

Abstract
Obesity and the associated state of subchronic inflammation are risk factors for numerous pathologies, including carcinogenesis. Recently, Schulz et al. (2014) demonstrated that high-fat diet-induced intestinal dysbiosis promotes cancer development in K-rasG12Dint mice without inducing obesity or mucosal inflammation, positioning microbial activities as a central component of diet-induced carcinogenesis.
The intestinal tract is a dynamic milieu where 100 trillion bacteria interact daily with lumenal contents, intestinal epithelial cells, and underlying immune cells. The interplay between these factors represents a key balance point between homeostasis and disease, as aberration in any of their activities can lead to inflammation and even cancer (Irrazábal et al., 2014). With its impact on host and microbes, nutrition represents an important factor able to influence the homeostatic circuitry present in the gut. For example, diet-induced obesity has been associated with a 30%–70% increased risk of colon cancer in men (Bardou et al., 2013) and is linked to an altered intestinal microbiota (Ley et al., 2006). However, the contribution of obesity-associated low-grade inflammation in diet-induced carcinogenesis and the role of microbes in this pathological cascade are unclear.
A recent publication by Schulz et al. contributes an important piece to this puzzle by demonstrating that the dysbiosis caused by consumption of a lard-based high-fat diet (HFD) enhances adenocarcinoma development and metastasis in K-rasG12Dint mice, without promoting overt inflammation or causing obesity (Schulz et al., 2014). This observation suggests that HFD-induced cancer is not preceded by obesity or inflammation, but rather depends on microbial activity. The cancer-promoting microbiota seems to depend on both the HFD and host genetics. Indeed, the authors showed that K-ras oncogene activation results in decreased Paneth cell function (cryptdin expression), while HFD attenuates mucin expression (Muc2), two components critical in controlling intestinal microbial load and geographical distribution. Therefore, these changes likely act in concert to promote the progression from low-level dysplasia in regular chow-fed mice to small bowel carcinogenesis and metastasis in mice consuming HFD.
The provocative observation that K-rasG12Dint mice fed HFD developed cancer while remaining significantly leaner, without evidence of metabolic syndrome, challenges the dogmatic sequence of diet inducing obesity and the subsequent inflammation leading to cancer. Indeed, the authors were able to transfer the carcinogenic phenotype by fecal transplants, demonstrating the primary role of bacteria in cancer development in these mice. However, this phenotype is only seen in fecal-transplanted K-rasG12Dint mice, not in transplanted LSL-K-rasG12D/+ littermate controls, supporting the notion that cancer arises from an interaction between genes and the environment (bacteria). Importantly, while inducing carcinogenesis, the microbiota transfer did not promote obesity or insulin desensitization (Schulz et al., 2014). Therefore, changes in microbial factors drive cancer development in these mice rather than dietary differences, body mass index, or low-grade intestinal inflammation. As few models are able to dissociate these consequences of HFD, this study provides a revelation regarding the interplay between environment factors such as nutrition and the path to carcinogenesis. While there is likely still a role for direct dietary effects on other aspects of cancer, it is clear that microbiota play a critical, central role in absorbing the impact of various dietary insults and passing the consequences on to the host (Figure 1).
Figure 1. High-Fat Diet-Induced Dysbiosis Promotes Cancer Development in K-rasG12Dint Mice.
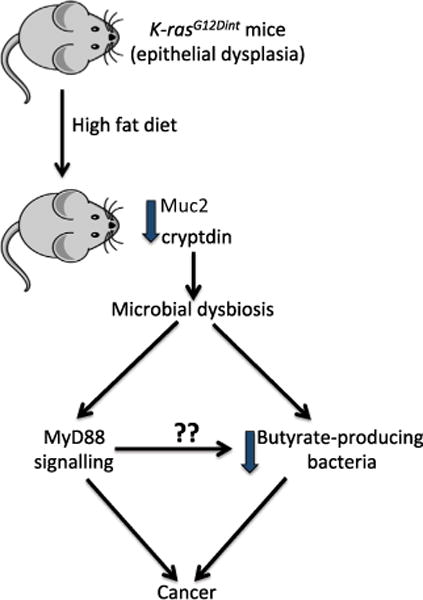
Decreased cryptdin expression caused by K-ras oncogene activation, coupled with decreased Muc2 expression from high-fat diet consumption, leads to a distinct shift in the microbiota. This dysbiosis is characterized by decreased butyrate production and leads to increased cancer occurrence. MyD88 signaling is implicated in the disease progression, perhaps by both promoting a proliferative signal and influencing the population of butyrate-producing bacteria, as this short-chain fatty acid prevents proliferation of tumor-initiated epithelial cells.
Interestingly, short-chain fatty acid production by the microbiota was impaired in HFD-fed K-rasG12Dint mice, and butyrate supplementation prevented carcinogenesis in this model by normalizing recruitment of dendritic cells to the gut (Schulz et al., 2014). Butyrate administration was also found to be protective in the Apcmin/+ mouse model of intestinal cancer (Singh et al., 2014), but detrimental in Apcmin/+Msh2−/−mice (Belcheva et al., 2014). In the latter model, a low-carbohydrate diet was associated with decreased abundance of butyrate-producing bacteria and reduced intestinal colonic polyps. This highlights the complex interplay between nutrition-derived microbial metabolites such as butyrate and host genetic status.
Additionally, the authors implicated host innate recognition of bacteria in cancer development in K-rasG12Dint mice, since ablation of Myd88 completely blocked tumor formation. These findings suggest that MyD88 may transmit a carcinogenic signal from the dysbiotic microbiota, although the nature of this signal has not been defined. However, since Schulz et al. reported that MyD88 deletion changes microbial composition in K-rasG12Dint; Myd88−/− mice, including enhanced abundance of butyrate-producing Ruminococcaceae, it is likely that host-derived MyD88 signaling also influences the microbiome. To expand on these results, it would have been interesting to measure levels of SCFA in K-rasG12Dint; Myd88−/− mice and to expose these mice to a low-fiber diet to reduce the ability of the microbiota to produce butyrate.
In summary, Schulz et al. have exposed an intriguing relationship between the intestinal microbiota, diet, and cancer development. While small bowel cancers are rare (comprising only 1%–2% of all gastrointestinal cancers), this model provides evidence for a causative role of diet-induced microbial dysbiosis in carcinogenesis. Although inflammation represents an important environmental condition fostering the capacity of microbes to induce carcinogenesis (Arthur et al., 2014; Elinav et al., 2013), Schulz et al. demonstrated that HFD may alternatively alter microbial activities leading to tumorigenesis in K-rasG12Dint mice. It will be important to extend the observation made with the K-rasG12Dint mice to more robust colorectal cancer models. For example, HFD also enhances tumor formation in Apcmin/+ mice (Wasan et al., 1997), but it is unknown if this is directly due to obesity.
The findings reported by Schulz et al. not only shift our expectations of how diet can cause cancer, but also opens up many intriguing questions. Is the obesity-independent, cancer-promoting activity of the K-rasG12Dint microbiota selective to this particular host mutation? Or is this a common feature of diet-induced carcinogenesis? Since cryptdin and mucin expression were only measured at the experimental endpoint of 22 weeks, do these host alterations result from or lead to the development of dysbiosis? In addition, although the intestinal epithelium is hyperplastic in K-rasG12Dint mice fed a normal diet, carcinogenesis developed only in the duodenum of HFD-exposed mice, suggesting a geographical impact of bacteria. This is reminiscent of the location-specific tumors observed in mice expressing two oncogenic transgenes throughout their intestines (cytomegalovirus chemokine receptor US28 and heparin-binding EGF-like growth factor) that developed serrated polyps only in the cecum. These mice showed tumor-specific microbial dysbiosis and decreased barrier function, which was associated with bacterial invasion and cancer promotion (Bongers et al., 2014). Therefore, it would be important to specifically assess the microbiome in mucosal-associated tumors in HFD K-rasG12Dint mice compared to similar locations in regular diet-fed K-rasG12Dint mice.
Understanding the sequence of events leading to dysbiosis and cancer is critical for the development of potential therapies. In addition, further studies will be needed to unveil not just the composition of nutrition-dependent cancer-promoting microbiota but also its functional impact using transcriptomic and metabolomic approaches. This kind of detailed analysis could ultimately lead to personalized therapies where information on host genetic and microbial activities could be merged to define the optimal therapeutic regimen matching a defined cancer type and individual.
References
- Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Nat Commun. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardou M, Barkun AN, Martel M. Gut. 2013;62:933–947. doi: 10.1136/gutjnl-2013-304701. [DOI] [PubMed] [Google Scholar]
- Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, Moriyama EH, Copeland JK, Kumar S, Green B, et al. Cell. 2014;158:288–299. doi: 10.1016/j.cell.2014.04.051. [DOI] [PubMed] [Google Scholar]
- Bongers G, Pacer ME, Geraldino TH, Chen L, He Z, Hashimoto D, Furtado GC, Ochando J, Kelley KA, Clemente JC, et al. J Exp Med. 2014;211:457–472. doi: 10.1084/jem.20131587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Irrazábal T, Belcheva A, Girardin SE, Martin A, Philpott DJ. Mol Cell. 2014;54:309–320. doi: 10.1016/j.molcel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Schulz MD, Atay Ç, Heringer J, Romrig FK, Schwitalla S, Aydin B, Ziegler PK, Varga J, Reindl W, Pommerenke C, et al. Nature. 2014;514:508–512. doi: 10.1038/nature13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasan HS, Novelli M, Bee J, Bodmer WF. Proc Natl Acad Sci USA. 1997;94:3308–3313. doi: 10.1073/pnas.94.7.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]