

Abstract
The protective effect of many HLA class I alleles on HIV-1 pathogenesis and disease progression is, in part, attributed to their ability to target conserved portions of the HIV-1 genome that escape with difficulty. Sequence changes attributed to cellular immune pressure arise across the genome during infection, and if found within conserved regions of the genome such as Gag, can affect the ability of the virus to replicate in vitro. Transmission of HLA-linked polymorphisms in Gag to HLA-mismatched recipients has been associated with reduced set point viral loads. We hypothesized this may be due to a reduced replication capacity of the virus. Here we present a novel method for assessing the in vitro replication of HIV-1 as influenced by the gag gene isolated from acute time points from subtype C infected Zambians. This method uses restriction enzyme based cloning to insert the gag gene into a common subtype C HIV-1 proviral backbone, MJ4. This makes it more appropriate to the study of subtype C sequences than previous recombination based methods that have assessed the in vitro replication of chronically derived gag-pro sequences. Nevertheless, the protocol could be readily modified for studies of viruses from other subtypes. Moreover, this protocol details a robust and reproducible method for assessing the replication capacity of the Gag-MJ4 chimeric viruses on a CEM-based T cell line. This method was utilized for the study of Gag-MJ4 chimeric viruses derived from 149 subtype C acutely infected Zambians, and has allowed for the identification of residues in Gag that affect replication. More importantly, the implementation of this technique has facilitated a deeper understanding of how viral replication defines parameters of early HIV-1 pathogenesis such as set point viral load and longitudinal CD4+ T cell decline.
Keywords: Infectious Diseases, Issue 90, HIV-1, Gag, viral replication, replication capacity, viral fitness, MJ4, CEM, GXR25
Introduction
Determining both the host and viral characteristics that influence HIV-1 pathogenesis and disease progression is paramount for rational vaccine design. The cellular immune response is a key component of the human immune response to HIV-1 infection. Cytotoxic T lymphocytes (CTL) are necessary for the initial control of acute viremia, and allow the host to establish a steady state (set point) viral load1,2. Experimental depletion of these effector cells results in loss of viral control3,4. Despite this, escape mutations arise within the viral genome that subvert CTL recognition of virally infected cells5-9.
Certain HLA alleles have been associated with lower viral loads and slower disease progression including B*57, B*27 and B*8110-15. Part of the protective benefit of HLA class I alleles can be attributed to the fact that they target functionally constrained regions of the genome such as Gag and select for escape mutations that decrease the ability of the virus to replicate in vitro16-21. Although escape from the cellular immune system is beneficial to the virus in the context of the selecting HLA class I allele, the effect of these mutations may have differential consequences for the host upon transmission to an HLA-mismatched individual22,23. Therefore, understanding the effects of transmitted HLA-associated escape mutations on viral replication capacity will be important to further our understanding of early HIV-1 pathogenesis.
While much progress has been made to identify and characterize the fitness defects of individual escape mutations associated with specific HLA class I alleles24-29, naturally occurring HIV-1 isolates have unique and complex footprints of HLA-associated polymorphisms, likely arising from the HLA-mediated immune pressure of different immunogenetic backgrounds30. In a previous analysis, Goepfert et al. showed that an accumulation of HLA-associated mutations in the transmitted Gag sequences derived from 88 acutely infected Zambians was associated with a reduction in set point viral load31. This suggested that the transmission of deleterious escape mutations, specifically in Gag, to HLA-mismatched recipients provides a clinical benefit, and may be due to attenuated viral replication. Moving forward, it is imperative to study how complex combinations of Gag polymorphisms within naturally occurring isolates work in concert to define characteristics of the transmitted virus such as replication capacity, and how early replication might in turn affect HIV-1 clinical parameters and late-stage pathogenesis.
Brockman et al. first demonstrated a link between the replication capacity of gag-pro sequences isolated during chronic stage infection and viral load in both subtype C and B infections32-35. The experimental approach presented in these studies, although appropriate for examining the in vitro replication capacity of sequences derived from chronically infected individuals, has several technical caveats and limitations that make studying HIV-1 replicative capacity in subtype C acutely infected individuals difficult. This method relies on the recombination of population based PCR-amplified sequences into the subtype B NL4-3 provirus, which was derived in part from LAV, a laboratory adapted virus stock36. Virus generation was accomplished by co-transfection of a CEM-based T-cell line37 with PCR amplicons and digested delta-gag-pro NL4-3 DNA. This method requires the outgrowth of virus over a period of weeks to months, potentially skewing the nature of the recovered virus stock in relation to the viral quasispecies in vivo, and therefore altering the measurement of replication capacity in vitro. This method is more appropriate for studying chronically infected individuals, where it effectively selects for virus with the highest replicative capacity, and where cloning numerous different viral variants from a large number of chronically infected individuals is quite labor intensive and therefore not feasible. However, within an acutely infected individual, there are generally one to two variants present, and thus eliminating the risk of skewing the nature of the recovered virus stock, through in vitro selection pressures, allows for a more accurate assessment of in vitro replication capacity. Secondly, this method requires recombining subtype C gag-pro sequences into a subtype B derived backbone, and could introduce backbone incompatibility biases into the analysis. Due to these limitations, large numbers of sequences must be analyzed in order to overcome any potential biases introduced.
Here we describe an alternative experimental approach appropriate for studying sequences derived from subtype C acutely infected individuals. We use a restriction enzyme based cloning strategy to introduce the gag gene derived from acute infection time points of HIV-1 subtype C infected individuals into the subtype C proviral backbone, MJ4. The use of MJ4 as a common backbone in which to clone gag genes is crucial for the analysis of subtype C derived sequences. MJ4 is derived from a primary isolate38, and thus would be less likely to introduce bias due to subtype incompatibility between the backbone and gag gene. In addition, the approach of using enzyme based restriction cloning allows for the proviral constructs to be transfected directly into 293T cells, and for the recovery of a clonal virus stock identical to the cloned gag sequence.
The method presented below is a high throughput method for assessing the replication capacity of subtype C derived Gag-MJ4 chimeric viruses. Transfection into 293T cells is straightforward and recovery of virus takes only three days. In vitro replication capacity is assayed on the same CEM-CCR5 based T-cell line created by Brockman et al.37, but using important protocol modifications necessary for the successful replication of subtype C MJ4 chimeric viruses. The use of an appropriate T-cell line rather than PBMCs allows for large numbers of subtype C MJ4-chimeric viruses to be tested with high assay reproducibility. Finally, using a radiolabeled reverse transcriptase assay for quantification of virus in the supernatant is more cost effective than using commercially available p24 ELISA kits. It also gives a higher dynamic range, which was important for detecting both poorly and highly replicating viruses within the same assay and for detecting subtle differences in replication between isolates.
In conclusion, the method presented here has allowed for the in-depth study of the replication capacity of gag sequences derived from HIV-1 subtype C acutely infected individuals from Zambia, and as written, could also be expanded to study other subtype C infected populations. A high degree of variation in replication capacities between different Gag isolates was observed. In addition, we were able to show a statistical association between the replication capacity of the transmitted Gag and set point viral load as well as with CD4+ decline over a three-year period39. Such results highlight the importance of studying how transmitted viral characteristics, such as replication capacity, interact with the host immune system to influence pathogenesis during early infection and will be integral for developing effective vaccine interventions as well as treatment.
Protocol
1. Amplification of the HIV-1 gag Gene from Infected, Frozen Plasma
- Extract viral RNA from 140 µl thawed HIV-1 infected plasma using an extraction kit.
- When feasible, immediately proceed to cDNA synthesis after RNA extraction as unfrozen viral RNA yields the best amplification results. If possible, set up PCR master mix for first-round DNA amplification and store at 4 °C prior to viral RNA extraction.
- Reverse-transcribe cDNA from RNA and amplify first-round DNA products using reverse-transcriptase and a thermostable DNA polymerase in a one-step RT-PCR.
- Take RNA out of -80 °C freezer (if freezing was necessary) and briefly thaw at room temperature then place in cold block. Transfer 5 µl of RNA to each PCR tube containing 45 µl of master mix to give a final volume of 50 µl. Immediately place remaining RNA samples into -80 °C freezer.
- After enzyme is added to the first-round PCR master mix (Table 1A), aliquot 45 µl into each thin-walled PCR amplification tube for the number of reactions desired. Make sure to use PCR tubes with individual caps and not strip caps to ensure minimal cross-contamination between samples. Place PCR tubes in a cold block to protect the temperature sensitive RT enzyme and RNA template. Note: Samples should be run in triplicate in order to adequately sample the viral quasispecies. It is also important to utilize a product with a high fidelity DNA polymerase enzyme in order to minimize PCR-introduced misincorporation. Please refer to the reagents list for the recommended product.
- After RNA template has been added to all reaction tubes, transfer PCR tubes to a thermocycler for amplification using the cycler program described in Table 1B. Note: The product of this amplification will be used in a second-round nested PCR.
- Perform a nested second-round PCR amplification, using 1 µl of the first round PCR amplification (1.2) as the DNA template.
- Aliquot 49 µl of second-round PCR master mix (Table 2A) to each thin-walled PCR tube for the number of reactions desired. Transfer 1 µl from the first-round PCR amplification to each reaction tube to give a final volume of 50 µl. Note: This will serve as template DNA for the second-round amplification. It is also important to utilize a DNA polymerase with very high fidelity and proofreading capabilities in order to minimize PCR-introduced misincorporation. Please refer to the reagents list for the recommended product.
- Transfer second-round PCR reaction mix to thermocycler and run program described in Table 2B. Note: After completion of the program, the products of this reaction will be run on an agarose gel to confirm the production of product.
Add 3 µl of 5x loading dye to 5 µl of the 50 µl second-round reaction volume and run at 120 V on a 1% agarose-TAE gel containing a UV-fluorescent DNA stain until bands are resolved. Visualize 1.6 kb bands on a blue light illuminator (see Figure 1A).
- Run the remaining 45 µl reaction volume on a 1% agarose-TAE gel containing a DNA stain, and excise the appropriate bands with a clean razor blade.
- Extract DNA from the gel slice using a gel purification kit, elute in nuclease-free water, combine three positive reactions per individual, and freeze product at -20 °C for subsequent use.
2. Preparing gag Amplicons for Cloning by Introduction of the Necessary Restriction Sites
Amplify the Long Terminal Repeat (LTR)/5’ UTR portion of the MJ4 plasmid (see Table 3).
Visualize 1.3 kb PCR products on illuminator, excise positive amplicons, gel purify and freeze as previously described (1.4,1.5; see Figure 1B).
Create fused MJ4 LTR-gag amplicons via "splice-overlap-extension" PCR (see Table 4).
Visualize, excise, purify, and freeze the 3.2 kb amplicons as in 1.4,1.5 (see Figure 1C).
3. Cloning Amplified gag Genes into the MJ4, Subtype C, Infectious Molecular Clone
Digest 1.5 µg of MJ4 plasmid and 1.5 µg of purified LTR-gag PCR product with the BclI (methylation sensitive) restriction endonuclease for 1.5 hr at 50 °C (see Table 5A).
Add 1 µl of NgoMIV to the digestion reaction and incubate at 37 °C for 1 hr.
Add 5x loading dye to restriction digest reactions and slowly electrophorese the total volume on a 1% agarose-TAE gel containing a DNA gel stain for 1-2 hr at 100 V. Visualize, excise, and purify indicated bands for cloning as previously described (see Figure 2).
Prepare ligation reactions using purified LTR-gag insert and MJ4 vector DNA at a 3:1 insert to vector ratio. Incubate ligation reactions overnight at 4 °C (see Table 5B).
- Transform JM109 chemically competent bacteria with ligation products and spread on LB agar plates with 100 µg/ml ampicillin and grow at 30 °C for 22+ hr.
- Thaw JM109 competent cells on ice for 15 min. Label 1.5 ml microcentrifuge tubes and chill on ice.
- Aliquot 50-100 µl of JM109 cells to pre-chilled 1.5 ml microcentrifuge tubes. Add 2.5-5 µl of ligation reaction to JM109 cells, flicking tube lightly to mix and immediately returning to ice. Incubate competent cells with ligation product on ice for 30 min.
- Heat shock 1.5 ml microcentrifuge tubes containing JM109 competent cells and ligation reaction in a 42 °C heat block for 45 sec and return to ice for at least 3 min.
- Add 50 µl of SOC media to each microcentrifuge tube and plate the entire transformation reaction onto room temperature LB-agar plates supplemented with 100 µg/ml of ampicillin.
- Transfer plates to a 30 °C incubator and leave for 20 hr, or until colonies are clearly formed.
Pick isolated colonies and grow in 4 ml of LB with 100 µg/ml ampicillin at 30 °C for 22+ hr.
Spin down cultures at 3,200 x g for 15 min, pour off broth, and extract plasmid DNA.
To confirm cloning fidelity, cut miniprep DNA with NgoMIV and HpaI restriction enzymes in a double digest at 37 °C for 2 hr. Analyze on 1% agarose-TAE gel (see Table 5C).
Sequence the LTR-gag insert region of each plasmid using the following primers: GagInnerF1, GagF2, Rev1, Rev3, and GagR6 (Table 6) to confirm sequence identity.
Compare the cloned sequences obtained with the population sequences derived from the initial purified amplicon from 1.5.1. Note: It is important to ultimately assess the replication capacity of two independent clones in order to ensure that any in vitro replication phenotypes are not due to backbone errors introduced during the cloning process.
4. Generation and Titering of Replication Competent Gag-MJ4 Chimeric Viruses
Generate replication competent virus by transfecting 1.5 µg of the chimeric MJ4 plasmid miniprep DNA into 293T cells using a 4:1 ratio of Fugene HD as described by Prince et al39.
- Titer harvested virus stocks on TZM-bl cells as described below and in Prince et al39.
- Plate TZM-bl cells 24 hr before infection with virus in order to have a 30-40% confluent cell monolayer on the following day. This can usually be achieved by adding 5 x 104 cells in a total volume of 800 µl per well in a 24-well plate.
- After adding cells to the well, gently move the 24-well plate forward and back, then side to side in order to efficiently distribute cells. Never swirl – this will result in cells accumulating in the center of the plate.
- The following day, prepare 1% FBS in DMEM; this will be used to dilute DEAE-Dextran and to dilute virus stocks. Stock DEAE-Dextran is 10 mg/ml or 125x, and a final concentration of 80 µg/ml or 1x is desired.
- Take out virus stock to be tested from -80 °C freezer and place on shaker to thaw at room temperature.
- Using a multichannel pipette, serially dilute virus stocks in a round-bottomed tissue culture treated 96-well plate with lid. This is a 3-fold dilution protocol. See Table 7 for the dilution scheme.
- Remove media from seeded TZM-bl 24-well plates using a vacuum aspirator making sure not to disturb cell monolayer. Only remove the media from one 24-well plate at a time so that cells do not dry out.
- Add 150 µl of the 1x DEAE-Dextran + 1% FBS in DMEM mixture to each well using a 1,000 µl pipette. Do not use repeat pipette as this disrupts the monolayer.
- Add 150 µl of each virus dilution to the appropriate well. Add virus to the center of the well and swirl gently to evenly distribute virus across the cell monolayer. Incubate infected cells for 2 hr in a tissue culture incubator at 37 °C and 5% CO2.
- After the 2 hr incubation, add 0.5 ml 10% FBS in DMEM to each well and return plates to incubator for an additional 48 hr.
- After 48 hr, cells should be 100% confluent. Because MJ4 is a replication competent virus, there will be some cell death, but this is to be expected.
- Remove media from each well and add 400 µl/well of fixing solution (see Table 8A for recipe). Fix only one plate at a time. Add fixing solution using a 1,000 µl pipette, and let sit for 5 min at room temperature.
- Remove fixing solution and wash 3x with PBS with Ca2+/Mg2+. Use a squirt bottle to gently add PBS to the side of the well to avoid disrupting the monolayer. After the third wash, blot plate dry on absorbent paper.
- Add 400 µl/well of staining solution (see Table 8B for recipe) to each well of the 24-well plate, and incubate at 37 °C for at least 2 hr. Longer incubation times are acceptable, but incubation times should be kept consistent between titering experiments.
- Wash 2x with PBS with Ca2+/Mg2+ and score for infection, or add PBS and store at 4 °C for scoring later. Plates can be kept at 4 °C for up to four days, as long as the monolayer is kept hydrated.
- To score for infection, use a permanent marker to divide wells of the 24-well plate into quadrants. Count all blue cells within a field of view, using a total magnification of 200X, once in each of the four quadrants of a well.
- Compute Infectious Units per µl as follows: [(# blue cells/4) x 67] / (µl virus added) = IU/µl. Refer to Table 8C for volume in µl of virus added to each well. Average several wells together; the numbers should be relatively similar for an accurate titration.
5. Preparation of Culture Media and Propagation of GXR25 Cells for in vitro Replication Assay
Prepare complete RPMI (cRPMI) for propagation of GXR25 cells. NOTE: It is extremely important to culture GXR25 cells at least 4 months prior to planned replication experiments (see Table 9).
Split cells 1:10 twice per week and maintain cell density between 1 x 105 to 2 x 106 cells/ml.
6. In vitro Replication of Gag-MJ4 Chimeras in GXR25 (CEM-CCR5-GFP) Cells
The day before the infection, split cells to a concentration between 2-3 x 105 cells/ml so that they are in logarithmic growth on the day of the infection.
On the day of infection, remove virus stocks from -80 °C freezer and thaw. Calculate the volume of virus needed from each stock based on the IU/µl titer from TZM-bl cell assay in order to infect 5 x 105 GXR25 cells at a multiplicity of infection of 0.05, using the formula: Volume of virus for infection (µl) = 1 µl/X IU x 0.05 x 500,000 NOTE: We have chosen an MOI of 0.05 as this results in a logarithmic growth phase for all of the viruses that we have tested. It is important to conduct initial experiments in order to determine the optimal MOI to capture logarithmic growth for the virus to be tested.
Dilute virus in cRPMI to a volume of 100 µl (in order to achieve a 0.05 MOI) and add into a well of a V-Bottom tissue culture treated 96-well plate. Note: Include both a positive (MJ4 WT infected) and a negative (mock infected cells) control in each set of replication experiments. Set up the plate such that every other column is blank to limit cross-contamination between wells. The positive control is imperative, as it will be used later as a standard to normalize the replication slopes, which are a measure of the replication rate of experimental replicates.
Count GXR25 cells using an automated cell counter. Calculate the number of cells needed in total for all infections (5 x 105 x # infections). Aliquot the required volume into a 50 ml conical tube and centrifuge to pellet cells. Always calculate for 25% more infections than needed.
Aspirate media carefully and resuspend in cRPMI at a concentration of 5 x 105 cells/100 µl.
Pipette cells into a sterile trough and mix thoroughly. Using a multi-channel pipette, add 100 µl of cells into each well of the 96-well plate containing the diluted virus. Mix thoroughly.
Add 2 µl of 5 mg/ml (100x) solution of polybrene to each well and mix cells thoroughly.
Incubate at 37 °C in tissue culture incubator with 5% CO2 for 3 hr.
In order to wash infected cells, centrifuge 96-well V-bottom plate to pellet cells. Then carefully remove 150 µl of the medium and replace with fresh 150 µl of cRPMI without disturbing the cell pellet. Repeat 2 more times (including centrifugation) to sufficiently wash cells.
After the last centrifugation and addition of 150 µl cRPMI, resuspend the cell pellet with a multichannel pipette and add the entire cell/virus mixture (approximately 200 µl) from each well independently to 800 µl of cRPMI in a well of a 24-well tissue culture plate.
Place plate in 5% CO2 tissue culture incubator at 37 °C.
Every two days, remove 100 µl of supernatant from surface of culture well and transfer to a 96-well U-bottom plate and store frozen at -80 °C until running virus quantitation assay. NOTE: Careful arrangement of supernatants in 96-well plates will allow for multi-channel pipette transfer of culture supernatants during virion quantification via a radiolabeled reverse transcriptase (RT) assay. Every plate should contain samples from an infection standard in order to normalize inter-plate variation in the RT assay readout.
After removing each 100 µl sample, split cells 1:2 by thoroughly resuspending cells and removing half of the remaining volume (450 µl). Restore original volume (1 ml) by adding 550 µl of fresh cRPMI to each well.
7. Analysis of Reverse Transcriptase (RT) in Cell Culture Supernatants
Protocol adapted from Ostrowski et al40.
- Set up biological safety hood in a BSL-3 facility for use with radioactive materials.
- Place absorbent paper in hood and replace aspirator for aspirator designated for radioactive use. Note: All radioactive waste must be properly disposed of with accordance to safety regulations.
Add 1-2 µl of 10 mCi/ml of [α-33P] dTTP and 4 µl of 1 M dithiothreitol (DTT) to each 1 ml aliquot of RT master mix (see Table 10 and Ostrowski et al.40).
Carefully mix each 1.5 ml microcentrifuge tube of RT master mix, DTT, and [α-33P] dTTP with a 1,000 µl pipette and transfer to a sterile trough.
Dispense 25 µl of labeled RT mix into each well of 96-well thin-walled PCR plate. Note: Include space for a positive control (MJ4 WT infection) and negative control (Mock infection).
Add 5 µl of each supernatant to the PCR plate containing the RT master mix.
Seal PCR plate with adhesive foil cover and incubate for 2 hr at 37 °C in a thermocycler. For safety reasons, rinse pipette tips with Amphyl and dispose of tips in small container containing Amphyl waste, which will later be disposed of in radioactive waste.
After incubation, make small holes in the foil cover using a 200 µl multi-channel pipette. Note: If pipette tips touch liquid in well, replace tips before moving to the next column or row.
Mix samples 5x and transfer 5 µl of each sample to the DE-81 paper. Air dry 10 min.
- Wash blots 5x with 1x SSC (sodium chloride, sodium citrate), and then 2x with 90% ethanol at 5 min per wash. Allow to air dry.
- Place each blot in a separate washing container, such as a sandwich storage box, add enough wash buffer (1x SSC or 90% EtOH) to cover blot, and shake for 5 min.
- Pour off wash buffer into separate container and repeat. Note: The first 3 washes are considered radioactive and should be disposed of properly. The last 2 washes can be disposed of regularly.
Once dry, carefully wrap blot in Saran wrap and expose to a phosphoscreen in a tightly sealed cassette overnight at room temperature.
Analyze phosphoscreens with a phosphorimager and quantify radioactive transcripts.
Draw a circle around each radioactive signal using OptiQuant software. Graph the Digital Light Unit (DLU) values to generate replication curves.
In order to generate replication capacity scores, DLU values were log10-transformed and slopes were calculated using the day 2, 4, and 6 time points. Replication slopes are then divided by the slope of WT MJ4 in order generate replication capacity scores. It is important to normalize based on the MJ4 WT values obtained from the same RT plate to reduce intra-assay variability.
Representative Results
In order to properly execute this protocol, which creates a proviral plasmid capable of assembling fully functional, infectious Gag-MJ4 chimeras, great care must be taken to generate the appropriate PCR amplicons. Determining whether the PCR has generated the appropriately sized gag amplicon is crucial. Products should be within 100 base pairs (bp) of the approximately 1,700 bp amplicon depicted in Figure 1A. The exact length of this fragment will vary depending on the gag gene under study. Next, the 5’ long terminal repeat (LTR) portion of the MJ4 molecular clone must be amplified and spliced to the gag amplicon in order to make it suitable for subsequent cloning. The MJ4 LTR amplicon should be 1,474 bp in length. Figure 1B shows a representative gel image for which correct band sizes are indicated. After the splice-overlap-extension PCR41, combined LTR-gag products should be approximately 3,200 bp in length, as depicted in Figure 1C.
Once the gag gene has been made suitable for cloning by fusion to the 5’ LTR from MJ4, which contains the necessary NgoMIV restriction site, both vector and gag insert must be digested with NgoMIV and BclI restriction enzymes and excised from an agarose gel after electrophoretic separation. It is imperative to excise the appropriate vector and insert bands. A representative gel is shown in Figure 2. The vector portion of the MJ4 plasmid should be approximately 10,000 bp in length, while the LTR-gag insert should remain at approximately 3,000 bp in length, as the restriction sites are located at the extreme ends of the amplicon. Any significant decrease in size will indicate an additional cut-site within the gag gene under study.
Following ligation of the two fragments, bacterial transformation, and isolation of plasmid DNA, the Gag-MJ4 chimeras must be checked for appropriate size by performing a double digest with NgoMIV and HpaI restriction enzymes. Full-length Gag-MJ4 chimeras that have not incurred any deletion events during bacterial replication should have a restriction pattern similar to that depicted in Figure 3, with two bands of approximately 8,700 and 4,300 bp.
An important distinction of this protocol compared to previous approaches, is the use of the HIV-1 subtype C infectious molecular clone, MJ4, rather than the more common laboratory-adapted NL4-3 virus. However, the approaches described in the previous section could be modified for cloning of subtype B gag sequences into pNL4-3.
An optimization of the multiplicity of infection (MOI) for use in subsequent experiments was performed in order to select the ideal MOI that showed logarithmic growth of a majority of the viruses tested. Figure 4 depicts representative replication curves from three different MOIs (0.01, 0.05, and 0.25) for MJ4 (Figure 4A) as well as for NL4.3 (Figure 4B). MJ4 replicates much less efficiently in GXR25 cells than NL4-3, which is important to take into account, as an MOI appropriate for NL4-3 replication would likely be too low to detect efficient MJ4 replication. As seen in Figure 4A, an MOI of 0.05 as opposed to 0.01 or 0.25, was the ideal choice, because logarithmic growth was observed between days 2-6 for MJ4. For the lower MOI, 0.01, day 6 DLU values are barely detectable, and we anticipated the generation of Gag-MJ4 chimeric viruses that replicated lower than MJ4. Therefore this MOI would not capture the growth of the more attenuated Gag-MJ4 chimeras, which may also be the most biologically critical. Additionally, an MOI of 0.25 was not ideal, because the rapid kinetics of viral replication killed a substantial amount of cells even by day 4. This causes the replication curve to plateau, and calculating a slope based on a curve such as this would underestimate the replication capacity. Based on curves generated for NL4-3, even at an MOI of 0.01, available cell targets have been noticeably exhausted by day 6 post infection. In conclusion, an MOI of 0.05 was found to be optimal for a large panel of Gag-MJ4 chimeric viruses, all of which had diverse Gag sequences and varying degrees of replication.
A total of 149 Gag-MJ4 chimeric viruses derived from acute subtype C Gag sequences have been tested for in vitro replication using this assay. The normalized RC values ranged from 0.01 to over 3.5 with some viruses replicating more than 100 times more efficiently than wild-type MJ4. Figure 5 shows the replication curves from nine representative Gag-MJ4 chimeric viruses, with wild-type MJ4 depicted in red, and demonstrates the wide range of replication capacities observed. Thus, the sequence diversity within the gag gene alone can drastically impact the ability of the virus to replicate in vitro. While this is representative of Gag-MJ4 chimeric viruses derived from acutely infected Zambians, other subtype C sequences have not been extensively tested, and may exhibit different replication kinetics. Therefore, great care must be taken to optimize the MOI to suit the specific replication of the viruses of a particular study, because there can be a wide range in the levels of replication between different HIV-1 backbones and Gag isolates.
One of the advantages of using a T-cell line such as the GXR25 cell line, that supports the replication of MJ4, is the level of reproducibility observed relative to replication experiments using stimulated peripheral blood mononuclear cells as targets. In initial optimization experiments, MJ4 wild type exhibited an intra-assay variability of 8.7%, and different clones of the same Gag-MJ4 chimeric virus exhibited variability in replication of 8.5%. Because different master mixes and phosphoscreen exposures may give DLU values that differ slightly in magnitude, intra-assay variability can further be controlled by running the same virus standard (in our case wild-type MJ4) in each RT assay plate. Figure 6 graphs the DLU values derived from the same MJ4 infection, quantified in eight different RT plates. Normalizing to a virus that is common to all RT assays can help to mitigate potential error induced by these slight changes in signal magnitude between assays.
Inter-assay variably was also tested and replicates repeated on different days were highly correlated. Figure 7 plots the normalized RC score values from two independent experiments performed approximately one year apart. A high degree of correlation with the absence of major outliers (R2 = 0.873) was observed between the two independent replicates.
Although highly correlated, there is some variability in the overall magnitude of replication kinetics between the two independent experiments. This can be attributed in part to the difference in passage numbers between the GXR25 cells stocks used in each experiment. In general, GXR25 cells stocks that have been passaged for a period of time greater than 6 months tend to support more efficient replication of Gag-MJ4 chimeras. Therefore, it is advisable to assess replication capacity among groups of chimeras within a one-month time frame. When the previous steps are followed closely, this assay is capable of producing highly robust and reproducible results, which are applicable to a wide range of studies.
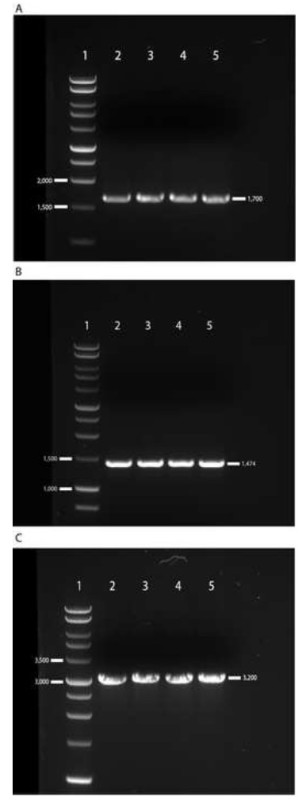 Figure 1. Representative gel images depicting electrophoretic separation of PCR products. For all PCR products, 5 µl of each 50 µl reaction was mixed with 3 µl of 5x loading dye, loaded into a 1% agarose-TAE gel supplemented with 1X SYBR-safe DNA gel stain, and separated by electrophoresis at 120 V for 45 min. The Promega 1 kb DNA ladder (Lane 1) was used to approximate amplicon sizes. A) The gag gene was amplified from viral RNA using a nested PCR approach. Due to insertions and deletions, the gag amplicon may vary from 1,600-1,700 bp in length and appears slightly above the 1,500 bp DNA ladder marker. Lanes 2-5 depict successful gag gene amplification. B) The 5’ LTR of MJ4 was amplified from the wild-type MJ4 plasmid and visualized via electrophoretic separation. Lanes 2-5 depict successful amplification of the 1,474 bp LTR product, which appears slightly below the 1,500 bp DNA ladder marker. C) The 5’ LTR derived from wild-type MJ4 and the gag gene amplified from patient plasma are fused together via splice-overlap-extension PCR and visualized via electrophoretic separation. Lanes 2-5 depict successfully fused amplicons that are approximately 3,200 bp in size, and which appear slightly above the 3,000 bp DNA ladder marker.
Figure 1. Representative gel images depicting electrophoretic separation of PCR products. For all PCR products, 5 µl of each 50 µl reaction was mixed with 3 µl of 5x loading dye, loaded into a 1% agarose-TAE gel supplemented with 1X SYBR-safe DNA gel stain, and separated by electrophoresis at 120 V for 45 min. The Promega 1 kb DNA ladder (Lane 1) was used to approximate amplicon sizes. A) The gag gene was amplified from viral RNA using a nested PCR approach. Due to insertions and deletions, the gag amplicon may vary from 1,600-1,700 bp in length and appears slightly above the 1,500 bp DNA ladder marker. Lanes 2-5 depict successful gag gene amplification. B) The 5’ LTR of MJ4 was amplified from the wild-type MJ4 plasmid and visualized via electrophoretic separation. Lanes 2-5 depict successful amplification of the 1,474 bp LTR product, which appears slightly below the 1,500 bp DNA ladder marker. C) The 5’ LTR derived from wild-type MJ4 and the gag gene amplified from patient plasma are fused together via splice-overlap-extension PCR and visualized via electrophoretic separation. Lanes 2-5 depict successfully fused amplicons that are approximately 3,200 bp in size, and which appear slightly above the 3,000 bp DNA ladder marker.
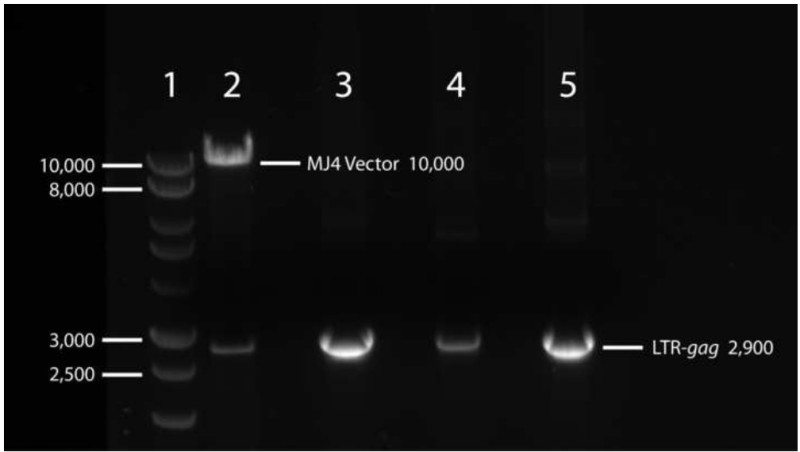 Figure 2. Representative gel image depicting electrophoretic separation of restriction digests for cloning patient gag genes into MJ4. Wild-type MJ4 plasmid and LTR-gag fusion products were digested with BclI for 1.5 hr at 50 °C and NgoMIV for 1 hr at 37 °C. Vector and insert fragments were visualized via electrophoretic separation on a 1% agarose-TAE gel supplemented with 1X SYBR-safe DNA gel stain at 100 V for 2 hr and using a blue light illuminator in order to reduce UV-induced DNA damage. The vector and insert fragments suitable for subsequent cloning steps appear at approximately 10,000 bp and 2,900 bp respectively.
Figure 2. Representative gel image depicting electrophoretic separation of restriction digests for cloning patient gag genes into MJ4. Wild-type MJ4 plasmid and LTR-gag fusion products were digested with BclI for 1.5 hr at 50 °C and NgoMIV for 1 hr at 37 °C. Vector and insert fragments were visualized via electrophoretic separation on a 1% agarose-TAE gel supplemented with 1X SYBR-safe DNA gel stain at 100 V for 2 hr and using a blue light illuminator in order to reduce UV-induced DNA damage. The vector and insert fragments suitable for subsequent cloning steps appear at approximately 10,000 bp and 2,900 bp respectively.
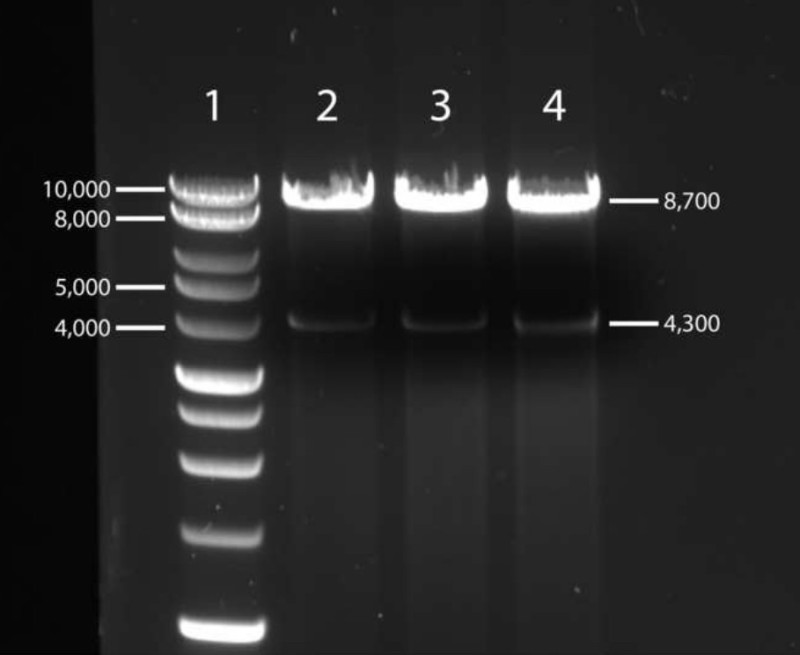 Figure 3. Representative gel image depicting electrophoretic separation of restriction digests of purified Gag-MJ4 chimera plasmid DNA. Purified Gag-MJ4 chimera plasmid DNA was double-digested with NgoMIV and HpaI restriction enzymes for 2 hr at 37 °C. Restriction digests were visualized via electrophoretic separation on a 1% agarose-TAE gel supplemented with 1X SYBR-safe DNA gel stain at 120 V for 45 min. Plasmids without large deletions will resolve to two distinct bands at approximately 8,700 and 4,300 bp.
Figure 3. Representative gel image depicting electrophoretic separation of restriction digests of purified Gag-MJ4 chimera plasmid DNA. Purified Gag-MJ4 chimera plasmid DNA was double-digested with NgoMIV and HpaI restriction enzymes for 2 hr at 37 °C. Restriction digests were visualized via electrophoretic separation on a 1% agarose-TAE gel supplemented with 1X SYBR-safe DNA gel stain at 120 V for 45 min. Plasmids without large deletions will resolve to two distinct bands at approximately 8,700 and 4,300 bp.
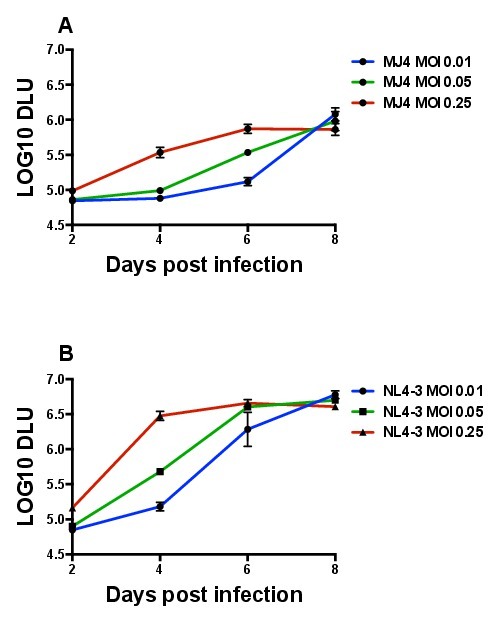 Figure 4. Replication of MJ4 and NL4-3 isolates of HIV-1 in the GXR25 cell line at different multiplicities of infection (MOI). 5 x 105 GXR25 cells were infected as described in the method protocol with 5-fold increasing MOI of each virus stock. Supernatants were collected on days 2, 4, 6, and 8 post infection and virion production was quantified via a radiolabeled reverse transcriptase assay. Infections were run in triplicate and error bars denote the standard deviation for the three replicates. (A) MJ4 (B) NL4-3.
Figure 4. Replication of MJ4 and NL4-3 isolates of HIV-1 in the GXR25 cell line at different multiplicities of infection (MOI). 5 x 105 GXR25 cells were infected as described in the method protocol with 5-fold increasing MOI of each virus stock. Supernatants were collected on days 2, 4, 6, and 8 post infection and virion production was quantified via a radiolabeled reverse transcriptase assay. Infections were run in triplicate and error bars denote the standard deviation for the three replicates. (A) MJ4 (B) NL4-3.
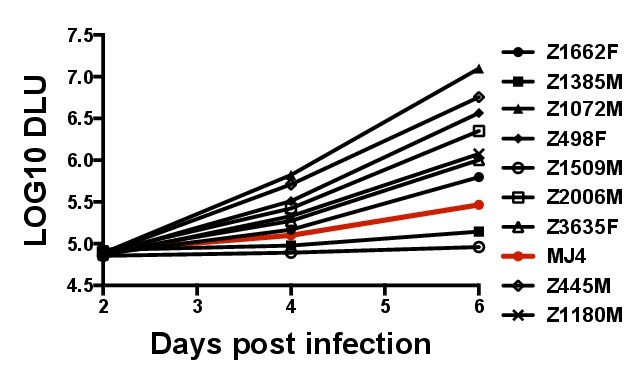 Figure 5. Representative range of replication for different Gag-MJ4 chimeras. As described in the method protocol, 5 x 105 GXR25 cells were infected with wild-type MJ4 or Gag-MJ4 chimeras at an MOI of 0.05, supernatants were collected at two day intervals post infection, and virion production quantified by a radiolabeled RT assay. Insertion of various subtype C derived gag genes can have a dramatic impact on the replication capacity of MJ4. Wild-type MJ4 replication is denoted in red.
Figure 5. Representative range of replication for different Gag-MJ4 chimeras. As described in the method protocol, 5 x 105 GXR25 cells were infected with wild-type MJ4 or Gag-MJ4 chimeras at an MOI of 0.05, supernatants were collected at two day intervals post infection, and virion production quantified by a radiolabeled RT assay. Insertion of various subtype C derived gag genes can have a dramatic impact on the replication capacity of MJ4. Wild-type MJ4 replication is denoted in red.
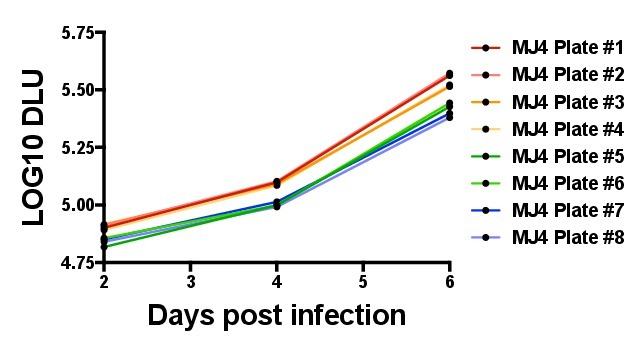 Figure 6. Comparison of intra-assay variation in the radiolabeled reverse transcriptase (RT) quantification assay. The graph depicts inherent intra-assay variability by plotting the DLU values of the same supernatants from a single wild-type MJ4 infection in eight different RT assay plates. The variation in curves reflects the slight changes in signal magnitude between plates, which can be corrected for by running a standard on each RT plate, which can be subsequently used to normalize the slopes of Gag-MJ4 chimeras assayed on the same plate.
Figure 6. Comparison of intra-assay variation in the radiolabeled reverse transcriptase (RT) quantification assay. The graph depicts inherent intra-assay variability by plotting the DLU values of the same supernatants from a single wild-type MJ4 infection in eight different RT assay plates. The variation in curves reflects the slight changes in signal magnitude between plates, which can be corrected for by running a standard on each RT plate, which can be subsequently used to normalize the slopes of Gag-MJ4 chimeras assayed on the same plate.
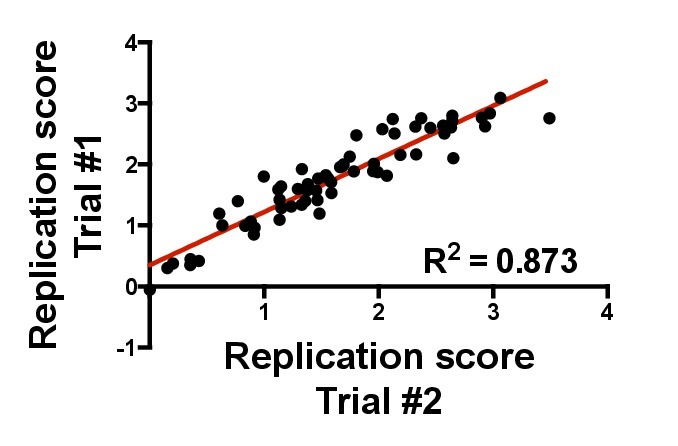 Figure 7. Reproducibility of the replication assay over time in the GXR25 cell line. The same Gag-MJ4 chimeric viruses were used to infect GXR25 cells in two independent experiments performed approximately one year apart. Replication scores were generated by calculating the slope of log-transformed DLU values and normalizing that slope to wild-type MJ4. Gag-MJ4 chimeras that replicate more efficiently than wild-type MJ4 have replication scores greater than 1, and those that replicate less efficiently than wild-type MJ4 have replication scores less than 1. The two independent measurements are strongly correlated (R2 = 0.87, linear regression) and highlight the reproducibility of assays performed at different times and with cells at different passages.
Figure 7. Reproducibility of the replication assay over time in the GXR25 cell line. The same Gag-MJ4 chimeric viruses were used to infect GXR25 cells in two independent experiments performed approximately one year apart. Replication scores were generated by calculating the slope of log-transformed DLU values and normalizing that slope to wild-type MJ4. Gag-MJ4 chimeras that replicate more efficiently than wild-type MJ4 have replication scores greater than 1, and those that replicate less efficiently than wild-type MJ4 have replication scores less than 1. The two independent measurements are strongly correlated (R2 = 0.87, linear regression) and highlight the reproducibility of assays performed at different times and with cells at different passages.
A)
| Reagent | Volume for 1x reaction (μl) |
| 2x Reaction Mix (Invitrogen) | 25 |
| Nuclease-free H2O | 17 |
| Forward primer GOF (20 μM concentration) | 1 |
| Reverse primer VifOR (20 μM concentration) | 1 |
| SuperScript III One-step Enzyme Mix | 1 |
| RNA template | 5 |
| Total volume | 50 |
B)
| Number of Cycles | Time (hr:min:sec) | Temperature (°C) |
| 1 | 1:00:00 | 50 |
| 1 | 2:00 | 94 |
| 10 | 0:15 | 94 |
| 0:30 | 56 | |
| 5:00 | 68 | |
| 40 | 0:15 | 94 |
| 0:30 | 56 | |
| 5:00 + 5 sec/cycle | 68 | |
| 1 | 12:00 | 68 |
| 1 | ∞ | 4 |
| END |
Table 1. A) Master mix and B) thermocycler conditions for first-round gag amplification. *Note: GOF primer sequence: 5'-ATTTGACTAGCGGAGGCTAGAA-3'. VifOR primer sequence: 5'-TTCTACGGAGACTCCATGACCC-3'.
A)
| Reagent | Volume for 1x reaction (μl) |
| Nuclease-free H2O | 35.5 |
| 5x Phusion HF Buffer | 10 |
| dNTPs (40 mM deoxynucleotides) | 1 |
| Forward primer GagInnerF1 (20 μM concentration) | 1 |
| Reverse primer BclIDegRev2 (20 μM concentration) | 1 |
| Phusion Hot Start II Polymerase | 0.5 |
| First-round PCR as template | 1 |
| Total volume | 50 |
B)
| Number of Cycles | Time (hr:min:sec) | Temperature (°C) |
| 1 | 0:30 | 98 |
| 29 | 0:10 | 98 |
| 0:30 | 53 | |
| 1:00 | 72 | |
| 1 | 10:00 | 72 |
| 1 | ∞ | 4 |
| END |
Table 2.A) Master mix and B) thermocycler conditions for nested second-round gag amplification. *Note: GagInnerF1 primer sequence: 5'-AGGCTAGAAGGAGAGAGATG-3'. BclIDegRev2 primer sequence: 5'-AGTATTTGATCATAYTGYYTYACTTTR-3'.
A)
| Reagent | Volume for 1x reaction (μl) |
| Nuclease-free H2O | 35.5 |
| 5x Phusion HF Buffer | 10 |
| dNTPs (40 mM deoxynucleotides) | 1 |
| Forward primer MJ4For1b (20 μM concentration) | 1 |
| Reverse primer MJ4Rev (20 μM concentration) | 1 |
| Phusion Hot Start II Polymerase | 0.5 |
| MJ4 plasmid as template (10 ng/ul) | 1 |
| Total volume | 50 |
B)
| Number of Cycles | Time (hr:min:sec) | Temperature (°C) |
| 1 | 0:30 | 98 |
| 29 | 0:10 | 98 |
| 0:30 | 58 | |
| 0:45 | 72 | |
| 1 | 10:00 | 72 |
| 1 | ∞ | 4 |
| END |
Table 3. A) Master mix and B) thermocycler conditions for 5’ MJ4 LTR amplification. *Note: MJ4For1b primer sequence: 5'-CGAAATCGGCAAAATCCC-3'. MJ4Rev primer sequence: 5'-CCCATCTCTCTCCTTCTAGC-3'.
A)
| Reagent | Volume for 1x reaction (μl) |
| Nuclease-free H2O | 34.5 |
| 5x Phusion HF Buffer | 10 |
| dNTPs (40 mM deoxynucleotides) | 1 |
| Forward primer MJ4For1b (20 μM concentration) | 1 |
| Reverse primer BclIRev (20 μM concentration) | 1 |
| MJ4 LTR 1.3 kb amplicon (Gel purified, ~50 ng) | 1 |
| Phusion Hot Start II Polymerase | 0.5 |
| Gag amplicon (Gel purified, ~100 ng) | 1 |
| Total volume | 50 |
B)
| Number of Cycles | Time (hr:min:sec) | Temperature (°C) |
| 1 | 0:30 | 98 |
| 29 | 0:10 | 98 |
| 0:30 | 58 | |
| 1:30 | 72 | |
| 1 | 10:00 | 72 |
| 1 | ∞ | 4 |
| END |
Table 4. A) Master mix and B) thermocycler conditions for splice-overlap-extension PCR to generate LTR-gag inserts. *Note: BclIRev primer sequence: 5'-TCTATAAGTATTTGATCATACTGTCTT-3'
A)
| Reagent | Volume for 1x reaction (μl) | Incubation Time (hr) | Incubation Temperature (°C) |
| 1.5 μg of 3 kb LTR-gag amplicon or MJ4 plasmid | x μl for 1.5 μg | ||
| NEB CutSmart Buffer (previously NEB Buffer #4) | 2 | ||
| BclI restriction enzyme | 1 | ||
| Nuclease-free H2O | x | ||
| Total volume | 19 | 1.5 | 50 |
| NgoMIV restriction enzyme | 1 | ||
| Total volume | 20 | 1 | 37 |
B)
| Reagent | Volume for 1x reaction (μl) | Incubation Time (hr) | Incubation Temperature (°C) |
| 50 ng cut MJ4 plasmid vector | x μl for 50 ng | ||
| 45 ng cut LTR-gag insert (3:1 ratio) | x μl for 45 ng | ||
| Roche 10x ligase buffer | 2 | ||
| Roche T4 DNA ligase (5 U/μl) | 1 | ||
| Nuclease-free H2O | x | ||
| Total volume | 20 | 18+ (overnight) | 4 |
C)
| Reagent | Volume for 1x reaction (μl) | Incubation Time (hr) | Incubation Temperature (°C) |
| 450 ng of Gag-MJ4 plasmid | x μl for 450 ng | ||
| NEB CutSmart Buffer (previously NEB Buffer #4) | 2 | ||
| NgoMIV restriction enzyme | 0.5 | ||
| HpaI restriction enzyme | 0.5 | ||
| Nuclease-free H2O | x | ||
| Total volume | 20 | 2 | 37 |
Table 5. A) Restriction digest master mix and B) ligation reaction for generation of Gag-MJ4 chimeric provirus. C) Diagnostic restriction digest to ensure Gag-MJ4 cloning fidelity.
| Primer Name | Nucleotide Sequence (5' - 3') |
| GagInnerF1 | AGGCTAGAAGGAGAGAGATG |
| GagF2 | GGGACATCAAGCAGCCAT |
| For3 | CTAGGAAAAAGGGCTGTTGGAAATG |
| GagR6 | CTGTATCATCTGCTCCTG |
| Rev3 | GACAGGGCTATACATTCTTACTAT |
| Rev1 | AATTTTTCCAGCTCCCTGCTTGCCCA |
Table 6. List of sequencing primers necessary to confirm 5’ LTR and gag sequence identity of Gag-MJ4 chimeric provirus.
| Well A | 8 µl virus + 232 µl 1% FBS in DMEM |
| Well B | 80 µl from Well A + 160 µl 1% FBS in DMEM |
| Well C | 80 µl from Well B + 160 µl 1% FBS in DMEM |
| Well D | 80 µl from Well C + 160 µl 1% FBS in DMEM |
| Well E | 80 µl from Well D + 160 µl 1% FBS in DMEM |
| Well F | 80 µl from Well E + 160 µl 1% FBS in DMEM |
Table 7. Dilution scheme (3-fold) for titering infectious viruses on TZM-bl cells.
A)
| Reagent | Volume |
| PBS without Ca2+ or Mg2+ | 500 ml |
| Gluteraldehyde | 4 ml |
| Formaldehyde | 11 ml |
*Note: Store at 4 °C.
B)
| Reagent | Volume |
| PBS without Ca2+ or Mg2+ | 4.75 ml |
| Potassium ferricyanide (0.2 M) | 100 μl |
| Potassium ferrocyanide (0.2 M) | 100 μl |
| Magnesium chloride (1 M) | 20 μl |
| X-gal (50 mg/ml) | 40 μl |
*Note: Make fresh and store away from light until use.
C.
| Original Dilution well | A | B | C | D | E | F |
| Volume of virus (µl) added to the wells of a 24-well plate row | 5 | 1.6667 | 0.5556 | 0.1852 | 0.06173 | 0.02057 |
Table 8. A) Staining and B) fixing solutions for titering of Gag-MJ4 chimeric viruses on the TZM-bl indicator cell line. C) The volume of virus added per well for calculating infectious units/µl.
| Reagent | Volume |
| Fetal bovine serum (FBS), defined | 55 ml |
| Penicillin, Streptomycin, Glutamine (100x) | 6 ml |
| HEPES buffer (1 M) | 6 ml |
Table 9. Recipe for complete RPMI medium for propagation of GXR25 cells.
| Reagent | Volume (ml) |
| Nuclease-free H2O | 419.5 |
| Tris-Cl, pH 7.8 (1 M) | 30 |
| Potassium chloride (1 M) | 37.5 |
| Magnesium chloride (1 M) | 2.5 |
| Nonidet P-40 (10%) | 5 |
| EDTA (0.5 M) | 1.02 |
| Polyadenylic acid, potassium salt (2 mg/ml) | 1.25 |
| Oligo-dT primer (25 μg/ml) | 3.25 |
| Total volume | 500 |
Table 10. Recipe for radiolabeled reverse-transcriptase assay master mix. *Note: Store as 1 ml aliquots at -20 °C.
Discussion
Due to the length and technical nature of this protocol, there are several steps that are critical for both the successful construction of chimeric Gag-MJ4 plasmids as well as for quantification of viral replication capacity. Although the restriction enzyme based cloning strategy for the introduction of foreign gag genes into MJ4 outlined in this protocol has numerous advantages over previously used recombination based methods, the protocol can be technically challenging if critical steps are not followed precisely.
First, it is absolutely essential to use MJ4 plasmid DNA that has been generated in a competent bacterial strain lacking the dcm and dam DNA methylases. This is necessary as the enzymatic activity of the BclI restriction endonuclease, which is used to clone gag genes into the MJ4 backbone, is dam/dcm methylation sensitive. The JM110 and SCS110 (an endA negative JM110 derivative) E. coli strains are suitable for generating unmethylated MJ4 plasmid DNA. Additionally, for the excision of vector and insert bands, it is highly recommended that a blue light illuminator be used for visualization. This will reduce UV wavelength-dependent DNA damage and drastically increase cloning efficiency. If a blue light illuminator is unavailable, cloning efficiency can be maximized by visualizing DNA with SYBR Safe DNA gel stain instead of ethidium bromide and minimizing UV exposure time.
Finally, molecular cloning with large (>10kb) and/or retroviral plasmids such as MJ4 has traditionally been difficult for a variety of reasons. Large plasmids reduce transformation efficiency of competent bacterial strains42 while retroviral inserts, which contain long terminal repeat (LTR) sequences, reduce stability of the plasmid and compromise replication fidelity of the plasmid DNA within the bacterial host leading to deletions of the retroviral genome43. Bacteria transformed with MJ4 or Gag-MJ4 plasmid products must be grown at 30 °C rather than the traditional 37 °C for most protocols. Recovery steps after heat shock transformation, growth of transformed bacteria on agar plates, and growth of bacterial colonies in liquid culture should all be performed at 30 °C. This lower temperature reduces the growth rate of the bacteria and thus helps to ensure replication fidelity of the MJ4 plasmid. Additionally, replication of the MJ4 plasmid is more stable when using the JM109 E. coli strain over the DH5α strain, in our hands. Due to unstable nature of the plasmid, purified plasmid products should always be checked for correct plasmid size by restriction enzyme digestion; here, a double digest with the NgoMIV and HpaI restriction endonucleases at 37 °C for 2 hr.
Once successful generation of chimeric Gag-MJ4 plasmids has been accomplished, virus is generated via transfection of 293T cells, titered on an indicator cell line, TZM-bl cells, and replication capacity is measured using a CEM-based T cell line. The CEM-based GXR25 cell line used for these replication studies is one of the few established T cell lines able to support entry and replication of CCR5-tropic strains of HIV-1. This has been achieved by retroviral transduction to allow stable expression of human CCR537. This cell line naturally expresses CXCR4 and will support replication of CXCR4-tropic HIV-1, such as the laboratory-adapted strain NL4-3. However, in order to support efficient replication of CCR5-tropic strains, such as MJ4, the GXR25 cells must be propagated for no less than 4 months prior to infection. Properly passaged cultures can support replication even after passaging for up to 1 year. Careful monitoring of CCR5-tropic replication throughout passaging is essential for successful experiments.
As with any technique, there are limitations to the protocol that must be considered. Due to the location of restriction sites in the MJ4 plasmid as well as the availability of conserved restriction sites in naturally occurring HIV-1 isolates, the 3’ distal restriction site, BclI, is located 137 nucleotides from the gag stop codon. Although this generates a chimeric protease gene, this region is 96.5% conserved in this cohort, and we did not observe an abundance of dead or defective chimeric viruses.
One of the advantages of using the MJ4 subtype C infectious molecular clone with subtype C derived sequences is that it reduces the risk of suboptimal gene pairing between gag genes and backbone vectors of different subtypes. However, a certain amount of within-clade diversity exists as evidenced by the clustering of HIV-1 sequences by country or region even when found within the same subtype31. This could contribute to suboptimal pairing between the gag genes derived from acutely infected Zambians and the MJ4 infectious molecular clone backbone, which was derived from a chronically infected individual from Botswana38. However, a majority of the analyzed constructs produced infectious progeny virus. As different HIV-1 subtype C infectious molecular clones become more widely available, it will be important to further validate this system by cloning in these HIV-1 clade C gag genes into other backbones in order to ensure that there is a minimal bias introduced due to backbone incompatibilities.
The GXR25 cell line is a unique cell line, specifically due to its ability to support both CXCR4 and CCR5-tropic strains and its HIV-1 inducible GFP reporter37. However, some limitations exist and should be carefully considered before using this cell line for experiments adapted from this protocol. The GXR25 cell line does not appear to support entry of a majority of subtype C or A, CCR5-tropic, primary isolates we have tested. Additionally, the parent CEM cell line from which the GXR25 cell line was derived exhibits high levels of cyclophilin A, up to 2 to 4-fold higher expression than the Jurkat cell line44. Due to high levels of cyclophilin A, the replication defect normally associated with the canonical HLA-B*57 associated escape mutation, T242N, which is attributed to a decreased ability of capsid to bind cyclophilin A, cannot be easily detected in this particular cell line25. Thus the CEM-based GXR25 cell line is not ideal for studying replication defects associated with mutations in the HIV-1 capsid cyclophilin-binding loop.
Finally, while this protocol is ideal for analyzing the replication capacity of gag sequences derived from acute time points, modifications must be made in order to study the replication capacity of gag genes derived from chronically infected individuals. This protocol involves the amplification of population sequences from acute time points (median 45 days post estimated date of infection) when viral diversity is limited. The gag gene from each chimera is then sequenced and compared to the initial population PCR amplicon to ensure cloning fidelity. Due to limited sequence diversity at acute time points, cloning from population PCR products is possible. However, sequence diversity exists within the viral quasispecies of a chronically infected individual; therefore, in order to accurately assess the replication capacity of the chronic quasispecies, single genome amplification must be employed to capture several representative variants. Each of these variants must then be assayed for in vitro replication.
This technique has several broader applications, which stem from its advantages over existing methods. Since this process results in a clonal replication competent plasmid, it is simple to use constructs for additional mutagenesis studies, which can help to elucidate the contributions of specific residues to viral replication. Furthermore, by cloning in gag genes from longitudinal time points, one can assess the evolution of viral replication capacity over time and how these changes may affect pathogenesis in an HIV-1 infected individual.
This technique can be modified in order to expand its utility for different applications. Additional HIV-1 viral proteins can be engineered into the MJ4 plasmid in order to assess their effects on viral replication or their interactions with host proteins. This can be accomplished by engineering additional restrictions sites at desired regions in the genome through the introduction of silent nucleotide changes. However, special consideration must be taken when engineering novel restriction sites into the 3’ half of the HIV-1 genome as many accessory proteins are encoded in alternate reading frames, and silent changes in one protein could lead to amino acid substitutions in another. In this instance, restriction site independent cloning methods such as those discussed by Dudley et al.45 may overcome this limitation. Viral sequences derived from different populations may have varying ranges of replication capacities, and the MOI can be adjusted accordingly in order to capture the majority of viral isolates within their logarithmic phase of growth. Finally, the GXR25 cell line has been stably transfected with GFP under an LTR-driven, Tat-inducible promoter, and viral spread can be measured as a function of GFP positive cells via flow cytometry37 as an alternative to the RT assay described here or a traditional p24 ELISA.
In conclusion, this protocol provides an efficient and powerful technique for assessing HIV-1 viral replication as conferred by the gag gene, which encodes a conserved structural protein necessary for proper virion formation, budding, maturation, and disassembly46-48. Furthermore, this technique generates a replication competent clonal plasmid, which is ideal for mutagenesis studies and, thus, provides a method for elucidating the specific amino acid determinants of viral fitness. Studies such as these are imperative to enhance the understanding of how immune-driven viral evolution affects pathogenesis and disease progression in HIV-1 infected individuals.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The investigators thank all the volunteers in Zambia who participated in this study and all the staff at the Zambia Emory HIV Research Project in Lusaka who made this study possible. The investigators would like to thank Jon Allen, Smita Chavan, and Mackenzie Hurlston for technical assistance and sample management. We would also like to thank Dr. Mark Brockman for his discussions and generous donation of the GXR25 cells.
This study was funded by R01 AI64060 and R37 AI51231 (EH) and the International AIDS Vaccine Initiative. This work was made possible in part by the generous support of the American people through the United States Agency for International Development (USAID). The contents are the responsibility of the study authors and do not necessarily reflect the views of USAID or the United States Government. This work also was supported, in part, by the Virology Core at the Emory Center for AIDS Research (Grant P30 AI050409). DC and JP were supported in part by Action Cycling Fellowships. This work was supported in part by the Yerkes National Primate Research Center base grant (2P51RR000165-51). This project was also funded in part by the National Center for Research Resources P51RR165 and is currently supported by the Office of Research Infrastructure Programs/OD P51OD11132.
References
- Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. Journal of virology. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koup RA, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. Journal of virology. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. The Journal of experimental medicine. 1999;189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JE, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Brumme ZL, et al. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. Journal of virology. 2008;82:9216–9227. doi: 10.1128/JVI.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AJ, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nature medicine. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- Phillips RE, et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature. 1991;354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- Price DA, et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulder PJ, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nature. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- Tang J, et al. Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1. Journal of virology. 2002;76:8276–8284. doi: 10.1128/JVI.76.16.8276-8284.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice HA, et al. HLA-B*57 versus HLA-B*81 in HIV-1 infection: slow and steady wins the race. Journal of virology. 2013;87:4043–4051. doi: 10.1128/JVI.03302-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migueles SA, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2709–2714. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A, et al. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. Journal of virology. 2010;84:9879–9888. doi: 10.1128/JVI.00320-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaslow RA, et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nature medicine. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- Altfeld M, et al. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS. 2003;17:2581–2591. doi: 10.1097/00002030-200312050-00005. [DOI] [PubMed] [Google Scholar]
- Kiepiela P, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nature medicine. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- Mothe B, et al. Definition of the viral targets of protective HIV-1-specific T cell responses. Journal of translational medicine. 2011;9:208. doi: 10.1186/1479-5876-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyerl FW, Barouch DH, Letvin NL. Structural constraints on viral escape from HIV- and SIV-specific cytotoxic T-lymphocytes. Viral immunology. 2004;17:144–151. doi: 10.1089/0882824041310658. [DOI] [PubMed] [Google Scholar]
- Rolland M, et al. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PloS one. 2008;3:e1424. doi: 10.1371/journal.pone.0001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, et al. Molecular and functional analysis of a conserved CTL epitope in HIV-1 p24 recognized from a long-term nonprogressor: constraints on immune escape associated with targeting a sequence essential for viral replication. J Immunol. 1999;162:3727–3734. [PubMed] [Google Scholar]
- Wang YE, et al. Protective HLA class I alleles that restrict acute-phase CD8+ T-cell responses are associated with viral escape mutations located in highly conserved regions of human immunodeficiency virus type 1. Journal of virology. 2009;83:1845–1855. doi: 10.1128/JVI.01061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopera DR, et al. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS pathogens. 2008;4:e1000033. doi: 10.1371/journal.ppat.1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford H, et al. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. The Journal of experimental medicine. 2009;206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutwell CL, et al. Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. Journal of virology. 2013;87:3952–3965. doi: 10.1128/JVI.03233-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. Journal of virology. 2007;81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford H, et al. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. Journal of virology. 2007;81:8346–8351. doi: 10.1128/JVI.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Picado J, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. Journal of virology. 2006;80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidewind A, et al. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. Journal of virology. 2007;81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JK, et al. Impact of HLA-B*81-associated mutations in HIV-1 Gag on viral replication capacity. Journal of virology. 2012;86:3193–3199. doi: 10.1128/JVI.06682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima Y, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goepfert PA, et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. The Journal of experimental medicine. 2008;205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, et al. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. Journal of virology. 2010;84:11937–11949. doi: 10.1128/JVI.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KH, et al. Progression to AIDS in South Africa is associated with both reverting and compensatory viral mutations. PloS one. 2011;6:e19018. doi: 10.1371/journal.pone.0019018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JK, et al. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. Journal of virology. 2010;84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JK, et al. Influence of Gag-protease-mediated replication capacity on disease progression in individuals recently infected with HIV-1 subtype. C. Journal of virology. 2011;85:3996–4006. doi: 10.1128/JVI.02520-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. Journal of virology. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, Tanzi GO, Walker BD, Allen TM. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. Journal of virology. 2006;131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Ndung'u T, Renjifo B, Essex M. Construction and analysis of an infectious human Immunodeficiency virus type 1 subtype C molecular clone. Journal of virology. 2001;75:4964–4972. doi: 10.1128/JVI.75.11.4964-4972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince JL, et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS pathogens. 2012;8:e1003041. doi: 10.1371/journal.ppat.1003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski MA, Chun TW, Cheseboro B, Stanley SK, Tremblay M, et al. Detection assays for HIV proteins. Current protocols in immunology / edited by John E. Coligan ... [et al.] 12(Unit 12 15) doi: 10.1002/0471142735.im1205s70. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Bichara M, Pinet I, Schumacher S, Fuchs RP. Mechanisms of dinucleotide repeat instability in Escherichia coli. Genetics. 2000;154:533–542. doi: 10.1093/genetics/154.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerson B, Rey O, Canon J, Krogstad P. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. Journal of virology. 1998;72:303–308. doi: 10.1128/jvi.72.1.303-308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DM, et al. A novel yeast-based recombination method to clone and propagate diverse HIV-1 isolates. BioTechniques. 2009;46:458–467. doi: 10.2144/000113119. [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. Journal of virology. 2004;78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. Journal of virology. 2002;76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]