsN1–72 (SPOR and E- domains deleted) or FtsN1–140 (SPOR domain deleted) or wild type FtsN. These FtsN constructs differ in the extent of the periplasmic domain but interact with FtsA and insert in the membrane similar to wild type FtsN. In the presence of pMG20, a plasmid expressing a periplasmic version of EFtsN at a level sufficient to complement an ftsN-null strain (induced with 1% arabinose) (Gerding et al., 2009), both FtsN and FtsNΔSPOR were able to suppress ZipA1Ts. Here again, and as observed in Fig. 2, both constructs, even if they have a different efficiency in complementing the ftsN-null strain (see Fig. 3), suppress ZipA1Ts at the same concentration of IPTG. This suggests that the important factor for suppression is the number of the N-terminal cytoplasmic peptides of FtsN that are produced. However, the FtsN1–72 construct, which also expresses FtsN’s N-terminal domain, but is truncated before the E-domain and is unable to complement an ftsN-null strain, does not suppress ZipA1Ts. Since FtsN’s E-domain is expressed in trans in this strain it reduces the likelihood that FtsN1–72 behaves as an inhibitor of cell division by competing with the resident FtsN. Neither overexpression of FtsN1–72 (truncated before the E-domain) or FtsN’s periplasmic domain (including the E-domain) is able to suppress ZipA1Ts (Fig. S4 and Fig. S7), but overexpression of FtsN1–140 can (Fig. 2, Fig. 4 and Fig. S7). Because of this, we suspect that suppression of ZipA1Ts requires that the FtsN construct, which must interact with FtsA, must also be linked to the E-domain. Together, it suggests that it is actually the increase in the number of “active FtsN” molecules (aka FtsN molecules able to attach to a divisome by the periplasmic interaction with the EFtsN), recruited to the Z ring before constriction initiates (as it is independent of the SPOR domain) that is important for the suppression of ZipA1Ts and probably zipA-null.
Discussion
FtsA is a member of the actin family with an unusual domain structure, but can assemble into actin-like filaments (Szwedziak et al., 2012, van den Ent & Lowe, 2000). In our model for the role of FtsA in cell division, FtsA monomers (or at least FtsA molecules with their domain IC not involved in self interaction) recruit downstream division proteins and ZipA’s essential role is to promote these monomeric FtsA at the Z ring (Pichoff et al., 2012). We then speculated that overproduction of a downstream division protein that interacts with FtsA could tip the balance in favor of FtsA monomers (this interaction with FtsA’s IC domain blocks it for participating in FtsA’s self-interaction) making ZipA dispensable. In this report we discover and characterize the overexpression of FtsN as a means to suppress zipA1Ts or zipA-null phenotypes. Since overexpression of other cell division proteins and proteins (FtsP or DapE) that have been shown to suppress multiple cell division defects (Samaluru et al., 2007) do not bypass ZipA, this property is fairly unique to FtsN. We also show that FtsN absolutely requires its demonstrated ability to interact with FtsA in order to suppress the need for ZipA. We identified a conserved motif at position 4–6 (R/KDY) in FtsN’s cytoplasmic N-terminus and provide evidence that this sequence directly mediates the interaction of FtsN with FtsA. A modest change at residue 5 (FtsND5N) causes significant impairment of the interaction with FtsA and the ability for FtsN’s N-terminus to localize to the septal ring independently of the SPOR domain, localization of which, has been shown in an earlier report to be directly dependent on interaction of the N-terminus of FtsN with FtsA (Busiek & Margolin, 2014).
A possible analogy for the interaction between FtsA and FtsN comes from the study of pilus assembly in Thermus thermophilus. PilM, a protein structurally related to FtsA, binds to a peptide present in the short cytoplasmic N-terminal domain of the bitopic PilN protein (Karuppiah & Derrick, 2011). Interestingly, the peptide binds to a well-conserved binding site in PilM proteins from Gram-negative bacteria, which is located in a narrow channel between domains 1A and 1C. The peptide is located at the N-terminus of PilN about 20 residues away from PilN’s transmembrane domain; a similar distance from the membrane as the conserved sequence we identified in FtsN that interacts with FtsA. Possibly, this conserved sequence binds in the equivalent channel between the 1A and 1C domains of FtsA. Interestingly, it has been suggested that domain IC of FtsA is flexible in relation to the rest of the molecule (van den Ent & Lowe, 2000)), and we speculate that the binding of FtsN’s peptide in FtsA’s channel between domains 1A and 1C greatly reduces the flexibility of domain 1C. The binding of FtsN could lock FtsA’s 1C in a position where it is less able to participate in FtsA’s self-interaction, hence locking FtsA in the monomer state (Fig. 11).
Figure 11.

Model for the recruitment of late cell division proteins by FtsA. ZipA (pink) and FtsA (blue) both bind to the membrane and the conserved C-terminal motif of FtsZ (teal). FtsA domain 1C can participate either in FtsA’s self-interaction or FtsA’s interaction with the late cell division proteins (represented in green [unidentified protein] and red [FtsN]) and these two interactions compete with one another. When domain 1C of FtsA is involved in FtsA self-interaction it is unable to interact with the late cell division proteins. ZipA modulates the ability of FtsA to self-interact at the Z ring causing FtsA’s 1C domain to become available for interaction with the late cell division proteins, leading to the hierarchical recruitment of the late cell division protein to the divisome. Once FtsA interacts with the conserved cytoplasmic sequence of FtsN, FtsN “commits” FtsA to the monomer state by binding FtsA’s IC domain and blocking FtsA self-interaction. When FtsN is overexpressed it interacts with FtsA earlier, bypassing the normal hierarchy of recruitment of the late cell division proteins. In this scenario, ZipA’s role to modulate FtsA’s self-interaction at the Z ring becomes dispensable because FtsN cause FtsA’s 1C domain to become available for other late cell division proteins leading to the formation of a complete divisome.
It was a little surprising that FtsN is the only late cell division protein that bypasses ZipA when overexpressed. In the hierarchical sequential recruitment of the late essential cell division proteins to the septum, FtsN is last and depends on the prior recruitment of many other proteins (Addinall et al., 1997, Goehring et al., 2006, Chen & Beckwith, 2001). Because of this, we expected that at least one other late cell division protein, also interacting with the IC domain of FtsA, but recruited earlier than FtsN, would also bypass ZipA when overexpressed. There are many potential explanations why we did not identify such a protein. One possibility is that we did not test the overexpression of that specific protein because it has not been identified yet. Our genetic screen for multicopy suppressors of ZipA’s depletion only gave us plasmids carrying ftsN or sdiA (Fig. S3), making this possibility somewhat less likely. Another possibility is that the proteins tested needed to be overexpressed along with others as they form complexes outside the septum (FtsQBL for example (Buddelmeijer & Beckwith, 2004)). Finally, a possibility that we favor in our model (Fig. 11) is that the interaction of FtsA with another late cell division protein(s) may not fully commit FtsA to a monomeric form. Only the putative interaction of FtsN in FtsA’s channel between domains IC and IA, as we hypothesized above, locks FtsA’s IC domain into a position that ensures FtsA is a monomeric species. It is interesting to note that an allele of ftsA has been previously isolated as a suppressor of FtsN deficiency (Bernard et al., 2007). That allele contained multiple mutations but it was later shown that a single mutation (E124A) could suppress just as well and could also bypass ZipA. This mutation is unusual among ftsA mutations that bypass ZipA because it did not result in a significant decrease in the ability of FtsA to self-interact (Pichoff et al., 2012). The E124 residue is located close to the hinge region between the IC and 1A domains, near the channel where we hypothesize that FtsN’s peptide binds. Possibly, this mutation causes a structural change in FtsA that more effectively recruits one of the other division proteins.
In this study, we also find that when FtsN is impaired for its interaction with FtsA, it cannot function efficiently and has to be expressed more than the physiological level to complement an ftsN-null strain. This result is consistent with previous reports showing that FtsN’s periplasmic domain can substitute for full length FtsN, provided it is expressed above the wild type FtsN level (~25 fold) (Gerding et al., 2009, Dai et al., 1996). It seems logical that when the EFtsN is not targeted efficiently to the divisome to initiate constriction, it needs to be overexpressed to overcome this localization deficiency.
Finally, we show that suppression of many cell division defects by FtsN’s overexpression that have been characterized previously and also in this report, are strictly dependent on FtsN’s interaction with FtsA and as a consequence, on the early recruitment of EFtsN to the divisome. Overall, as discussed below, we find that all of our results are in accordance and provide additional support for our previously published model about the role of FtsA in the recruitment of the late cell division proteins to the divisome (Fig. 11).
Role of FtsN in our model for the recruitment of late cell division proteins by FtsA
In our model (Pichoff et al., 2012, Lutkenhaus et al., 2012) the essential role of ZipA is to regulate the ability of FtsA to polymerize at the Z ring possibly by competing with FtsA for binding to the conserved C-terminal tail of FtsZ. Domain IC of FtsA is involved in FtsA’s self-interaction and also involved in the interaction with one or more late cell division proteins and our results are consistent with a competition between these two interactions. Previously, we described many ftsA alleles that reduce FtsA’s self-interaction but are still functional and able to support cell division (Pichoff et al., 2012). In addition, these alleles have gained the ability to bypass ZipA in the E. coli septation process. Based on this, we clearly identified a decrease in the ability of FtsA to self-interact as the basis for the mechanism that allows the bypass of ZipA for E. coli cytokinesis. The results presented here combined with previous results suggest that lowering the interaction of FtsA with itself helps with its ability to interact directly with at least FtsN and maybe other components of the divisome. It should be noted though that only FtsN, among all the cell division proteins tested, was able to bypass ZipA suggesting it, or more likely, the way it interacts with FtsA is unique. In our model we proposed the interaction of FtsA with FtsN occurs at the expense of FtsA’s self-interaction. Thus, overexpression of FtsN would promote formation of (or trap) FtsA monomers at the Z ring, mimicking the effect of the FtsA mutants impaired for self-interaction.
Suppression of ftsQ1 and ΔftsEX or ΔftsK by overexpression of FtsN
Because of the competition between FtsA’s self-interaction and interaction with FtsN, both requiring FtsA’s IC domain, our model proposes that if FtsN is overexpressed, it will cause FtsA to be more monomeric at the Z ring which is necessary for recruitment of the late cell division proteins leading to septation. In other words by promoting FtsA becoming monomeric at the Z ring FtsN’s overexpression promotes the formation of the complete divisome. Even if there is an apparent hierarchy in the recruitment of proteins downstream of FtsA at the divisome, these components also rely on a complex network of interactions to be recruited. This is evident in previous studies using ZapA fusions to late cell division proteins such as FtsQ or FtsW (Goehring et al., 2006, Goehring et al., 2005). These fusions artificially recruit the proteins directly to the Z ring, earlier than their normal timing, and independent of the upstream proteins usually required. These studies show that a late cell division protein that is localized early to the Z ring by being fused to ZapA, not only recruits proteins downstream of it, but can also back-recruit proteins upstream of it and on which it normally depends for localization. However, in these situations, FtsN cannot be recruited to the divisome if FtsA or FtsQ are not already present, even if all the other essential cell division proteins directly upstream (FtsI for example) are present. Finally, upon FtsK depletion, proteins like FtsQ, FtsL or FtsI are not recruited to the septum, but they localize to the divisome provided that GFP-FtsN is overexpressed (Goehring et al., 2007) and this recruitment is sufficient to allow cells to divide. Taken together, these results indicate that even if there is a hierarchy in the recruitment pathway, there is also what has been termed a “back recruitment” process where one of the late cell division proteins is recruited to the divisome by a protein that normally comes after it.
In the suppression of ftsK-null by overexpression of FtsN, it is important to note that the cells are still elongated (Geissler & Margolin, 2005, Goehring et al., 2007). This implies that the cell division defect is not fully suppressed and suggests that the back recruitment of the other cell division proteins is not as efficient as the regular hierarchical recruiting. By increasing FtsN expression in the cells, one might force its normally weak interaction with FtsA, allowing FtsN to be recruited earlier to the divisome. By doing this, FtsN could back-recruit other cell division proteins that are normally recruited before it. In addition, FtsN has been shown to interact with FtsA, FtsQ and FtsI and by interacting with the thermosensitive variants of these proteins it might stabilize the mutant proteins at non-permissive temperature. Overexpressed FtsN might also directly recruit FtsI or FtsQ mutant proteins to the septum through its direct interaction with FtsA. It might also stabilize the FtsA mutant by direct interaction so that it functions in cell division. In accordance with this idea, we have reported previously that a mutant FtsA (FtsAE69R) which complements ftsA12 did not allow the ftsA-null strain to grow (Pichoff et al., 2012), indicating that even at non-permissive temperature the mutant protein encoded by ftsA12 is still partly functional. Finally in the case where an early cell division protein such as FtsK or FtsEX, is missing, the regular sequential and hierarchical recruitment process is disrupted but the back recruitment of all the other cell division proteins to the Z ring through FtsN interaction with FtsA (when FtsN is overexpressed) is apparently enough for the cell to divide. Under these conditions the N-terminal cytoplasmic domain of FtsN becomes essential as the D5N mutant cannot suppress these null mutants. In other words, whenever the normal hierarchical recruitment pathway is disrupted the N-terminal domain of FtsN appears to become essential because it might be required for the back-recruitment of all the other division proteins.
FtsN’s E domain has to be linked to cytoFtsN in order to bypass ZipA
The bypass of ZipA and the suppression of ΔftsEX by overexpressed of FtsN required that the E domain of FtsN be linked to the cytoFtsN domain. Overexpression of FtsN1–71 in the presence of an excess of the exported EFtsN domain failed to bypass ZipA (Fig. S7). This was somewhat surprising since the cytoFtsN domain is not absolutely required for growth and the EFtsN domain alone is sufficient for growth when it is overexpressed and exported to the periplasm. What then is the basis for the requirement that these two domains of FtsN to be physically linked? Clearly under some conditions they do not have to be linked. For example, an untethered EFtsN domain is not very efficient and has to be overexpressed 40 fold to suppress ΔftsN. However, under these conditions ZipA is present and FtsA monomers are more abundant increasing the likelihood that the untethered EFtsN domain contacts an otherwise complete divisome complex (the complex of [FtsE, X, K, I, Q, L. B and W] formed by the hierarchical recruitment of downstream proteins to an FtsA monomer). In the absence of ZipA, a Z ring is formed but downstream proteins are not recruited because FtsA is not monomeric. Although overexpression of FtsN1–71 might enhance the formation of FtsA monomers, a complete divisome may not be attached to an FtsA monomer or the FtsN1–71 may not be very efficient. In contrast, when FtsN or an FtsNΔSPOR is overexpressed and a ctyoFtsN domain contacts an FtsA monomer, the additional physical linkage of EFtsN domain ensures that a complete divisome is bound to that FtsA monomer, facilitating activation of the linked divisome complex by this EFtsN domain. In the case of suppression of ΔftsEX the overexpressed FtsN not only causes FtsA monomers to form, but the disruption of the hierarchical recruitment, means that the FtsN bound to the FtsA monomer may back recruit all the downstream division proteins to form a complete divisome complex attached to the FtsA monomer.
Maintenance of FtsA at the Z ring during the septation process
We also predict that the interaction between FtsN and FtsA plays an important role in the maintenance of the late cell division proteins at the Z ring up to the end of cytokinesis. As septation ensues, the dynamic Z ring constricts and FtsZ is removed from the ring so there are fewer FtsZ tails available at the ring to interact with all of its partners (FtsA, ZipA, ZapD, etc). It is also well known that the ratio of FtsZ/FtsA/ZipA at the ring is important for the cytokinesis process to work as too much of any one of these proteins hinders septation (Hale & de Boer, 1997, Pichoff & Lutkenhaus, 2001, Rueda et al., 2003, Begg et al., 1998, Dai & Lutkenhaus, 1992, Dewar et al., 1992). The ratio of these proteins in the cells is about 1 FtsA to 2 ZipA to 5 FtsZ (Rueda et al., 2003). Also, FtsN recruitment is self-enhanced at the septum once constriction begins, meaning that as a cell constricts there is a lot more FtsN at the divisome than before the beginning of constriction (Gerding et al., 2009). Finally, earlier work (Shen & Lutkenhaus, 2009) clearly showed that the in vivo affinity of ZipA for the FtsZ’s C-terminal peptide motif (“CCTP”) is stronger than FtsA’s affinity for that motif since overexpression of MinCD, which also binds the “CCTP” and competes with these two other proteins for binding, displaces FtsA before ZipA. So, as the septum constricts it should become more and more difficult for FtsA to be maintained at the Z ring if it only relied on its ability to interact with the FtsZ’s “CCTP”. One could imagine that FtsZ would interact with ZipA instead of FtsA, causing FtsA to leave the divisome before the end of constriction. But a recent study indicates that FtsA remains to the end of constriction (Soderstrom et al., 2014). Because of FtsN’s self-enhanced recruitment at the divisome, it mimics FtsN overexpression, and FtsN would interact with FtsA at the Z ring and promote FtsA’s monomeric state, hence promoting the ability of FtsA to be maintained at the septum and to recruit the late cell division proteins.
Experimental Procedures
Strains, plasmids, primers and media
All strains and plasmids used in this study are listed in Table S2 in the supplemental material. Unless indicated otherwise cells were grown in LB (Luria Broth) medium containing 0.5% NaCl (with 15 g agar per liter for plates) at the noted temperatures. When needed ampicillin, kanamycin, spectinomycin, chloramphenicol, arabinose and glucose were added at a final concentration of 100 µg/ml, 25 µg/ml, 50 µg/ml, 20 µg/ml, 0.2% and 0.2% respectively.
Plasmid constructions
PCR fragments carrying the genes of interest were obtained by PCR from W3110 chromosomal DNA using adequate pairs of oligos as indicated Table S3. The PCR fragments were cut at the indicated restriction sites and ligated into pDSW208 cut at the same sites as follows: PCR fragments carrying ftsN, ftsN1–140, ftsN1–72, ftsI, ftsL, ftsQ, ftsE, ftsX, ftsEX, ftsW, and ftsB were cloned between the NcoI and HindIII sites to give pSEB416, pSEB417, pSEB419, pSEB420, pSEB422, pSEB425, pSEB426, pSEB427, pSEB428, pSEB429 and pSEB430, respectively. PCR fragments carrying ftsP, ftsK, ftsK1–235 were inserted between the NcoI and SalI sites to give pSEB421, pSEB423 and pSEB424 respectively. A PCR fragment carrying dapE was cloned between the BamHI and HindIII sites to give pDSW208-DapE. PCR fragments carrying ftsN and ftsN1–140 were cloned into pDSW210 between the NcoI and HindIII sites to give pSEB413 and pSEB414, respectively. A PCR fragment carrying ZipA from pBAD-ZipA (Pichoff & Lutkenhaus, 2002), obtained using the oligos 5’NcoI ZipA and 3’Amp-PstI (see Table S3), was cloned into pDSW210 between the NcoI and HindIII sites to give pSEB370. All the above expression vectors are constructed the same way and expressed native proteins (untagged). These plasmids were checked for their abilities to complement a defect of the corresponding gene (depletion or thermosensitive mutation for example) with just the basal expression (no induction by IPTG) and also for causing similar suppression or toxicity as previously reported when the gene they carry is overexpressed (Fig. S1 and data not shown).
PSEB436 was obtained by cloning a version of ftsN with an ATG start codon obtained by PCR using the oligos 5’ NcoI ATG FtsN and 3’FtsN HindIII cut with NcoI and HindIII and ligated into pDSW208 at the same sites to give pSEB433. The pSEB433 fragment containing ftsN or the pSEB306-R300E (Pichoff & Lutkenhaus, 2007) fragment containing ftsAR300E were cut with SphI and ScaI and ligated to the SphI–EcoRV fragment of pACYC184 containing the OriP15A and the chloramphenicol resistance gene to give pSEB436 or pSEB440-R300E respectively. All the plasmid constructs were done in the JS238 strain (Pichoff et al., 1995).
Mutagenesis
Mutagenesis was done by site-directed mutagenesis using the Quickchange II kit (Agilent) following the protocol provided with the kit. The oligonucleotides used for the mutagenesis reactions are provided in Table S3. All mutations were confirmed by sequencing. Plasmids carrying a mutated gene were named after the original plasmid carrying the wild type version of the gene by adding the residue that was changed to the name, such as pSEB413-D5N, which expresses FtsND5N.
Protein Localization and microscopy
Localization of the FtsN N-terminus in W3110 was done using Margolin’s lab plasmids and protocol as described in Busiek and Margolin (2014). Mutations were introduced by site directed mutagenesis and confirmed by sequencing as described above. Cells were observed and photographed as described earlier (Pichoff & Lutkenhaus, 2005).
BACTH Experiments
These experiments were done using protocols and plasmids provided by Margolin’s laboratory as described in Busiek et al. (2012) but instead of streaking colonies on plates to observe the color change, colonies were touched with a toothpick and the attached cells were re-suspended in 500 µl of LB, then 5 µl of suspension was spotted onto selective plates. Mutations in plasmids were introduced by site directed mutagenesis and checked by sequencing as described above. β-galactosidase measurements were done following the Margolin’s lab procedures (Shiomi & Margolin, 2007) but instead of starting from re-suspended colonies we used mid exponential phase cultures.
Western Blotting
Western blotting was performed as described (Pichoff & Lutkenhaus, 2001). Proteins were separated by SDS-PAGE using 12% acrylamide gels and transferred to nitrocellulose membranes. FtsN was detected by an indirect immunostaining procedure with a rabbit polyclonal antiserum against a MalE-FtsN fusion (1:500) (Dai et al., 1996) or affinity-purified antiserum against FtsN (1:250) (Addinall et al., 1997) [in this case MalE is not detected anymore] and a goat anti-rabbit immunoglobulin G antibodies coupled to alkaline phosphatase (BioRad #170–6518) diluted 1:3000.
Supplementary Material
Acknowledgements
We thank Kim Busiek and Bill Margolin as well as Piet De Boer and David Weiss for providing us with some strains and plasmids necessary for this study. This work was supported by NIH grant GM29764 and a KUMC LIED grant.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Aarsman ME, Piette A, Fraipont C, Vinkenvleugel TM, Nguyen-Disteche M, den Blaauwen T. Maturation of the Escherichia coli divisome occurs in two steps. Molecular microbiology. 2005;55:1631–1645. doi: 10.1111/j.1365-2958.2005.04502.x. [DOI] [PubMed] [Google Scholar]
- Addinall SG, Cao C, Lutkenhaus J. FtsN, a late recruit to the septum in Escherichia coli . Molecular microbiology. 1997;25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- Begg K, Nikolaichik Y, Crossland N, Donachie WD. Roles of FtsA and FtsZ in activation of division sites. Journal of bacteriology. 1998;180:881–884. doi: 10.1128/jb.180.4.881-884.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard CS, Sadasivam M, Shiomi D, Margolin W. An altered FtsA can compensate for the loss of essential cell division protein FtsN in Escherichia coli . Molecular microbiology. 2007;64:1289–1305. doi: 10.1111/j.1365-2958.2007.05738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli . Molecular cell. 2005;18:555–564. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Lutkenhaus J. Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. Journal of bacteriology. 1993;175:1118–1125. doi: 10.1128/jb.175.4.1118-1125.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi EF, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia coli . Nature. 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- Buddelmeijer N, Beckwith J. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Molecular microbiology. 2004;52:1315–1327. doi: 10.1111/j.1365-2958.2004.04044.x. [DOI] [PubMed] [Google Scholar]
- Busiek KK, Eraso JM, Wang Y, Margolin W. The early divisome protein FtsA interacts directly through its 1c subdomain with the cytoplasmic domain of the late divisome protein FtsN. Journal of bacteriology. 2012;194:1989–2000. doi: 10.1128/JB.06683-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busiek KK, Margolin W. A role for FtsA in SPOR-independent localization of the essential Escherichia coli cell division protein FtsN. Molecular microbiology. 2014;92:1212–1226. doi: 10.1111/mmi.12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camberg JL, Hoskins JR, Wickner S. ClpXP protease degrades the cytoskeletal protein, FtsZ, and modulates FtsZ polymer dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10614–10619. doi: 10.1073/pnas.0904886106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Beckwith J. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Molecular microbiology. 2001;42:395–413. doi: 10.1046/j.1365-2958.2001.02640.x. [DOI] [PubMed] [Google Scholar]
- Corbin BD, Geissler B, Sadasivam M, Margolin W. Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. Journal of bacteriology. 2004;186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Lutkenhaus J. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli . Journal of bacteriology. 1992;174:6145–6151. doi: 10.1128/jb.174.19.6145-6151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Xu Y, Lutkenhaus J. Cloning and characterization of ftsN, an essential cell division gene in Escherichia coli isolated as a multicopy suppressor of ftsA12(Ts) Journal of bacteriology. 1993;175:3790–3797. doi: 10.1128/jb.175.12.3790-3797.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Xu Y, Lutkenhaus J. Topological characterization of the essential Escherichia coli cell division protein FtsN. Journal of bacteriology. 1996;178:1328–1334. doi: 10.1128/jb.178.5.1328-1334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer PA. Advances in understanding E. coli cell fission. Current opinion in microbiology. 2010;13:730–737. doi: 10.1016/j.mib.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer PA, Crossley RE, Rothfield LI. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli . Cell. 1989;56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- Dewar SJ, Begg KJ, Donachie WD. Inhibition of cell division initiation by an imbalance in the ratio of FtsA to FtsZ. Journal of bacteriology. 1992;174:6314–6316. doi: 10.1128/jb.174.19.6314-6316.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lallo G, Fagioli M, Barionovi D, Ghelardini P, Paolozzi L. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology. 2003;149:3353–3359. doi: 10.1099/mic.0.26580-0. [DOI] [PubMed] [Google Scholar]
- Draper GC, McLennan N, Begg K, Masters M, Donachie WD. Only the N-terminal domain of FtsK functions in cell division. Journal of bacteriology. 1998;180:4621–4627. doi: 10.1128/jb.180.17.4621-4627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du S, Lutkenhaus J. SlmA antagonism of FtsZ assembly employs a two-pronged mechanism like MinCD. PLoS genetics. 2014;10:e1004460. doi: 10.1371/journal.pgen.1004460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Heredia J, Rivkin E, Fan G, Morales J, Janakiraman A. Identification of ZapD as a cell division factor that promotes the assembly of FtsZ in Escherichia coli . Journal of bacteriology. 2012;194:3189–3198. doi: 10.1128/JB.00176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Elraheb D, Margolin W. A gain-of-function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli . Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Margolin W. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Molecular microbiology. 2005;58:596–612. doi: 10.1111/j.1365-2958.2005.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Shiomi D, Margolin W. The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring. Microbiology. 2007;153:814–825. doi: 10.1099/mic.0.2006/001834-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MA, Liu B, Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. Self-enhanced accumulation of FtsN at division sites and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. Journal of bacteriology. 2009;191:7383–7401. doi: 10.1128/JB.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Beckwith J. Diverse paths to midcell: assembly of the bacterial cell division machinery. Current biology : CB. 2005;15:R514–R526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Goehring NW, Gonzalez MD, Beckwith J. Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Molecular microbiology. 2006;61:33–45. doi: 10.1111/j.1365-2958.2006.05206.x. [DOI] [PubMed] [Google Scholar]
- Goehring NW, Gueiros-Filho F, Beckwith J. Premature targeting of a cell division protein to midcell allows dissection of divisome assembly in Escherichia coli . Genes & development. 2005;19:127–137. doi: 10.1101/gad.1253805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Robichon C, Beckwith J. Role for the nonessential N terminus of FtsN in divisome assembly. Journal of bacteriology. 2007;189:646–649. doi: 10.1128/JB.00992-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PA. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli . Cell. 1997;88:175–185. doi: 10.1016/s0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- Hale CA, de Boer PA. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ and independent of FtsA. Journal of bacteriology. 1999;181:167–176. doi: 10.1128/jb.181.1.167-176.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PA. ZipA is required for recruitment of FtsK, FtsQ, FtsL, and FtsN to the septal ring in Escherichia coli . Journal of bacteriology. 2002;184:2552–2556. doi: 10.1128/JB.184.9.2552-2556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, Shiomi D, Liu B, Bernhardt TG, Margolin W, Niki H, de Boer PA. Identification of Escherichia coli ZapC (YcbW) as a component of the division apparatus that binds and bundles FtsZ polymers. Journal of bacteriology. 2011;193:1393–1404. doi: 10.1128/JB.01245-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney SA, Glasfeld E, Hale C, Keeney D, He Z, de Boer P. Genetic analysis of the Escherichia coli FtsZ.ZipA interaction in the yeast two-hybrid system. Characterization of FtsZ residues essential for the interactions with ZipA and with FtsA. The Journal of biological chemistry. 2001;276:11980–11987. doi: 10.1074/jbc.M009810200. [DOI] [PubMed] [Google Scholar]
- Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. Journal of bacteriology. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppiah V, Derrick JP. Structure of the PilM-PilN inner membrane type IV pilus biogenesis complex from Thermus thermophilus . The Journal of biological chemistry. 2011;286:24434–24442. doi: 10.1074/jbc.M111.243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Mukherjee A, Lutkenhaus J. Recruitment of ZipA to the division site by interaction with FtsZ. Molecular microbiology. 1999;31:1853–1861. doi: 10.1046/j.1365-2958.1999.01322.x. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annual review of biochemistry. 2007;76:539–562. doi: 10.1146/annurev.biochem.75.103004.142652. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J. FtsN-trigger for septation. Journal of bacteriology. 2009;191:7381–7382. doi: 10.1128/JB.01100-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkenhaus J, Pichoff S, Du S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton. 2012;69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll A, Thanbichler M. FtsN-like proteins are conserved components of the cell division machinery in proteobacteria. Molecular microbiology. 2009;72:1037–1053. doi: 10.1111/j.1365-2958.2009.06706.x. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Escherichia coli division inhibitor MinCD blocks septation by preventing Z-ring formation. Journal of bacteriology. 2001;183:6630–6635. doi: 10.1128/JB.183.22.6630-6635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli . The EMBO journal. 2002;21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Molecular microbiology. 2005;55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Identification of a region of FtsA required for interaction with FtsZ. Molecular microbiology. 2007;64:1129–1138. doi: 10.1111/j.1365-2958.2007.05735.x. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Shen B, Sullivan B, Lutkenhaus J. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA’s self-interaction competes with its ability to recruit downstream division proteins. Molecular microbiology. 2012;83:151–167. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Vollrath B, Touriol C, Bouche JP. Deletion analysis of gene minE which encodes the topological specificity factor of cell division in Escherichia coli . Molecular microbiology. 1995;18:321–329. doi: 10.1111/j.1365-2958.1995.mmi_18020321.x. [DOI] [PubMed] [Google Scholar]
- Reddy M. Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional SufI or high osmotic strength. Journal of bacteriology. 2007;189:98–108. doi: 10.1128/JB.01347-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico AI, Garcia-Ovalle M, Mingorance J, Vicente M. Role of two essential domains of Escherichia coli FtsA in localization and progression of the division ring. Molecular microbiology. 2004;53:1359–1371. doi: 10.1111/j.1365-2958.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- Rueda S, Vicente M, Mingorance J. Concentration and assembly of the division ring proteins FtsZ, FtsA, and ZipA during the Escherichia coli cell cycle. Journal of bacteriology. 2003;185:3344–3351. doi: 10.1128/JB.185.11.3344-3351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaluru H, SaiSree L, Reddy M. Role of SufI (FtsP) in cell division of Escherichia coli: evidence for its involvement in stabilizing the assembly of the divisome. Journal of bacteriology. 2007;189:8044–8052. doi: 10.1128/JB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P. Bacterial cell division: a swirling ring to rule them all? Current biology : CB. 2014;24:R157–R159. doi: 10.1016/j.cub.2014.01.025. [DOI] [PubMed] [Google Scholar]
- Shen B, Lutkenhaus J. The conserved C-terminal tail of FtsZ is required for the septal localization and division inhibitory activity of MinC(C)/MinD. Molecular microbiology. 2009;72:410–424. doi: 10.1111/j.1365-2958.2009.06651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Lutkenhaus J. Examination of the interaction between FtsZ and MinCN in E. coli suggests how MinC disrupts Z rings. Molecular microbiology. 2010;75:1285–1298. doi: 10.1111/j.1365-2958.2010.07055.x. [DOI] [PubMed] [Google Scholar]
- Shiomi D, Margolin W. Dimerization or oligomerization of the actin-like FtsA protein enhances the integrity of the cytokinetic Z ring. Molecular microbiology. 2007;66:1396–1415. doi: 10.1111/j.1365-2958.2007.05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderstrom B, Skoog K, Blom H, Weiss DS, von Heijne G, Daley DO. Disassembly of the divisome in Escherichia coli: evidence that FtsZ dissociates before compartmentalization. Molecular microbiology. 2014;92:1–9. doi: 10.1111/mmi.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwedziak P, Wang Q, Freund SM, Lowe J. FtsA forms actin-like protofilaments. The EMBO journal. 2012;31:2249–2260. doi: 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursinus A, van den Ent F, Brechtel S, de Pedro M, Holtje JV, Lowe J, Vollmer W. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli . Journal of bacteriology. 2004;186:6728–6737. doi: 10.1128/JB.186.20.6728-6737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Ent F, Lowe J. Crystal structure of the cell division protein FtsA from Thermotoga maritima . The EMBO journal. 2000;19:5300–5307. doi: 10.1093/emboj/19.20.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Khattar MK, Donachie WD, Lutkenhaus J. FtsI and FtsW are localized to the septum in Escherichia coli . Journal of bacteriology. 1998;180:2810–2816. doi: 10.1128/jb.180.11.2810-2816.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, de Boer PA, Rothfield LI. A factor that positively regulates cell division by activating transcription of the major cluster of essential cell division genes of Escherichia coli . The EMBO journal. 1991;10:3363–3372. doi: 10.1002/j.1460-2075.1991.tb04900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. Journal of bacteriology. 1999;181:508–520. doi: 10.1128/jb.181.2.508-520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissel MC, Weiss DS. Genetic analysis of the cell division protein FtsI (PBP3): amino acid substitutions that impair septal localization of FtsI and recruitment of FtsN. Journal of bacteriology. 2004;186:490–502. doi: 10.1128/JB.186.2.490-502.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Georgopoulos C, Ang D. The essential Escherichia coli msgB gene, a multicopy suppressor of a temperature-sensitive allele of the heat shock gene grpE, is identical to dapE . Journal of bacteriology. 1992;174:5258–5264. doi: 10.1128/jb.174.16.5258-5264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Van Den Ent F, Neuhaus D, Brevier J, Lowe J. Solution structure and domain architecture of the divisome protein FtsN. Molecular microbiology. 2004;52:651–660. doi: 10.1111/j.1365-2958.2004.03991.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.