

Abstract
Small-molecule inhibitors of urea transporter (UT) proteins in kidney have potential application as novel salt-sparing diuretics. The urea analog dimethylthiourea (DMTU) was recently found to inhibit the UT isoforms UT-A1 (expressed in kidney tubule epithelium) and UT-B (expressed in kidney vasa recta endothelium) with IC50 of 2-3 mM, and was shown to have diuretic action when administered to rats. Here, we measured UT-A1 and UT-B inhibition activity of 36 thiourea analogs, with the goal of identifying more potent and isoform-selective inhibitors, and establishing structure-activity relationships. The analog set systematically explored modifications of substituents on the thiourea including alkyl, heterocycles and phenyl rings, with different steric and electronic features. The analogs had a wide range of inhibition activities and selectivities. The most potent inhibitor, 3-nitrophenyl-thiourea, had an IC50 of ∼0.2 mM for inhibition of both UT-A1 and UT-B. Some analogs such as 4-nitrophenyl-thiourea were relatively UT-A1 selective (IC50 1.3 vs. 10 mM), and others such as thioisonicotinamide were UT-B selective (IC50 >15 vs. 2.8 mM).
Keywords: Urea transporter, kidney, diuretic, thiourea, structure-activity relationships
Introduction
Urea transporter (UT) proteins facilitate the passive transport of urea across cell plasma membranes [1-4]. UTs are expressed mainly in kidney, where they are required for the formation of a concentrated urine by countercurrent multiplication and exchange mechanisms [2,5,6]. UT-A isoforms encoded by the SLc14A2 gene are expressed in kidney tubule epithelial cells and UT-B encoded by the SLc14A1 gene is expressed in kidney vasa recta endothelial cells [7,8]. X-ray crystal structures have been solved for a bacterial UT homolog produced by Desulfovibrio vulgaris [9] and for bovine UT-B [10]. Studies in UT knockout mice [11-15] and in rodents administered UT inhibitors [16-18] support the conclusion that UT-A1, the UT isoform expressed at the luminal membrane of tubule epithelial cells in inner medullary collecting duct, is the primary target for diuretic development.
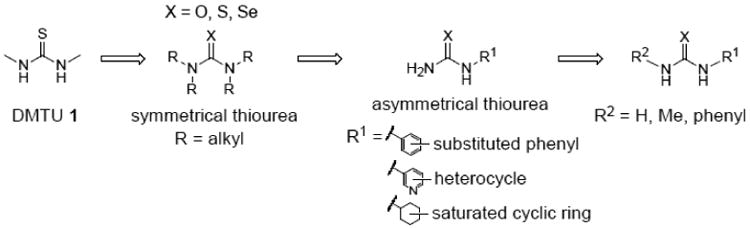
Small-molecule screening from our laboratory has identified inhibitors of UT-A1 and UT-B with a wide range of selectivities [16,19,20], which are being characterized and optimized as potential drug candidates. Interestingly, the small urea analog dimethylthiourea (DMTU), which has been administered at high doses in experimental animal models as a hydroxyl radical scavenger [21,22], inhibits both UT-A1 and UT-B (IC50 2-3 mM) and has been shown to reduce urinary osmolality in rats and produce a salt-sparing diuresis [23,24]. Motivated by the DMTU findings, the study here was done to establish structure-activity relationships of thiourea analogs for inhibition of UT-A1 and UT-B urea transport, with the goal of identifying more potent and UT-A1-selective analogs for use as research tools and as potential development candidates. Our strategy, as diagrammed in Fig. 1, was to initially screen symmetrical, close structural analogs of DMTU, which was followed by screening of mono-N-substituted, asymmetrical thioureas with greater structural diversity. Further refinement to improve potency was done by modification of the position and nature of substitutions. Compounds were identified with > 10-fold improved urea transport inhibition potency over DMTU, as well as compounds with high UT-A1 or UT-B selectivity.
Figure 1. Strategy for identification of thiourea analogs with improved potency.
Material and methods
Compounds
Unless otherwise specified, urea/thiourea analogs were purchased from Sigma-Aldrich (St. Louis, MO). Compounds 4, 26 and 27 were purchased from Acros Organics-Fisher Scientific (Pittsburgh, PA); 6 from TCI (Portland, OR); 5, 9, 18, 21 and 25 from Alfa Aesar (Ward Hill, MA); 10 and 28 from Santa Cruz Biotechnology (Dallas, TX) and 25 and 30 from ChemBridge (San Diego, CA). Compound 8 was synthesized as follows [25]: To a solution of N, N′-dimethylthiourea (200 mg, 1.92 mmol) in methanol (2 mL) was added iodomethane (0.24 mL, 3.84 mmol) dropwise and the resulting mixture was refluxed for 1 h and then cooled to 0 °C. The resulting white solid was dissolved in anhydrous methanol (5 mL) and reacted with freshly prepared NaSeH (300 mg, 2.88 mmol) in ethanol (2 mL) overnight. The reaction mixture was neutralized with diluted acetic acid and the solvent was removed under reduced pressure. The resulting mixture was diluted with water, extracted with dichloromethane and the solution dried over anhydrous sodium sulfate. The product was purified by crystallization from dichloromethane solution and afforded 8 as a colorless solid (98 mg, 34%). 1H NMR (chloroform-d) δ 6.60 (brs, 2H), 3.04 (brs, 6H); 13C NMR (chloroform-d) δ 180.2, 31.5. 1H and 13C NMR spectra were obtained in chloroform (CDCl3) using a 300 MHz Varian spectrometer referenced to TMS. Chemical shifts are expressed in units of Hertz (Hz). Splitting pattern is designated as brs (broad singlet).
Cell culture
Madin-Darby canine kidney (MDCK) cells stably expressing rat UT-A1, yellow fluorescent protein (YFP)-H148Q/V163S and human aquaporin-1 (AQP1), as described previously [19], were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum, penicillin G (100 U/mL), streptomycin (100 μg/mL), zeocin (500 μg/mL), geneticin (600 μg/mL) and hygromycin B (500 μg/mL) at 37 °C, 5% CO2.
UT-A1 inhibition assay
UT-A1 inhibition was measured as described [19]. The stably transfected MDCK cells were plated in 96-well plates at a density of 15,000 cells/well at 37 °C for 24 hours prior to assay. Briefly, MDCK- UT-A1-AQP1-YFP cells were incubated for 20 min with test compounds and then subjected to a 800 mM urea gradient during continuous measurement of the YFP fluorescence with a plate reader (model Infinite M1000, Tecan Trading AG, Switzerland). The addition of urea to the triply transfected cells produces a rapid cell shrinking (reduced fluorescence) followed by UT-A1-facilitated cell swelling (fluorescence increase). UT-A1 inhibition increases the initial fluorescence decrease and slows recovery. Percentage UT-A1 inhibition was computed as 100% (Fneg − Ftest)/(Fneg − Fpos), where F is fluorescence measured 7 s after urea addition of the negative control (Fneg), test compound (Ftest) and positive control (Fpos).
Functional studies
Reversibility of UT-A1 inhibition was tested by pre-incubation of MDCK cells with inhibitor at a concentration that inhibits urea transport by ∼50% and then washing the cells with PBS prior to assay. The kinetics of UT-A1 inhibition was measured by adding inhibitor (at the same concentration used for the reversibility studies) at different times prior to assay. The urea concentration-dependence of UT-A1 inhibition was studied from inhibitor concentration-response data using different of urea gradients from 80 to a 1,600 mM.
UT-B inhibition measurements
As described [20], whole rat blood was diluted to a hematocrit of ∼1.5% in PBS containing 1.25 M acetamide and 5 mM glucose. Red blood cell suspensions were added to microplates containing the test compounds and incubated for 15 min. Cells were then subjected to a rapid hypotonic gradient and their lysis was quantified by absorbance at 710 nm. Phloretin was used as a positive control and percentage erythrocyte lysis was computed as: % lysis = 100% (Aneg − Atest)/(Aneg − Apos), where A is absorbance for the negative control (Aneg), test compound (Atest) and positive control (Apos) at 710 nm. In some studies, urea permeability in rat erythrocytes was assayed by stopped-flow light scattering using a Hi-Tech Sf-51 instrument (Wiltshire, UK) as described [20]. Briefly, a 200-fold dilution of rat blood cells in PBS was incubated with test compound for 10 min, and then subjected to inwardly directed urea gradient (250-500 mM). After an initial osmotic shrinking phase, the kinetics of increasing cell volume caused by urea influx was measured as the time course of 90° scattered light intensity at 530 nm, with increasing cell volume resulting in reduced scattered light intensity.
Results
Fig. 2A shows concentration-inhibition data for reference compound DMTU, 1. UT-A1 inhibition was measured by a cell-based fluorescence assay in which cell volume, measured using an intracellular fluorescent indicator, is measured in response to extracellular DMTU addition (top, left). UT-B inhibition was measured using an erythrocyte lysis assay (20). IC50 values for inhibition of UT-A1 and UT-B urea transport were 2-3 mM with near complete inhibition seen at higher concentrations.
Figure 2. Urea transport inhibition by thiourea analogs.
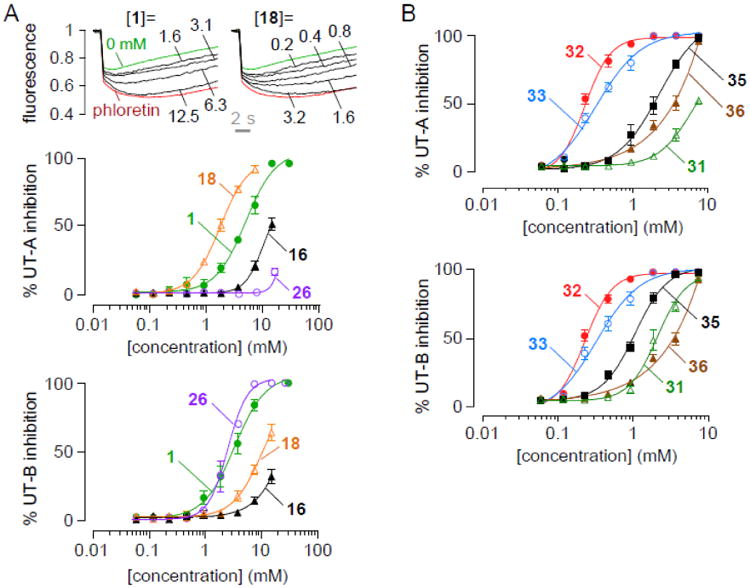
A. (top) UT-A1 concentration-inhibition curves for compounds 1 and 18. (bottom) Concentration-inhibition data for UT-A and UT-B of thiourea analogs 1, 16, 18 and 26. B. Concentration-inhibition data for UT-A and UT-B of thiourea analogs 31, 32, 33, 35 and 36.
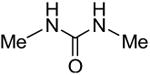
Symmetrical, bis-alkyl thiourea analogs with structural similarity to DMTU were tested for UT-A1 and UT-B inhibition activity (Table 1). Minor changes in the DMTU structure, such as replacing dimethyl (DMTU, 1) with hydrogen (2) or diethyl (4), resulted in significant loss of UT-A1 and UT-B inhibition activity. Tetramethylthiourea 5, which contains two additional methyl groups, and cyclic thioureas 6 and 7, were also inactive. Replacing the sulfur atom in DMTU with oxygen (dimethylurea 3) resulted in loss of activity, as did replacement with selenium (dimethylselenourea 8). The loss of UT inhibition activity in dimethylurea may be related to the significant differences between oxygen and sulfur in electronegativity (O = 3.5 Pauling unit, S = 2.5), carbon-oxygen/sulfur bond length (C=O = 1.2 Å, C=S = 1.6 Å) and van der Waals radius (O = 1.52 Å, S = 1.8 Å). The loss of inhibition activity of 8 was unexpected, as sulfur and selenium have similar electronegativity (S = 2.5, Se = 2.4), and van der Waals radius (S = 1.8 Å, Se = 1.9 Å); however, the significant difference in polarizability of sulfur and selenium (S = 2.9 Å, Se = 3.8 Å) and slightly elongated carbon-selenium bond length (C=S = 1.6 Å, C=Se = 1.86 Å) [27] could account for its loss of activity.
Table 1. Structure-activity analysis of symmetrical DMTU analogs.
| Inhibitor | Structure | UT-A1 IC50 (mM) | UT-B IC50 (mM) |
|---|---|---|---|
| 1 |
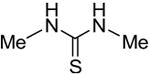
|
6.6 ± 0.4 | 3.9 ± 0.6 |
| 2 |
|
>25 | >25 |
| 3 |
|
22 ± 1 | 14 ± 2 |
| 4 |
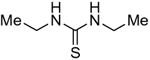
|
>25 | 17 ± 2 |
| 5 |
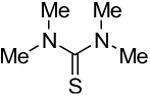
|
>25 | >25 |
| 6 |
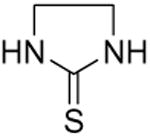
|
>15 | 13 ± 2 |
| 7 |
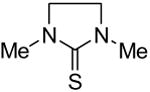
|
>25 | >25 |
| 8 |
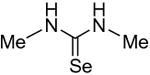
|
>25 | >25 |
As the symmetrical thiourea analogs examined above were not active, we next tested a series of asymmetrical mono-substituted thioureas. IC50 values are summarized in Table 2 and concentration-inhibition data for selected compounds is shown in Fig. 2A. This included phenylthiourea 9, heterocyclic thioureas 10-11, and unsaturated cyclohexylthiourea 12, which each inhibited UT-A1 and UT-B with potencies comparable to DMTU. Encouraged by these results, we further tested phenyl rings with different steric and electronic properties, including halide (13-15), electron-donating (16-18), electron-withdrawing (19-20) and sterically crowded di- or tri-substitutions (21-24). Analogs with electron-withdrawing groups, 4-nitrophenylthiourea 18 and 3,4-dichlorophenylthiourea 21, gave the best IC50 of 1.3 and 1.5 mM, respectively. Substitution at the 2-position on the phenyl ring resulted in loss of activity as seen in thiourea analogs 22-24. Analogs with electron-donating groups, such as methoxy and dimethylamino (19-20), were inactive. Also, an additional methylene group between the amino group and phenyl ring resulted in loss of activity (compare 14 and 25). Interestingly, thionicotinamide 25 and thioisonicotinamide 26, which lack the second amino group found in thiourea, were selective for UT-B over UT-A1.
Table 2.
Structure-activity analysis of non-symmetrical DMTU analogs.
| Inhibitor | Structure | R | UT-A1 IC50 (mM) | UT-B IC50 (mM) |
|---|---|---|---|---|
| 9 |
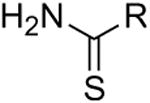
|
aniline | 8.8 ± 0.3 | 14 ± 1 |
| 10 | 2-amino-benzimidazole | 2.2 ± 0.3 | 4.7 ± 1.4 | |
| 11 | 4-amino-pyridine | 5.3 ± 0.6 | 1.6 ± 0.3 | |
| 12 | 4-amino-cyclohexyl | 3.7 ± 0.2 | 5.2 ± 0.6 | |
| 13 | 4-fluoro-aniline | 13 ± 3 | >15 | |
| 14 | 4-chloro-aniline | 6.3 ± 1.3 | >15 | |
| 15 | 4-bromo-aniline | 3.8 ± 0.6 | 2.4 ± 0.2 | |
| 16 | 4-trifluoromethyl-aniline | 15 ± 0.3 | >15 | |
| 17 | 4-carboxy-aniline | 7.5 ± 1.0 | 3.6 ± 0.6 | |
| 18 | 4-nitro-aniline | 1.3 ± 0.3 | 10 ± 1 | |
| 19 | 4-methoxy-aniline | >15 | >15 | |
| 20 | 4-dimethylamino-aniline | >15 | >15 | |
| 21 | 3,4-dichloro-aniline | 1.5 ± 0.4 | 0.4 ± 0.1 | |
| 22 | 2,3-dichloro-aniline | >15 | >15 | |
| 23 | 2,6-dichloro-aniline | >15 | >15 | |
| 24 | 2,4,6-trichloro-anilne | 5.9 ± 1.1 | >15 | |
| 25 | 4-chloro-benzylamine | >15 | >15 | |
| 26 | 4-pyridine | >15 | 2.8 ± 0.5 | |
| 27 | 3-pyridine | >25 | 10 ± 2 |
We next focused on the most potent inhibitors, 4-nitrophenylthiourea 18 and 3,4-dichlorophenyl thiourea 21. N-phenyl and N-methyl substitution on 18 and 21, which gave thioureas 28-30, resulted in similar or reduced potency (Table 3, data for selected compounds shown in Fig. 2B). Changing the position of the nitro-group on phenylthiourea from para- to meta- (31) decreased activity while changing to ortho- (3-nitrophenylthiourea 32) gave an IC50 of 0.2 mM for inhibition of both UT-A1 and UT-B, ∼6-fold better than 18. We sought to further improve the potency of 32 by N-substitution on the thiourea moiety, including methyl, phenyl, 3-nitrophenyl and semicarbazide groups (33-36). However, these substitutions reduced inhibition activity for UT-A1 and UT-B.
Table 3. Structure-activity analysis of nitrophenyl and chlorophenyl thiourea analogs.
| Inhibitor | Structure | UT-A1 IC50 (mM) | UT-B IC50 (mM) |
|---|---|---|---|
| 28 |
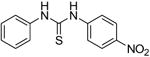
|
2.5 ± 0.5 | >15 |
| 29 |
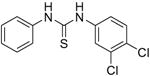
|
>15 | >15 |
| 30 |
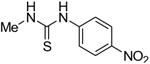
|
2.0 ± 0.6 | 2.3 ± 0.2 |
| 31 |
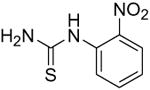
|
9.3 ± 0.7 | 4.8 ± 1.3 |
| 32 |
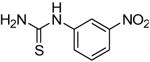
|
0.2 ± 0.1 | 0.2 ± 0.1 |
| 33 |
|
0.3 ± 0.2 | 0.3 ± 0.1 |
| 34 |
|
0.8 ± 0.2 | 4.4 ± 0.6 |
| 35 |
|
1.6 ± 0.9 | 1.1 ± 0.4 |
| 36 |
|
3.4 ± 0.7 | 3.4 ± 0.8 |
Selectivity of the thiourea analogs for UT-A1 vs. UT-B inhibition is summarized in Fig. 3A, which compares reciprocal IC50 values for inhibition of UT-A1 (abscissa) and UT-B (ordinate) urea transport. Several compounds were UT-A1 selective (red squares), including 14, 18, 24 and 28, while 26 and 31 were UT-B selective (green triangles). The most potent thioureas, 32 and 33, were non-selective.
Figure 3. Determinants of compound potency and selectivity.
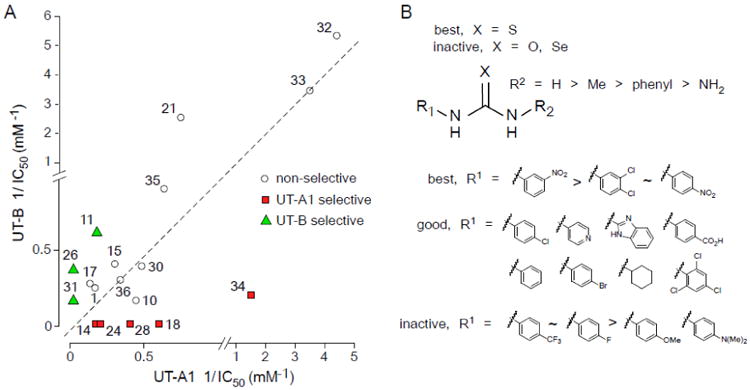
A. Summary of reciprocal IC50 values for inhibition of rat UT-A1 and rat UT-B for indicated compounds. B. Structural determinants of thioureas for UT-A1 inhibition activity.
Fig. 3B summarizes the structural determinants of compound potency and selectivity. Only thiourea analogs (X = S) were active, as urea (X = O) and selenourea (X = Se) were inactive. Substitution at R1 in thioureas with an aryl ring bearing electron-withdrawing substituents such as 3,4-dichloro 4-nitro were most active, with 3-nitro giving the best inhibition. In general, various cyclic rings at the R1 position, such as pyridine, benzimidazole, cyclohexyl and substituted phenyls, also gave good activity. Phenyl rings at R1 containing electron-donating substituent resulted in loss of activity. For R2, unsubstitution (R2 = H) gave the best inhibition, while increasing the size of the substituent decreased activity (R2 = methyl, phenyl)
The most potent compound, 32, was further characterized. Fig. 4A shows similar percentage of inhibition for UT-B and UT-A1, as measured by stopped-flow and plate reader assays, respectively. UT-A1 inhibition by 32 was fully reversible (Fig. 4B) and very rapid (Fig. 4C). The deduced IC50 using a wide range of urea gradients (80 to 1,600 mM) showed a small increase IC50 for both 32 and 33, suggesting a competitive inhibition mechanism (Fig. 4D). The water solubility of 32 and 33 was measured as 0.64 and 0.38 mM, respectively, substantially lower than that of DMTU (7.9 M). Compounds 32 and 33 showed no cytotoxicity to MDCK cells at their solubility limit.
Figure 4. Characterization of 32.
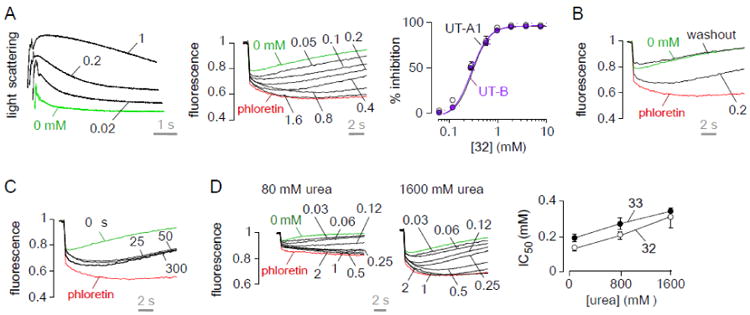
A. Inhibition of rat UT-B urea transport measured in erythrocytes by stopped-flow light scattering (left). UT-A1 concentration-inhibition curves (middle panel). Concentration-inhibition of rat UT-A1 and UT-B (right) (mean ± S.E., n = 3). B. Reversibility. Compounds were incubated (at 0.2 mM) with the triple transfected MDCK cells for 15 min, then washed twice and subjected to UT-A1 inhibition assay. C. Kinetics of UT-A1 inhibition. UT-A1 urea transport measured at different times after addition of 0.2 mM of 32. D. Concentration-inhibition data for 32 and 33 using 80 and 1600-mM urea gradients (left). IC50 as a function of urea gradient (right).
Discussion
This study was done to identify more potent and selective thiourea analogs than DMTU for inhibition of urea transporters. Minor symmetrical modifications of DMTU resulted in loss of activity. We found that some non-symmetrical substituted arylthiourea analogs gave better inhibition activity than DMTU. Inhibition activity was dependent on the stereo-electronic properties of the substituent, with electron-withdrawing substituent at the meta-position on the aryl ring giving greatest potency.
Two prior studies examined UT-B inhibition by small urea analogs. In an early report, Mayrand and Levitt [28] studied inhibition of erythrocyte urea transport by urea analogs using [14C] urea and a fast-flow system. UT-B inhibition was reported of various thiourea analogs at low millimolar concentrations, including phenylthioureas or thionicotinamides, in agreement with the data here. Zhao et al. [29] subsequently reported that various urea analogs permeate UT-B, albeit less well than urea itself, and that some analogs were weak inhibitors of urea transport.
There are several prior reports on the pharmacological properties of thioureas. DMTU is a hydroxyl radical scavenger that has been used in animals to reduce cellular injury associated with production of hydroxyl radicals. Pre-treatment of rats with DMTU (500 mg/kg) reduced hyperoxia-induced lung edema [30], and DMTU given orally 30 minutes prior to acute ethanol injury reduced stomach injury [31]. Pharmacological data for other thioureas have also been reported. The ability to taste the bitterness of phenylthiourea 9 (commonly known as phenylthiocarbamide) is a simple genetic trait governed by a pair of alleles, dominant T for tasting and recessive t for non-tasting [32]. Interestingly, 9 has been used to generate transparent zebrafish by blocking tyrosinase-dependent steps in the melanin pathway [33]. These reports suggest that thioureas are well-tolerated, with minimal cytotoxicity in animals. In general, thioureas are easily synthesized in 2-3 steps from commercially available starting materials for further structure-activity analysis.
One of the reasons for investigating the inhibition potency and selectivity of thiourea analogs was to improve on DMTU, which was shown to have diuretic efficacy in rats and hence potentially be used for proof-of-concept studies in clinically relevant models of edema. The diuretic action of DMTU requires intraperitoneal injection at a very high dose (500 mg/kg in saline) to reach stable levels above its IC50 (>3 mM) in plasma and urine [23,24]. In preliminary studies, we investigated the potential use of 32 and 33 in rodents. Due to their low water solubility compared to DMTU (soluble to >50 mg/mL in saline) it was necessary to use a different delivery vehicle (5% DMSO, 2.5% polyethylene glycol 400 and 2.4% Tween80 in saline; or 5% dimethylacetamide and 10% solutol or kolliphorHS15 in saline); however, it was not possible to obtain plasma or urine concentrations greater than 0.4 mM. Thus, though several thioureas identified here had up to 10-fold improved UT inhibition potency compared to DMTU, their low solubility precluded their use as UT inhibitors in animal models.
Highlights.
Thiourea analogs inhibit kidney UT-A and UT-B with a wide range of selectivies
UT inhibitors may be useful as novel, salt-sparing diuretics to treat edema
Thiourea analogs inhibit UT by a competitive mechanism
Acknowledgments
This study was supported by grants DK101373, DK35124, DK72517, EB00415 and EY13574 from the National Institutes of Health. The authors acknowledge OpenEye Scientific (Santa Fe, NM, USA) for its Academic Site License program.
Abbreviations
- UT
urea transporter
- DMTU
dimethylthiourea
- MDCK
Madin-Darby canine kidney
- AQP1
aquaporin-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sands JM. Renal urea transporters. Curr Opin Nephrol Hypertens. 2004;13:525–532. doi: 10.1097/00041552-200409000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Smith CP. Mammalian urea transporters. Exp Physiol. 2009;94:180–185. doi: 10.1113/expphysiol.2008.043042. [DOI] [PubMed] [Google Scholar]
- 3.Klein JD, Blount MA, Sands JM. Urea transport in the kidney. Compr Physiol. 2011;2:699–729. doi: 10.1002/cphy.c100030. [DOI] [PubMed] [Google Scholar]
- 4.Esteva-Font C, Anderson MO, Verkman AS. Urea transport proteins as drug targets. Nat Rev Nephrol. 2014 doi: 10.1038/nrneph.2014.219. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein JD, Blount MA, Sands JM. Molecular mechanisms of urea transport in health and disease. Pflugers Arch. 2012;464:561–572. doi: 10.1007/s00424-012-1157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenton RA. Essential role of vasopressin-regulated urea transport processes in the mammalian kidney. Pflugers Arch. 2009;458:169–177. doi: 10.1007/s00424-008-0612-4. [DOI] [PubMed] [Google Scholar]
- 7.Doran JJ, Klein JD, Kim YH, Smith TD, Kozlowski SD, Gunn RB, Sands JM. Tissue distribution of UT-A and UT-B mRNA and protein in rat. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1446–R1459. doi: 10.1152/ajpregu.00352.2004. [DOI] [PubMed] [Google Scholar]
- 8.Shayakul C, Clemencon B, Hediger MA. The urea transporter family (SLC14): physiological, pathological and structural aspects. Mol Aspects Med. 2013;34:313–322. doi: 10.1016/j.mam.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Levin EJ, Quick M, Zhou M. Crystal structure of a bacterial homologue of the kidney urea transporter. Nature. 2009;462:757–761. doi: 10.1038/nature08558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levin EJ, Cao Y, Enkavi G, Quick M, Pan Y, Tajkhorshid E, Zhou M. Structure and permeation mechanism of a mammalian urea transporter. Proc Natl Acad Sci U S A. 2012;109:11194–11199. doi: 10.1073/pnas.1207362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci U S A. 2004;101:7469–7474. doi: 10.1073/pnas.0401704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenton RA, Flynn A, Shodeinde A, Smith CP, Schnermann J, Knepper MA. Renal phenotype of UT-A urea transporter knockout mice. J Am Soc Nephrol. 2005;16:1583–1592. doi: 10.1681/ASN.2005010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchida S, Sohara E, Rai T, Ikawa M, Okabe M, Sasaki S. Impaired urea accumulation in the inner medulla of mice lacking the urea transporter UT-A2. Mol Cell Biol. 2005;25:7357–7363. doi: 10.1128/MCB.25.16.7357-7363.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein JD, Frohlich O, Mistry AC, Kent KJ, Martin CF, Sands JM. Transgenic mice expressing UT-A1, but lacking UT-A3, have intact urine concentration ability. FASEB J. 2013;27:1111.1117. abstract. [Google Scholar]
- 15.Yang B, Bankir L, Gillespie A, Epstein CJ, Verkman AS. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J Biol Chem. 2002;277:10633–10637. doi: 10.1074/jbc.M200207200. [DOI] [PubMed] [Google Scholar]
- 16.Esteva-Font C, Cil O, Phuan PW, Su T, Lee S, Anderson MO, Verkman AS. Diuresis and reduced urinary osmolality in rats produced by small-molecule UT-A-selective urea transport inhibitors. FASEB J. 2014 doi: 10.1096/fj.14-253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao C, Anderson MO, Zhang J, Yang B, Phuan PW, Verkman AS. Triazolothienopyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J Am Soc Nephrol. 2012;23:1210–1220. doi: 10.1681/ASN.2011070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li F, Lei T, Zhu J, Wang W, Sun Y, Chen J, Dong Z, Zhou H, Yang B. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013;83:1076–1086. doi: 10.1038/ki.2013.62. [DOI] [PubMed] [Google Scholar]
- 19.Esteva-Font C, Phuan PW, Anderson MO, Verkman AS. A small molecule screen identifies selective inhibitors of urea transporter UT-A. Chem Biol. 2013;20:1235–1244. doi: 10.1016/j.chembiol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin MH, de la Fuente R, Verkman AS. Urearetics: a small molecule screen yields nanomolar potency inhibitors of urea transporter UT-B. FASEB J. 2007;21:551–563. doi: 10.1096/fj.06-6979com. [DOI] [PubMed] [Google Scholar]
- 21.Cil O, Ertunc M, Gucer KS, Ozaltin F, Iskit AB, Onur R. Endothelial dysfunction and increased responses to renal nerve stimulation in rat kidneys during rhabdomyolysis-induced acute renal failure: role of hydroxyl radical. Ren Fail. 2012;34:211–220. doi: 10.3109/0886022X.2011.643389. [DOI] [PubMed] [Google Scholar]
- 22.Cil O, Ertunc M, Onur R. The diuretic effect of urea analog dimethylthiourea in female Wistar rats. Hum Exp Toxicol. 2012;31:1050–1055. doi: 10.1177/0960327112459205. [DOI] [PubMed] [Google Scholar]
- 23.Cil O, Esteva-Font C, Tas ST, Su T, Lee S, Anderson MO, Ertunc M, Verkman AS. Salt-sparing diuretic action of a water-soluble urea analog inhibitor of urea transporters UT-A and UT-B in rats. Kidney Internatl. 2014 doi: 10.1038/ki.2015.138. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esteva-Font C, Cil O, Phuan PW, Su T, Lee S, Anderson MO, Verkman AS. Nanomolar-potency, UT-A-selective inhibitors of kidney tubule urea transporters produce salt-sparing diuresis in rats. 2014 ASN abstract in press. [Google Scholar]
- 25.Arca M, Demartin F, Devillanova FA, Isaia F, Lelj F, Lippolis V, Verani G. An experimental and theoretical approach to the study of the properties of parabanic acid and related compounds: synthesis and crystal structure of diethylimidazolidine-2-selone-4,5-dione. Can J Chem. 2000;78:1147–1157. [Google Scholar]
- 26.Liu Y, Esteva-Font C, Yao C, Phuan PW, Verkman AS, Anderson MO. 1,1-Difluoroethyl-substituted triazolothienopyrimidines as inhibitors of a human urea transport protein (UT-B): new analogs and binding model. Bioorgan Med Chem Lett. 2013;23:3338–3341. doi: 10.1016/j.bmcl.2013.03.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
- 28.Mayrand RR, Levitt DG. Urea and ethylene glycol-facilitated transport systems in the human red cell membrane. Saturation, competition, and asymmetry. J Gen Physiol. 1983;81:221–237. doi: 10.1085/jgp.81.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao D, Sonawane ND, Levin MH, Yang B. Comparative transport efficiencies of urea analogues through urea transporter UT-B. Biochim Biophys Acta. 2007;1768:1815–1821. doi: 10.1016/j.bbamem.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Fox RB. Prevention of granulocyte-mediated oxidant lung injury in rats by a hydroxyl radical scavenger, dimethylthiourea. J Clin Invest. 1984;74:1456–1464. doi: 10.1172/JCI111558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith GS, Barreto JC, Schmidt KL, Tornwall MS, Miller TA. Protective effect of dimethylthiourea against mucosal injury in rat stomach. Implications for hydroxyl radical mechanism. Dig Dis Sci. 1992;37:1345–1355. doi: 10.1007/BF01296002. [DOI] [PubMed] [Google Scholar]
- 32.Wooding S. Phenylthiocarbamide: a 75-year adventure in genetics and natural selection. Genetics. 2006;172:2015–2023. doi: 10.1093/genetics/172.4.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karlsson J, Von Hofsten J, Olsson PE. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Mar Biotechnol (NY) 2001;3:522–527. doi: 10.1007/s1012601-0053-4. [DOI] [PubMed] [Google Scholar]