

Abstract
Mammary gland involution involves a process that includes one of the most dramatic examples of cell death in an adult mammalian organism. We have previously shown that Stat3 regulates a lysosomal pathway of cell death in the first 48 hours of involution and induces lysosome leakiness in mammary epithelial cells. Interestingly, Stat3 is associated also with the striking induction of autophagy which occurs concomitantly with cell death, presumably as a transient survival mechanism. The PI3 Kinase regulatory subunits p55α and p50α are dramatically and specifically upregulated at the transcriptional level by Stat3 at the onset of involution. We show here that ablation of either Stat3 or p55α/p50α in vivo affects autophagy during involution. We used two different cell culture models, normal mammary epithelial cells and mouse embryonic fibroblasts, to further investigate the role of p55α/p50α in autophagy regulation. Our results demonstrate a direct role for p55α/p50α as inhibitors of autophagy mediated by p85α. Thus Stat3 and its downstream targets p55α/p50α are key regulators of the balance between autophagy and cell death in vivo.
Keywords: Stat3, p55α/p50α, autophagy, mammary gland, PI3K
INTRODUCTION
Post-lactational regression, or involution, of the mammary gland is characterised by one of the most dramatic examples of cell death in an adult mammalian organism and occurs with every pregnancy/lactation/involution cycle [1]. Alveolar mammary epithelium, which develops during pregnancy, is removed upon cessation of lactation by a programme of cell death coupled with tissue remodelling that returns the gland to its pre-pregnant state. This provides an ideal experimental system in which to study cell death in a physiological context in vivo. We have shown that conditional deletion of the transcription factor Stat3 severely delays involution and abrogates cell death [2].
Although assumed to be apoptosis, recent work from our laboratory has demonstrated that cell death during involution is not classical apoptosis but occurs rather by a lysosomal mediated, and executioner caspase independent, pathway of cell death (LM-PCD) [3].
One intriguing aspect of cell death during involution is that it occurs in two phases; an initial reversible phase that lasts for up to 48h in the mouse and is initiated by LIF mediated activation of Stat3 [4] and a second irreversible phase from 48h that is regulated by oncostatin M (OSM) activation of Stat3 [1, 5]. Interestingly, expression of the receptor for OSM is regulated by Stat3 [6] and conditional deletion of Stat3 extends the reversible phase to at least 6 days [7]. During 1st phase involution, dying cells detach from the alveolar epithelial wall and are shed into the lumen, presumably to maintain the integrity of the alveoli and their functionality. Shed cells are then phagocytosed by the viable epithelium. Cell death during this phase is a stochastic event and it seems likely that a subpopulation of the alveolar cells could use a survival mechanism in order to provide the reversibility of this phase. In 2nd phase involution, the remaining alveolar epithelial cells die rapidly in situ in concert with extensive tissue remodelling. A previous study highlighted the induction of autophagy at 24h involution [8] but the mechanism of induction and the role of autophagy are not clear. Here we propose a pro-survival role of autophagy in the 1st phase of involution.
Stat3 has been implicated in regulating autophagy although its precise role is not clear and depends on the localisation of Stat3. While Stat3 phosphorylation in neuroblastoma H4 cells has been shown to inhibit autophagy [9], cytosolic unphosphorylated Stat3 inhibits autophagy by binding to PKR in U20S cells [10]. Thus, the function of Stat3 seems to be context and phosphorylation dependent.
Stat3 regulates the expression of a number of genes during involution and we have demonstrated that unique binding sites for Stat3 are present in the first intron of the pik3r1 gene which encodes three regulatory subunits of the class IA PI(3)Kinases, p85α, p55α and p50α. The small subunits, p55α and p50α, are strikingly upregulated at 24h involution and mediate cell death [11]. Although Stat3 and Stat5 can bind the same genomic sequences [12] there is no overlap between Stat3 and Stat5 target genes in mammary epithelial cells [6] and Stat5 has recently been shown to regulate only the p85α subunit [13]. These subunits all share a common carboxy-terminal domain which encompasses the p110 catalytic subunit binding site but have unique amino-terminal sequences [14]. While that of p85α comprises a SH3 and a BCR-homology (BH) domain which bind to PTEN, the negative regulator of PI3K, enhancing its lipid phosphatase activity [15] the p55α and p50α subunits have much smaller unique N-terminal sequences of 34aa for p55α and 6aa for p50α. Specific ablation of these subunits by deleting exons IB (p50α) and 1C (p55α) results in improved insulin sensitivity and protection from obesity-induced insulin resistance [16]. However, the function of these subunits is still somewhat enigmatic.
A role for Stat3 in autophagy induction in mammary gland has not been investigated. This, coupled with our previous demonstration of a role for Stat3 as an inducer of p55α and p50α expression specifically at the onset of involution, prompted us to investigate the role of Stat3 and these subunits in autophagy regulation during involution.
RESULTS
Mammary gland involution is accompanied by an early induction of autophagy
Autophagosomes are characterised by double membranes [17] that are evident by electron microscopy (EM). Although evidence that autophagy occurs in mammary gland has been presented previously [8, 18] the mechanism of induction has not been addressed. Initially we confirmed the induction of autophagy at early stages of mammary gland involution, by examining thin sections of 10d lactation and 24h involution mammary tissue by EM and noted a significant increase in the number of autophagosomes in involuting tissue sections (Fig. 1A and B). We next measured changes in RNAs associated with autophagy and observed that Lc3a, Lc3b, p62 and Beclin1 were all upregulated at least 3-fold at 24h involution (Fig. 1C). This is accompanied by appearance of p62 aggregates at 24h involution (Fig. 1D), which could reflect this increase in transcription. The induction of autophagy is further supported by the increase in LC3B lipidation (LC3B II), another marker of autophagy, during the first 48h [17] (Fig. 1E). The decline in p62 aggregates and LC3B II levels from 48h involution, suggests that autophagy has a specific role in the reversible phase of involution. However the high levels of LC3B II, coupled with accumulation of p62 puncta, indicate that although autophagy is induced at the onset of involution, it could be accompanied by a defect in the resolution of the autophagic process possibly due to disruption of lysosomal function, which we have shown to occur during involution [3].
Figure 1.
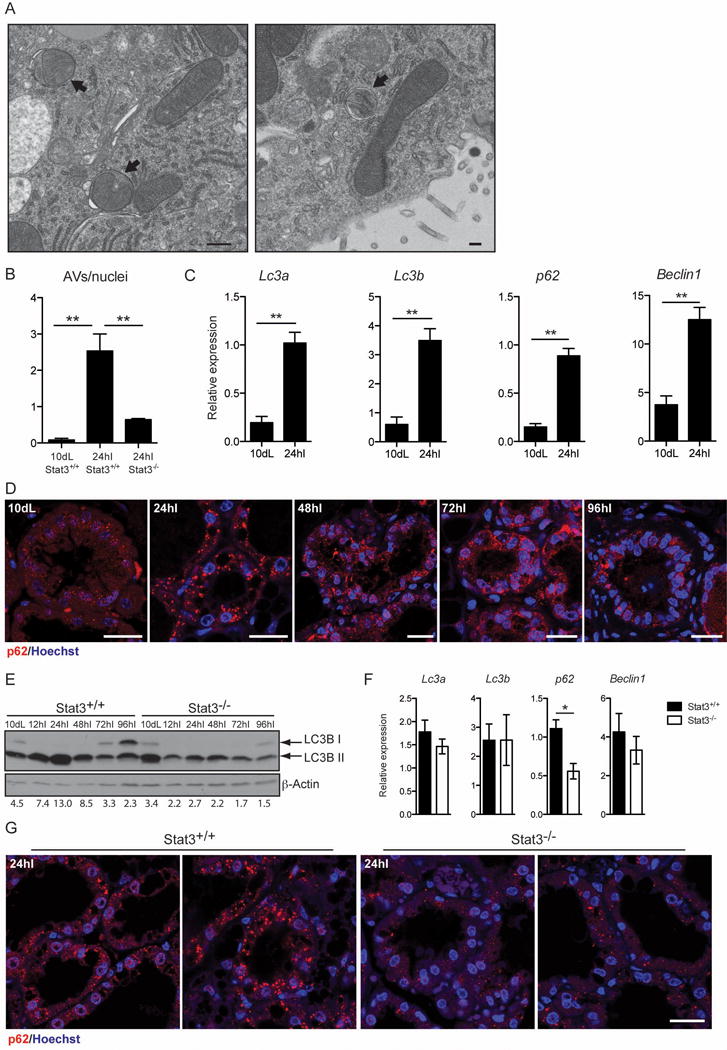
Mammary gland involution is accompanied by an early induction of autophagy, which is disrupted in the absence of Stat3. (A) Representative transmission electron micrograph of mammary epithelial cells in tissue from 24h involution time point, showing evidence of double membrane autophagic vesicles (black arrows; scale bar, 500 nm). (B) Quantification of autophagic vescicles (AVs) over nuclei as observed by transmission electron microscopy. Quantification has been performed on 3 independent biological replicates per group and is shown as means ± s.e.m. (** P<0.01, as determined by one-way ANOVA analysis with post-hoc Bonferroni′s correction). (C) Quantitative real-time PCR relative to cyclophilin a of the following genes: Lc3a, microtubule-associated protein 1 light chain 3 alpha; Lc3b, microtubule-associated protein 1 light chain 3 beta; p62, SQSTM1, sequestosome 1; Beclin1. All results are means ± s.e.m. of at least three independent biological repeats. 10dL, 10 days lactation; 24hI, 24 hours involution. (** P<0.01, as determined by Student’s t—test). (D) Immunohistochemical analysis of p62 (red) of lactation (Lac) and involution (Inv) time points in control mice, as indicated. Nuclei are stained with Hoechst. Scale bar, 20 μm. (E) Immunoblot showing lipidation of LC3B (LC3B II) in control and Stat3−/− during involution. dL, days of lactation; hI, hours of involution. β-Actin is the loading control. Quantification of LC3B II over β-Actin is shown below the Immunoblot. (F) Quantitative real-time PCR relative to cyclophilin a of the indicated genes at 24 hours involution. All results are means ± s.e.m. of at least four independent biological repeats (*P<0.05, as determined by Student’s t -test). (G) Reduced size and number of p62 puncta in the Stat3−/− glands, as shown by immunohistochemistry of p62 (red). Nuclei are stained with Hoechst. Scale bar, 20 μm.
Autophagy is affected by the absence of Stat3
We have previously shown that Stat3 is a master regulator of cell death in mammary gland involution [2] and that cell death occurs by lysosomal membrane permeabilisation (LMP), independently of executioner caspases [3]. Recent work from other laboratories has implicated Stat3 in the regulation of autophagy [9, 10]. To clarify the role of Stat3 in autophagy and to determine if Stat3 is required for the induction of autophagy observed at the onset of involution, we analysed glands from Stat3 controls (Stat3fl/fl;BLG-Cre−, hereafter referred to as Stat3+/+) and Stat3 depleted mice (Stat3fl/fl;BLG-Cre+, in which Stat3 is depleted from the epithelium during lactation, hereafter referred to as Stat3−/−) for autophagy markers. The increase in number of autophagosomes at 24h involution was severely abrogated in the absence of Stat3 (Fig. 1B). The p62 aggregates observed at 24h involution are reduced in both number and size in the Stat3−/− glands compared to controls (Fig. 1G) and this could reflect both reduced expression and/or increased autophagic flux. This latter contention is supported by the observation that LC3B lipidation is profoundly diminished in the Stat3 depleted glands (Fig. 1E). Interestingly gene expression was unaffected by Stat3 deletion apart from a small (2-fold) decrease in p62 expression (Fig. 1F). These data highlight a role for Stat3 in regulating autophagy in vivo in the mammary gland.
The Stat3 target genes p55α/p50α regulate autophagy during mammary gland involution
We have shown previously that Stat3 upregulates expression of the PI3K regulatory subunits p55α/p50α at the onset of involution [19]. Accordingly, expression of the two small subunits p55α/p50α but not p85α is markedly reduced in Stat3−/− glands at the time of autophagy induction (Fig. 2A). We therefore sought to determine if the small subunits play a role in regulating autophagy by analysing p55α−/−/p50α−/− mice [16], which display delayed involution and diminished cell death [11]. Expression of autophagy regulatory genes was investigated and we found that only Lc3b was significantly downregulated in the absence of p55α/p50α expression (Fig. 2B). Interestingly, analysis of p55α−/−/p50α−/− mammary tissue revealed a substantial reduction of LC3B lipidation at 24h and 48h of involution with a similar pattern to the Stat3−/− tissues (Fig. 2C). This correlates with a striking decrease in the size and number of p62 puncta (Fig. 2D). We have previously shown that p55α/p50α are expressed predominantly in the epithelium and are not expressed in the adipose cells of the mammary gland [11]. Since infiltrating immune cells play a role mostly in the second phase of involution, we suggest that the phenotype observed is a consequence of the absence of the small subunits in the epithelial compartment.
Figure 2.
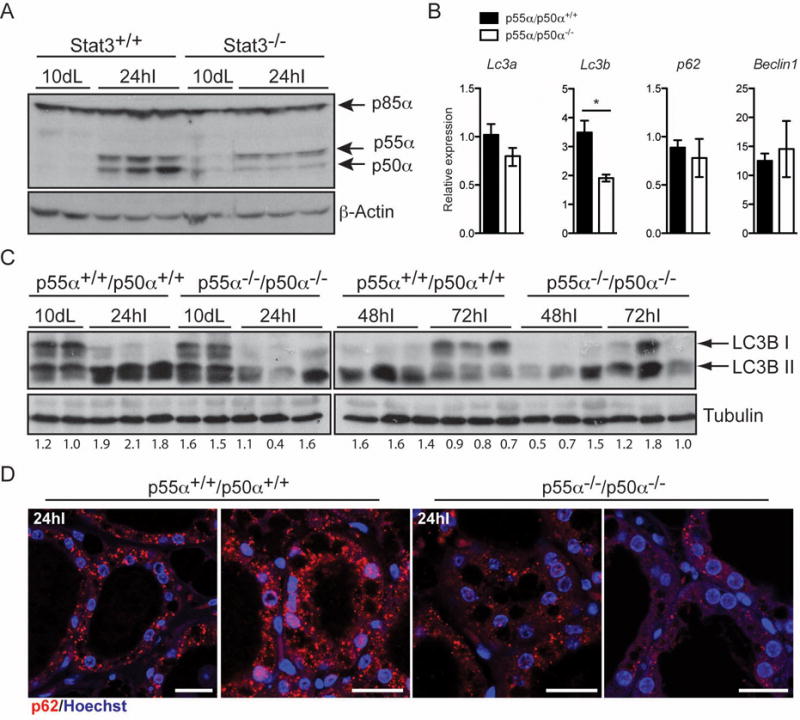
p55α/p50α deletion disrupts autophagy in the mammary gland. (A) Immunoblot showing reduced levels of p55α/p50α in the Stat3−/− glands at 24h of involution. β-Actin is the loading control. L, lactation; I, involution. (B) Quantitative real-time PCR relative to cyclophilin a of the indicated genes at 24 hours involution. All results are means ± s.e.m. of at least five independent biological repeats (*P<0.05, as determined by Student’s t -test). (C) Immunoblot showing lipidation of LC3B (LC3B II). Lanes represent independent biological replicates. Tubulin is the loading control. L, lactation; I, involution. Quantification of LC3B II over Tubulin is shown below the Immunoblot. (D) Reduced size and number of p62 puncta in the p55α−/−/p50α−/− glands, as shown by immunohistochemistry of p62 (red). Nuclei are stained with Hoechst. Scale bar, 20 μm.
There are two possible explanations for the results observed in vivo in the Stat3−/− and p55α−/−/p50α−/− glands: in the control glands, accumulation of LC3B II and p62 puncta could indicate induction of autophagy or could result from a failure to clear autophagosomes. We favour the latter interpretation because we have already shown that lysosomes become leaky during involution, which would abrogate the fusion with autophagosomes [3]. This suggests that p55α/p50α block the resolution of autophagy downstream of Stat3. However, it cannot be excluded that both induction of autophagy and a partial block in fusion with lysosomes/late endosomes contribute to the phenotype observed in involution and both could be modulated by the absence of p55α and p50α.
OSM-dependent upregulation of p55α/p50α increases autophagic flux while augmenting the number of p62 aggregates
Since autophagic flux is difficult to address in vivo we investigated the role of Stat3 and p55α/p50α in the EpH4 mammary epithelial cell line that we have previously shown accurately mimics LM-PCD mechanisms in vivo [3]. While LIF is the physiological inducer of p55α/p50α expression in the mammary gland [20], and other Stat3-activating cytokines could potentially induce the expression of these subunits, we observed that OSM activates Stat3 and induces high levels of p55α and p50α expression in EpH4 cells (Fig. 3A). OSM also induced an increase in LC3B II with levels reaching a maximum at 72h post-treatment (Fig. 3B and C). Interestingly treatment with bafilomycin A1, which blocks the function of the vacuolar ATPase and thus abolishes the ability of the lysosome to fuse with autophagosomes [17], induced rapid accumulation of LC3B II and had an additive effect on OSM treatment (Fig. 3B). This suggests that OSM induces autophagy. OSM treatment also resulted in the accumulation of numerous large puncta of p62 at 24h. Both OSM and vehicle-treated cells showed a similar increase upon bafilomycin A1 treatment (Fig. 3E) although protein levels were mostly unchanged (Fig. 3D). This is indicative of a partial block of lysosome function, a notion supported by our observation that OSM induces leakage of LysoTracker™ in EpH4 cells [3]. Since OSM treatment recapitulates the autophagy phenotype observed in vivo, we can speculate that involution is characterized by an induction of autophagy simultaneously with a partial block of autophagosome/lysosome fusion due to leaky lysosomes. However, we were unable to dissect the direct contribution of the small subunits in EpH4 cells due to an inability to specifically silence their expression in these cells as a consequence of the very short domains of isoform-specific sequence (data not shown).
Figure 3.
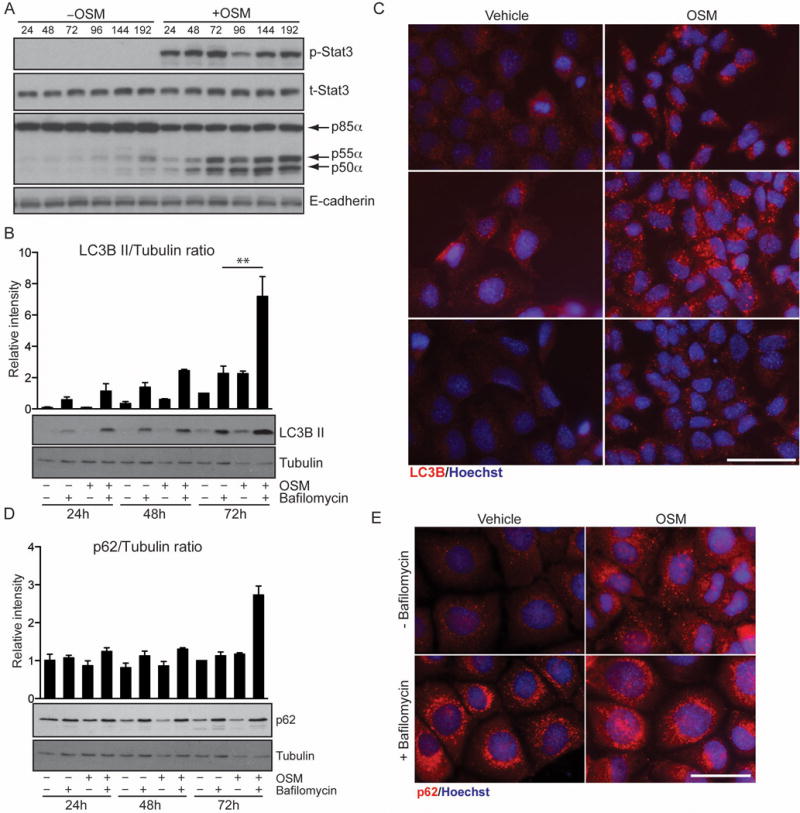
OSM-dependent upregulation of p55α/p50α increases autophagic flux while augmenting the number of p62 aggregates. (A) EpH4 cells were stimulated with vehicle or OSM for the indicated hours and immunoblotted for the indicated proteins to show the induction of p55α/p50α downstream of OSM. E-cadherin is shown as loading control. (B) Increased LC3B II in cells stimulated with OSM. EpH4 cells were treated with vehicle, OSM and/or bafilomycin Al as indicated. LC3B lipidation was then analysed by immunoblot, one time course representative of three independent experiments is shown. Tubulin is the loading control. (C) Immunohisotchemistry showing endogenous expression of LC3B (red) in EpH4 cells treated with vehicle or OSM for 48h. Nuclei are stained with Hoechst. Scale bar, 50 μm. (D) p62 protein levels are mostly unchanged in response to OSM. EpH4 cells were treated with vehicle, OSM and/or bafilomycin Al as indicated. p62 was analysed by immunoblot, one time course representative of three independent experiments is shown. Tubulin is the loading control. Graph above (B) and (D) is the quantification of the intensity of the bands of three independent biological replicates shown as means ± s.e.m. (** P<0.01, as determined by one-way ANOVA analysis with post-hoc Bonferroni’s correction). (E) Immunohistochemistry for p62 (red) in EpH4 cells stimulated with vehicle or OSM for 48h and/or bafilomycin Al showing increased accumulation of p62 puncta in response to OSM with additional increment upon Bafilomycin Al treatment. Scale bar, 50 μm.
p55α/p50α inhibit autophagy in primary MEFs by competition with p85α
To investigate a general role of p55α and p50α in the regulation of autophagy in other cell types, we analysed primary mouse embryonic fibroblasts (MEFs) obtained from control and p55α−/−/p50α−/− mice. Starvation of both control and p55α−/−/p50α−/− MEFs resulted in the induction of LC3B lipidation within 2h of treatment, as clearly shown by the accumulation of LC3B II after bafilomycin A1 treatment of starved cells as compared to controls (Fig. 4A). Interestingly however, the levels of LC3B II in the unstarved p55α−/−/p50α−/− MEFs are higher compared to the controls (Fig. 4A and quantification below), demonstrating that the primary role of the small subunits is to inhibit basal autophagy and the higher levels of LC3B II in bafilomycin Al treated starved cells reflect this higher basal level of autophagy in p55α−/−/p50α−/− cells. Analysis of immortalised control and pik3r1−/− MEFs, which are depleted of the longer p85α subunit as well as p55α and p50α, revealed no difference when comparing the two cell lines upon starvation (Fig. 4B). These data point to a role for the small subunits in regulating the extent of starvation-induced autophagy mediated by the longer subunit p85α.
Figure 4.
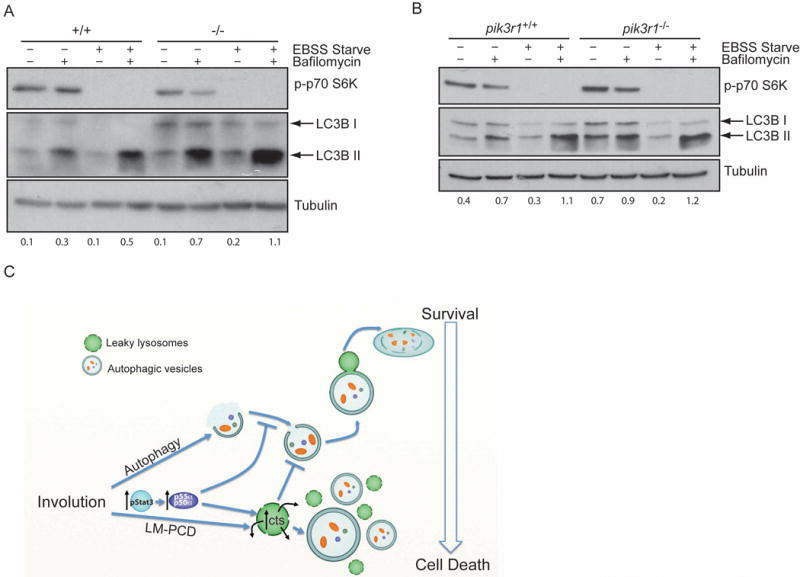
p55α/p50α inhibit autophagy in primary MEFs. (A, B) Primary MEFs derived from control (+/+) and p55α−/−/p50α−/− mice (−/−) (A) or pik3r1+/+ and pik3r1−/− 3T3 MEFs (B) were pre-treated with bafilomycin Al for 2h and then starved with EBSS medium for 2h in the presence of bafilomycin Al as indicated. LC3B lipidation was then analysed by immunoblot. Immunoblot of phospho-p70 S6K shows the efficient inhibition of mTOR upon starvation. Tubulin is the loading control. Quantification of LC3B II over Tubulin is shown below the Immunoblot. (C) Graphical summary of the interplay between autophagy and LM-PCD during mammary gland involution highlighting the roles of Stat3 and p55α/p50α subunits. Our data suggest that autophagy provides a transient survival signal which is overridden by Stat3-induced expression of p55α/p50α. Concomitantly, the induction of lysosomal cathepsins (cts) and lysosomal leakiness further inhibits autophagy by blocking its resolution.
DISCUSSION
Cell death during mammary gland involution is extensive and removes approximately 80% of the mammary epithelium in the space of a few days in the mouse. The programmed cell death occurring at the end of lactation is counteracted by a temporary survival mechanism to allow survival of sufficient alveolar cells that can recommence milk production if offspring are returned to the mother up to 48 hours after separation in the mouse or longer in larger mammals. We propose here that autophagy is such a survival mechanism during the early phase of mammary gland involution and that these two opposing events are co-ordinately regulated by Stat3 (Fig. 4C).
Recent work has demonstrated a role for Stat3 in regulating autophagy [9, 10]. On the one hand LIF-mediated activation of Stat3 in human neuroblastoma H4 cells inhibits LC3B II accumulation in an mTORCl independent manner [9]. On the other hand, genetic deletion of Stat3 in hepatocytes in vivo induces autophagy while retention of Stat3 in the cytosol resulted in autophagy suppression [10]. Thus, the function of Stat3 is phosphorylation and context dependent. Interestingly, starvation or rapamycin treatment of HeLa cells have been shown to lead to Stat3 activation, that is necessary for the production of IL-6, which in turn promotes survival and proliferation in cancer cells [21].
We, and others, have shown that autophagy is induced at the onset of involution. Autophagic vesicles are present in the mammary gland as early as 24h of involution (Fig. 1A–C), concomitantly with the appearance of p62 (Fig. 1D) and LC3B puncta [8]. If autophagy induction in the mammary gland represents a survival mechanism to guarantee the reversibility of involution and a halting of cell death events, it would be expected that disruption of autophagy mediators in the mammary gland would lead to increased cell death. Previous work has shown an accumulation of shed cells in the alveoli of mice carrying a monoallelic deletion of Beclinl or deficient for Atg7 in the first 72h of involution, which the authors interpreted as a compromised ability of mammary cells to phagocytose, and thus clear, dead cells present in the lumen [8]. This is indeed possible although we favour a different interpretation and suggest that the accumulation of dead cells observed in Beclin1+/− and Atg7-deficient glands indicates an increased rate of cell death in the 1st phase of involution. The class III PI3K Vps34 is involved in autophagy and has been shown to be fundamental in regulating phagosome maturation [22]. In the past few years, however, a number of studies have underlined a function for members of class IA PI3K in autophagy regulation. One catalytic subunit, pll0β, has been shown to positively regulate autophagy both in vitro and in vivo in liver and heart in a kinase-independent manner, p110β, associates with the Vps34-Vpsl5-Beclin l-Atgl4L complex to increase the generation of PtdIns(3)P, thus promoting autophagy [23]. A different catalytic subunit, p110α, is required for the delivery of the lysosomal membrane proteins LAMP-1 and LAMP-2 to Rab7-positive vesicles during vacuole maturation in human leukemic monocytes, although phagocytosis does not seem to be substantially affected by the absence of p110α in these cells [24]. In addition, degradation of the regulatory subunit p85β, through interaction with the F-box protein FBXL2, inhibits serum starvation-induced autophagy in NHF fibroblasts [25]. Furthermore, the p85α regulatory subunit has been shown to bind Rab5 [26], which is an important component of the vesicle trafficking machinery [27].
Given the role of Stat3 in regulating cell death and expression of the class I PI3K regulatory subunits p55α and p50α during mammary gland involution, we examined both Stat3 and p55α/p50α deficient involuting mammary glands for evidence of altered autophagy. Conditional deletion of Stat3 disrupted autophagy in the first 48h of involution (Fig. 1) and this phenotype was mirrored in the p55α/p50α deficient glands (Fig. 2). Only minor changes in p62 and Lc3b gene expression were detected suggesting that direct transcriptional regulation of these proteins by Stat3 and p55α/p50α is not a major factor in induction of autophagy. In both these strains, cell death is also abrogated [2, 3, 11]. In order to tease apart the mechanism, we utilised EpH4 cells treated with OSM to induce Stat3 activity as a surrogate for involution. Elevated levels of LC3B II were observed upon OSM stimulation, and a corresponding appearance of p62 puncta suggested an induction and/or a block of autophagy (Fig. 3). Since an additive effect was achieved with bafilomycin Al treatment this suggests that at least part of the response to OSM is an induction of autophagy. However, although a considerable number of p62 puncta were apparent upon treatment with OSM, we did not observe a notable difference with the addition of bafilomycin Al plus OSM compared to bafilomycin Al alone, suggesting a considerable block in the resolution of autophagy in the presence of OSM. Moreover, immunoblotting revealed a failed turnover of p62 further supporting an impairment of the later stages of autophagy, as lysosomes become leaky with prolonged treatment with OSM [3].
In order to look more generally at the role of p55α/p50α in autophagy, we investigated starvation- rather than stress-induced autophagy utilising MEFs derived from p55α−/−/p50α−/− mice (Fig. 4). Although starvation is able to induce LC3B lipidation in both control and p55α−/−/p50α−/− MEFs, the level of LC3B II was enhanced in the absence of the subunits, indicating their inhibitory effects on autophagy. As we have previously suggested, one mechanism of action of the small subunits is direct competition with p85α for binding to partner proteins [28]. Thus we utilized MEFs deleted for all three subunits and found that the enhancing effect of deletion of the small subunits is abrogated. This suggests that p55α/p50α compete with, or block the ability of p85α to contribute to autophagy.
Expression of the p55α and p50α regulatory subunits is not limited to mammary gland with variable levels of each expressed in a wide range of tissues (data not shown). This raises the possibility that autophagy in other tissues could be modulated by inducing either p55α or p50α. Given the very small unique N-terminal domains, and our inability to detect any binding partners other than the catalytic subunits (data not shown), we propose that the small subunits could function in autophagy regulation in response to starvation or other stressors that mediate Stat3 activity by competing with p85α. In mammary gland, we propose that Stat3 regulates involution by inhibiting the pro-survival, autophagic process through two mechanisms: inducing the expression of p55α and p50α and disrupting lysosomal function [3].
MATERIALS AND METHODS
Animal husbandry
p55α/p50α and Stat3 null mice have been generated previously (respectively [16] and [2]). C57BL/6 mice were purchased from Harlan labs (Indianapolis, IN, US). The mice were bred in regular cages with food and water ad libitum. Virgin female mice, 8–14 weeks old, were mated and males were subsequently removed before birth to avoid second pregnancies. Dams were killed at indicated time points. For involution studies, pups were removed at 10 days of lactation. Unless otherwise stated, at least three mice were used for each time point in every experiment. All animals were treated according to local ethical committee and the UK Home Office guidelines and killed through CO2 asphyxiation or dislocation of the neck.
Transmission electron microscopy
Transmission electron microscopy was carried out as previously described [3].
Cell culture
Embryos were dissected 13.5 days post coitum for MEF derivation and primary MEFs were used within the first 5 passages in culture. pi3kr1−/− 3T3 MEFs were a kind gift of Dr. David A. Fruman. EpH4, primary MEF and 3T3 MEF cells were grown in DMEM (Gibco, Life Technologies, Carlsbad, CA, US) containing 10% fetal calf serum (FCS, Sigma, St. Louis, MO, US). For protein extraction or immunofluorescence experiments, cells were seeded into 6-well plates containing glass slides as required at a density of 100,000 cells per well in DMEM 10% FCS. The next day media was replaced with DMEM media containing 1% FCS and either 25 ng/ml recombinant mouse OSM (495-MO, R&D Systems, Minneapolis, MN, US) or vehicle for the indicated times. Bafilomycin A1 (final concentration of 200 nM, Tocris-R&D, 1334) treatment was carried out four hours prior to harvesting cells. Starvation of primary and 3T3 MEFs was carried out in EBSS (24010-043, Life Technologies) for two hours. Bafîlomycin Al (Santa Cruz Biotechnology, Dallas, TX, US, final concentration of 200 nM) treatment was carried out four hours prior to harvesting cells.
Immunoblotting
Sample preparation and immunoblotting was carried out as previously described [29]. The following antibodies from Cell Signaling Technologies (Danvers, MA, US) were used: anti-phospho-Stat3 (Tyr705: 9131), anti-total-Stat3 (9132), anti-LC3B (2775) and anti-phospho-p70 S6K (9234). The following antibodies from Abcam (Cambridge, UK) were used: anti-Tubulin (ab6160) and anti-β-actin (ab8227). Other commercial antibodies used were: anti-pan-p85 (Millipore, Danmstadt, Germany, 06-49 6, also detects p50α/p55α subunits), anti-p62/SQSTMl (MBL International, Woburn, MA, US, PM045) and anti-E-Cadherin (BD biosciences, San Jose, CA, US, 610182). All antibodies were used at a standard dilution of 1:1,000. Secondary horseradish peroxidase (HRP)-conjugated antibodies were purchased from Dako (Ely, UK).
Immunohistochemistry
Tissue sections were prepared as previously described [29]. For tissue-culture samples, EpH4 cells were seeded on glass slides. After OSM treatment, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% saponin in PBS for the p62 staining and ice cold methanol for the LC3B staining, and blocked in 5% normal goat serum (Dako) in PBS, with 0.5% saponin for the p62 staining. The pictures of p62 and LC3B puncta on EpH4 cells were acquired on a Zeiss Axioplan 2 microscope. All the other pictures were acquired on a Zeiss LSM 700 confocal microscope.
Quantitative real-time PCR
RNA extraction, complementary DNA synthesis and quantitative real-time PCR were carried out as previously described [29]. Primers used were: cyclophilin A (housekeeping gene) forward, 5′-CCT TGG GCC GCG TCT CCT T-3′, reverse, 5′- CAC CCT GGC ACA TGA ATC CTG-3′; Lc3α forward, 5′- GAC CGC TGT AAG GAG GTG C -3′, reverse, 5′- CTT GAC CAA CTC GCT CAT GTT A-3′; Lc3b forward, 5′- TTA TAG AGC GAT ACA AGG GGG AG -3′, reverse, 5′- CGC CGT CTG ATT ATC TTG ATG AG -3′; p62 forward, 5′- AGG ATG GGG ACT TGG TTG C -3′, reverse, 5′- TCA CAG ATC ACA TTG GGG TGC -3′. Beclin1 forward, 5′- ATG GAG GGG TCT AAG GCG TC -3′, reverse, 5′- TCC TCT CCT GAG TTA GCC TCT -3′. Primers were designed using the PrimerBank website (http://pga.mgh.harvard.edu/primerbank/).
Acknowledgments
We thank Beth Levine for critically reading the manuscript and for her helpful suggestions, Jeremy Skepper for assistance with the transmission electron microscopy, Helen Skelton for tissue histology and Watson laboratory members for helpful discussions. This work was funded by BBSRC and MRC grants (BB/D012937/1 and MR/J001023/1) awarded to CJW. SP is the recipient of a Marie Curie IEF fellowship (EU Marie Curie grant no. 273365).
ABBREVIATIONS
- LM-PCD
lysosomal-mediated programmed cell death
- PI3Ks
phosphatidylinositol 3-kinases
- ctsB
cathepsin B
- ctsL
cathepsin L
- SH3
Src homology 3
- BH
breakpoint cluster region-homology
- OSM
oncostatin M
- LIF
leukemia inhibitory factor
- PTEN
phosphatase and tensin homolog
- EM
electron microscopy
- Stat3
signal transducer and activator of transcription 3
- LMP
lysosomal membrane permeabilisation
- MEFs
mouse embryonic fibroblasts
Footnotes
AUTHOR CONTRIBUTIONS
SP planned and performed experiment, analysed the data and wrote the paper; BL-L, TJS and HKR performed experiments and analysed the data; CRK provided the p55α−/−/p50α−/− mice and CJW planned experiments, analysed the data and wrote the paper.
References
- 1.Watson CJ. Post-lactational mammary gland regression: molecular basis and implications for breast cancer. Expert Rev Mot Med. 2006;8:1–15. doi: 10.1017/S1462399406000196. doi: S1462399406000196 [pii] 10.1017/S1462399406000196. [DOI] [PubMed] [Google Scholar]
- 2.Chapman RS, Lourenco PC, Tonner E, Flint DJ, Seibert S, Takeda K, Akira S, Clarke AR, Watson CJ. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson G. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13:303–309. doi: 10.1038/ncb2171. doi: ncb2171 [pii] 10.1038/ncb2171. [DOI] [PubMed] [Google Scholar]
- 4.Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130:3459–3468. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- 5.Tiffen PG, Omidvar N, Marquez-Almuina N, Croston D, Watson CJ, Clarkson RW. A dual role for oncostatin M signaling in the differentiation and death of mammary epithelial cells in vivo. Mol Endocrinol. 2008;22:2677–2688. doi: 10.1210/me.2008-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarkson RW, Boland MP, Kritikou EA, Lee JM, Freeman TC, Tiffen PG, Watson CJ. The genes induced by signal transducer and activators of transcription (STAT)3 and STAT5 in mammary epithelial cells define the roles of these STATs in mammary development. Mol Endocrinol. 2006;20:675–685. doi: 10.1210/me.2005-0392. [DOI] [PubMed] [Google Scholar]
- 7.Humphreys RC, Bierie B, Zhao L, Raz R, Levy D, Hennighausen L. Deletion of Stat3 blocks mammary gland involution and extends functional competence of the secretory epithelium in the absence of lactogenic stimuli. Endocrinology. 2002;143:3641–3650. doi: 10.1210/en.2002-220224. [DOI] [PubMed] [Google Scholar]
- 8.Teplova I, Lozy F, Price S, Singh S, Barnard N, Cardiff RD, Birge RB, Karantza V. ATG proteins mediate efferocytosis and suppress inflammation in mammary involution. Autophagy. 2013;9:459–475. doi: 10.4161/auto.23164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010;18:1041–1052. doi: 10.1016/j.devcel.2010.05.005. doi: S1534-5807(10)00213-3 [pii] 10.1016/j.devcel.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Marino G, Lachkar S, Senovilla L, et al. Cytoplasmic STAT3 Represses Autophagy by Inhibiting PKR Activity. Mol Cell. 2012;48:667–680. doi: 10.1016/j.molcel.2012.09.013. doi: S1097-2765(12)00789-7 [pii] 10,1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Pensa S, Neoh K, Resemann HK, Kreuzaler PA, Abell K, Clarke NJ, Reinheckel T, Kahn CR, Watson CJ. The PI3K regulatory subunits p55alpha and p50alpha regulate cell death in vivo. Celt Death Differ. 2014 doi: 10.1038/cdd.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang K, Robinson GW, Hennighausen L. Comprehensive meta-analysis of Signal Transducers and Activators of Transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genomics. 2013;14:4. doi: 10.1186/1471-2164-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt JW, Wehde BL, Sakamoto K, Triplett AA, Anderson SM, Tsichlis PN, Leone G, Wagner KU. Stat5 regulates the phosphatidylinositol 3-kinase/Akt1 pathway during mammary gland development and tumorigenesis. Molecular and cellular biology. 2014;34:1363–1377. doi: 10.1128/MCB.01220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biocbem Sei. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. doi: S0968-0004(05)00056-3 [pii] 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD, Anderson DH. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sei U S A. 2010;107:5471–5476. doi: 10.1073/pnas.0908899107. doi: 0908899107 [pii] 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen D, Mauvais-Jarvis F, Bluher M, Fisher SJ, Jozsi A, Goodyear U, Ueki K, Kahn CR. p50alpha/p55alpha phosphoinositide 3-kinase knockout mice exhibit enhanced insulin sensitivity. Mol Cell Biol. 2004;24:320–329. doi: 10.1128/MCB.24.1.320-329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zarzynska J, Gajkowska B, Wojewodzka U, Dymnicki E, Motyl T. Apoptosis and autophagy in involuting bovine mammary gland is accompanied by up-regulation of TGF-beta1 and suppression of somatotropic pathway. Pol J Vet Sci. 2007;10:1–9. [PubMed] [Google Scholar]
- 19.Abell K, Bilancio A, Clarkson RW, Tiffen PG, Altaparmakov Al, Burdon TG, Asano T, Vanhaesebroeck B, Watson CJ. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Blot. 2005;7:392–398. doi: 10.1038/ncb1242. doi: ncbl242 [pii] 10.1038/ncbl242. [DOI] [PubMed] [Google Scholar]
- 20.Abell K, Bilancio A, Clarkson RW, Tiffen PG, Altaparmakov Al, Burdon TG, Asano T, Vanhaesebroeck B, Watson G. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nature cell biology. 2005;7:392–398. doi: 10.1038/ncbl242. [DOI] [PubMed] [Google Scholar]
- 21.Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy. 2010;6:1125–1138. doi: 10.4161/auto.6.8.13547. [DOI] [PubMed] [Google Scholar]
- 22.Dall’Armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23:R33–45. doi: 10.1016/j.cub.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dou Z, Chattopadhyay M, Pan JA, Guerriero JL, Jiang YP, Ballou LM, Yue Z, Lin RZ, Zong WX. The class IA phosphatidylinositol 3-kinase p110-beta subunit is a positive regulator of autophagy. The Journal of cell biology. 2010;191:827–843. doi: 10.1083/jcb.201006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thi EP, Lambertz U, Reiner NE. Class IA phosphatidylinositol 3-kinase p110alpha regulates phagosome maturation. PLoS One. 2012;7:e43668. doi: 10.1371/journal.pone.0043668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuchay S, Duan S, Schenkein E, Peschiaroli A, Saraf A, Florens L, Washburn MP, Pagano M. FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nature cell biology. 2013;15:472–480. doi: 10.1038/ncb2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chamberlain MD, Berry TR, Pastor MC, Anderson DH. The p85alpha subunit of phosphatidylinositol 3′-kinase binds to and stimulates the GTPase activity of Rab proteins. J Biol Chem. 2004;279:48607–48614. doi: 10.1074/jbc.M409769200. [DOI] [PubMed] [Google Scholar]
- 27.Chua CE, Gan BQ, Tang BL. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol Life Sci. 2011;68:3349–3358. doi: 10.1007/s00018-011-0748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abell K, Watson CJ. The Jak/Stat pathway: a novel way to regulate PI3K activity. Cell Cycle. 2005;4:897–900. doi: 10.4161/cc.4.7.1837. [DOI] [PubMed] [Google Scholar]
- 29.Khaled WT, Read EK, Nicholson SE, Baxter FO, Brennan AJ, Came PJ, Sprigg N, McKenzie AN, Watson G. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 2007;134:2739–2750. doi: 10.1242/dev.003194. [DOI] [PubMed] [Google Scholar]