

Abstract
Purpose
Hemoglobin (Hb) depletion with nickel affinity chromatography has been shown to increase the number of proteins identified in proteomic studies of erythrocytes, but limited data exist on the application of this technique in depletion of Hb from plasma or serum required for clinical biomarker studies. The aim of this study was to explore the potential of using nickel-beads for Hb depletion of plasma.
Experimental design
Nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography was used to deplete Hb from hemolyzed plasma samples obtained from children with sickle cell disease (SCD, n=7) and normal human plasma (n=4). Ni-NTA bound proteins were analyzed by one-dimensional gel electrophoresis, followed by in-gel digestion for characterization using a LTQ-Orbitrap hybrid mass spectrometer. In addition, the loss of two non-hemoglobin related plasma proteins, thrombospondin1 (TSP1) and L-selectin, by Ni-NTA was determined by ELISA (SCD n=6, non-SCD controls n=2).
Results
Ni-NTA resulted in an average 60% decrease in plasma protein concentration, which was not hemolysis dependent. Specifically, Hb (7 peptides) and the top three proteins, alpha-2-macroglobulin (75 peptides), apolipoprotein B-100 (73 peptides), and albumin (42 peptides) were Ni-NTA bound. In addition, using an ELISA assay two non-hemoglobin associated plasma proteins TSP1 and L-selectin were decreased by Ni-NTA.
Conclusions and clinical relevance
Hb depletion with Ni-NTA is effective for Hb removal but is not specific. There is potential for deleterious depletion of potential biomarkers that may limit the applicability of this method. Consideration of alternate methods of Hb depletion for clinical proteomics may be warranted.
Keywords: Biomarkers, Hemoglobin depletion, Plasma proteomics, Sickle cell disease
Sickle cell disease (SCD) is an inherited hemoglobinopathy characterized by chronic hemolytic anemia, recurrent vaso-occlusive crisis, increased risk of cerebral infarction and other organ system damage, as well as early mortality. As a result of chronic intravascular hemolysis, patients can have plasma concentrations of free hemoglobin (Hb) as high as 90 mg/dL [1]. This predominance of free Hb can hinder proteomic studies by masking the detection of low abundance proteins and thus present a challenge for the identification of potential biomarkers. Previous studies have reported increased erythrocyte proteome coverage using depletion techniques that have taken advantage of the affinity of Hb for nickel [2, 3]. Furthermore, Hb depletion using nickel or cobalt chromatography is recommended by Ciphergen Biosystems for protein profiling of plasma using surface-enhanced laser desorption and ionization (http://lpg.nci.nih.gov/lpg_small/protocols/Ciphergen). However, the impact of binding non-hemoglobin proteins by nickel affinity chromatography, and the potential nonspecific binding to the agarose support, which would impact the effectiveness of downstream clinical proteomic analysis, has not been studied. The main focus of this study is to determine the consequences of using nickel-nitrilotriacetic acid (Ni-NTA) beads for Hb depletion on plasma obtained from children with SCD.
Seven baseline steady state plasma samples (defined as the absence of a manifest painful crisis, acute cerebrovascular disease, or an acute chest syndrome) from children aged 5 – 15 years with SCD enrolled in the Silent Infarct Transfusion Trial (SIT Trial, ClinicalTrials.gov identifier NCT00072761) were used in this study. Details of the study are previously described [4]. Steady state plasma from four age-matched healthy controls, without evidence of chronic or acute illness, was obtained through a separate Institutional Review Board-approved study and pooled prior to assaying. For all samples peripheral whole blood was collected in heparinized tubes and spun at 1500g for 8 minutes at room temperature. Plasma was stored at −80°C until assayed. All samples were aliquoted without additives into cryovials and were thawed only once at the time the samples were used. Plasma samples from children with SCD were divided into high (n=4) and low (n=3) hemolysis groups based on whether the values of known clinical laboratory markers of hemolysis, reticulocyte count and total bilirubin, fell above or below reported means for SCD (average reticulocyte 9%, average bilirubin 2.7 mg/dL)[5]. Ni-NTA agarose and NTA agarose beads, both with binding capacities of up to 50 mg/ml (Qiagen, Valencia, CA), were used to deplete Hb according to the following protocol suggested by Ciphergen Biosystems: Ni-NTA agarose and NTA agarose beads were washed with PBS/0.3 M NaCl, and a 50% nickel bead solution (50 μl beads in 50 μl PBS/0.3 M NaCl) was then added to 100 μl of each plasma sample. After a 20 minute rotation at 4°C, the Ni-NTA beads turned red, indicating hemoglobin removal, and were separated by centrifugation (2 min at 3000 rpm at room temperature). To elute bound proteins, beads were washed with PBST (0.05% Tween), incubated with 50 μl of 0.5M imidazole, subjected to rotation for 15 minutes at room temperature and separated from the eluted protein by centrifugation (2 min at 3000 rpm at room temperature). Protein concentrations were determined using the Coomassie Blue dye protein assay reagent (Bio-Rad Laboratories, Hercules, CA). Protein samples (30 μg) in 2X Laemmli buffer containing 62.5 mM Tris-HCL, pH 6.8/ 2% SDS/25% glycerol/5% β-mercaptoethanol/0.01% bromophenol blue, (Bio-Rad) were resolved by one-dimensional gel electrophoresis (4–12% 1-D SDS PAGE, Novex) according to Laemmli [6]. The gel was stained with Coomassie Blue (Simply Blue Safe Stain, Invitrogen, Carlsbad, CA). Twenty-four bands were excised (see Figure S1 for location of each band), trypsin-digested for protein identification as previously described [7] and analyzed using a LTQ-Orbitrap hybrid mass spectrometer (ThermoFisher, San Jose, CA) equipped with an on-line nano-HPLC (Agilent technologies, 1200 Series, Wilmington, DE). The peptides were separated on a reverse-phase analytical column packed with 10 cm of C18 beads (Biobasic C18 PicoFrit column, New Objective, Woburn, MA) with a 10-μm emitter tip (New Objective, Woburn, MA) attached. The HPLC gradient was 5–60% B for 28 min (A, 0.1% formic acid; B, 90% acetonitrile in 0.1% formic acid) and then quickly bumped up to 100% B before coming back to initial conditions. The flow rate was 300 nL/min. The ion-trap mass spectrometer was operated in a data-dependent mode in which a full MS scan was followed by MS/MS scans of the five most intensive ions. The ions were automatically selected for collision-induced dissociation. The data were analyzed using PASS (Integrated Analysis, Bethesda, MD) with X! Tandem searches (www.thegpm.org; version 2008.12.01) of the International Protein Index (IPI) peptide database (human, 3.19). The dataset was filtered to 90% sequence identity with CD-HIT. Sequence homologies, families, domains and functional sites were accessed from http://www.ncbi.nlm.nih.gov/BLAST/. The data were converted using PRIDE Converter (version 2.4.1) and are available in the PRIDE database (www.ebi.ac.uk/pride) under accession number 13275 [8].
Incubation of plasma with Ni-NTA resulted in an average 60% decrease in the plasma protein concentration. Average concentrations before and after incubation with Ni-NTA agarose were 44.7 and 19.3 μg/mL for human plasma and 24.9 and 10.4 μg/mL for sickle cell samples (n=7), respectively, after one incubation step. This decrease was equivalent in hemolyzed and non-hemolyzed samples, suggesting significant binding of proteins other than hemoglobin to the Ni-NTA beads. On average, there was a 9% decrease in plasma protein concentration after incubation with NTA agarose alone, suggesting that the majority of plasma protein were binding to the nickel and not to the NTA agarose support [Figure S2]. The capacity of the Ni-NTA agarose beads was not exceeded in these experiments as the flow through (depleted) fraction contained little or no Hb [Figure S3]. To investigate further, the proteins bound to the Ni-NTA beads were eluted and resolved by 1D SDS-PAGE and as shown in Figure 1, contained abundant high and low molecular weight proteins (15- >250 kDa) including alpha, beta and delta subunits of hemoglobin. Proteins from gel bands [Figure S1] representing the entire range of proteins bound to Ni-NTA agarose and identified by MS are shown in Figure 1 and summarized in Table 1. A detailed list of these proteins is provided in Supplemental Table 1. Several of the proteins identified have known nickel binding ability (haptoglobin, α2-macroglobulin, and complement C3) [9].
Figure 1.
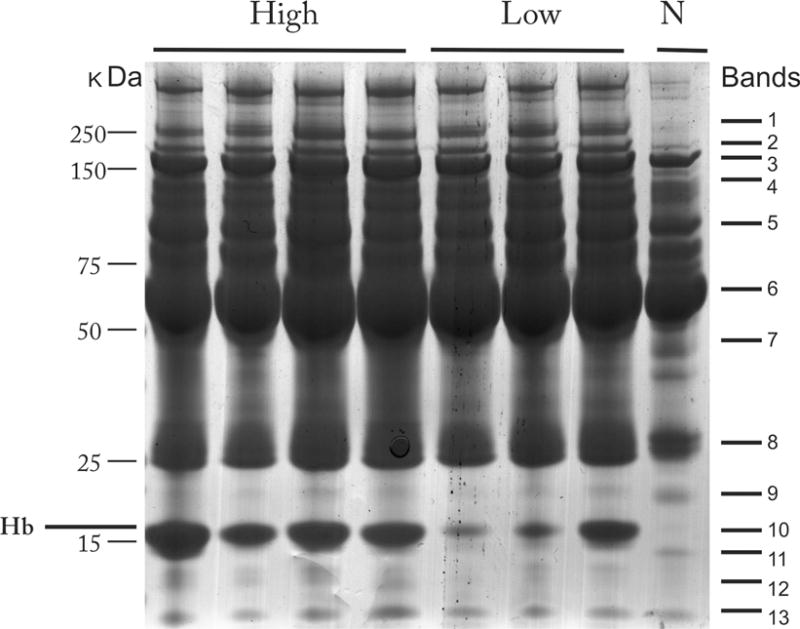
Coomassie-stained 1D SDS-PAGE of plasma proteins bound to Ni-NTA from individuals with sickle cell disease and hemolysis or normal control. High= increased hemolysis (average reticulocyte count 16%; average bilirubin 7.6 mg/dL); low = low hemolysis (avg. reticulocyte count 4.6%; average bilirubin 2.4 mg/dL); N= no hemolysis. Molecular weight markers (kDa) are indicated on left side and representative bands isolated for mass spectrometry identifications indicated on the right.
Table 1.
Representative proteins identified in the Ni-NTA bound plasma proteins by LTQ-Orbitrap MS. Average unique peptides = average of all peptides identified. A complete list of proteins identified in each of the excised gel bands are provided in the supplemental table online.
| Protein Name | Accession # | Avg Unique Peptides | Log(e) | 1D gel Band # |
|---|---|---|---|---|
| Apolipoprotein B-100 | Q9U | 73 | −201.53 | 1 |
| Desmin | P17661 | 26 | −55.72 | |
| Vimentin | P08670 | 4 | −13.98 | |
| Fibronectin | P02751 | 16 | −123.50 | 2 |
| Keratin, type I cytoskeletal 9 | P35527 | 8 | −9.67 | |
| Non-muscle myosin heavy chain | Q99529 | 2 | −7.30 | |
| Alpha-2-macroglobulin | Q9BQ22 | 75 | −139.26 | 3 |
| Ceruplasmin | Q6NSB2 | 3 | −4.50 | |
| Rhotekin | Q9BST9 | 2 | −5.90 | |
| Complement component C3 | Q6LDJ0 | 33 | −30.30 | 4 |
| Kininogen-1 | P01042 | 6 | −8.22 | |
| Fibrinogen alpha chain | P02671 | 2 | −2.96 | |
| ATP synthase subunit alpha | P25705 | 13 | −25.65 | 5 |
| Plasminogen | Q9UMI2 | 4 | −17.00 | |
| Complement component 7 | Q05CI3 | 3 | −12.20 | |
| Serum Albumin | Q56G89 | 42 | −119.84 | 6 |
| Vitamin D-binding protein | P02774 | 3 | −2.57 | |
| Serotransferrin | P02787 | 16 | −44.60 | 7 |
| Clusterin | P10909 | 2 | −7.70 | |
| Apolipoprotein AI | Q6LEJ8 | 15 | −14.41 | 8 |
| Serum amyloid P-component | P02743 | 2 | −9.10 | |
| Haptoglobin | Q14552 | 7 | −25.00 | 9 |
| Complement component C8 gamma chain | P07360 | 2 | −10.50 | |
| Transthyretin | P02766 | 9 | −21.20 | 10 |
| Hemoglobin alpha 1 globin chain | Q9BX83 | 7 | −6.44 | |
| Complement factor B (cDNA FLJ55673) | B4E1Z4 | 10 | −17.53 | 11 |
| Protein S100-A8 | P05109 | 3 | −14.00 | 12 |
| Apolipoprotein A-II | P02652 | 2 | −6.40 | 13 |
To quantify binding of non-hemoglobin proteins to Ni-NTA agarose and the NTA agarose control, we determined the concentrations of thrombospondin 1 (TSP1) and L-selectin before (naïve plasma) and after Ni-NTA agarose and NTA agarose chromatography, as well as in the 0.5M imidazole eluate. These proteins represent mid and lower abundant proteins known to be involved in SCD. TSP1 is an extracellular matrix glycoprotein [10,11] that has been shown to promote the adherence of sickle erythrocytes to the vascular endothelium [12]. L-selectin is an adhesion molecule that mediates leukocyte interaction with the vascular endothelium [13]. Assays for TSP1 and L-selectin were performed according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN) and all samples were assayed in duplicate. To account for dilution of the Hb-depleted sample, TSP and L-selectin ELISA values were adjusted using a correction factor of 1.25.
As shown in Figure 2, plasma TSP-1 and L-selectin levels were decreased after Ni-NTA incubation. This decrease was more substantial in samples with high hemolysis (n=3) when compared to the low hemolysis group (n=3) (54% vs. 42% for TSP1; 46% vs. 28% for L-selectin). For samples with high hemolysis, 35% of the total plasma TSP-1 and 28% of the total plasma L-selectin were eluted off of the Ni-NTA beads, compared to 30% of the total plasma TSP-1 and 22% of the L-selectin for the low hemolysis group. Less than 15% of TSP-1 and L-selectin were eluted off the agarose beads for the high and low hemolysis groups. These findings suggest that in samples with high levels of free Hb, especially those with high levels of hemolysis, there is potential for depletion of potential biomarkers to levels below the threshold of detection. In fact, Ringrose et. al. [3] demonstrated in their study that at least one of the 20 proteins found in their Ni-NTA fraction was not identified in their Hb-depleted erythrocyte protein fraction. As several clinically relevant biomarkers are found in the ng/mL or lower range by ELISA, even prior to depletion, the loss of a broad range of proteins to a range lower than ng/mL by Ni-NTA may make them undetectable by mass spectrometry [14]. With further depletion of other high abundant plasma proteins (e.g. albumin) using antibody columns, which are often used in clinical biomarker discovery studies, current technologies may be further challenged to make these protein identifications.
Figure 2.
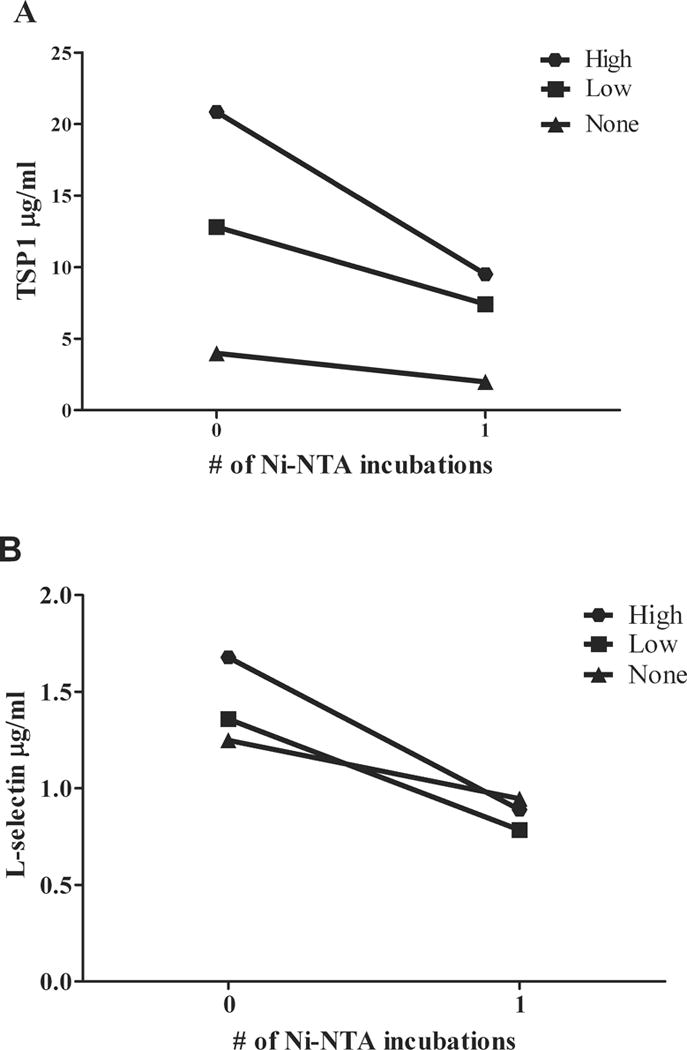
Ni-NTA decreased plasma concentrations of TSP1 (A) and L-selectin (B). A. Average plasma TSP1concentrations before and after depletion were 20.8 and 10.6 μg/mL for patients with high hemolysis (n=3), 12.8 and 8.3 μg/mL for low hemolysis (n=3) and 4.0 and 2.3 μg/mL for no hemolysis (n=2), respectively. B. Average Plasma L-selectin concentrations before and after depletion were 1.7 and 1.0 μg/mL for patients with high hemolysis (n=3), 1.4 and 1.1 μg/mL for low hemolysis (n=3) and 1.2 and 1.1 μg/mL for no hemolysis, respectively (n=2).
Many proteomic pipelines for biomarker discovery are based on differential protein concentration. The use of spectral counting (SC), defined as the total number of spectra identified for a protein, is a commonly used practical approach for label-free, quantitative proteomics [15]. SC correlate with absolute protein abundance and are therefore used to rank proteins by abundance estimates [16]. The predictive value of SC is greatest when mean values for SC are greater than 3. The use of Ni-NTA as a depletion strategy invalidates spectral counting, and alternative MS methods such as iTRAQ [17], as a means for quantitative proteomics because of the significant loss of both high and low abundant proteins. As a result, the use of complementary quantitative techniques may be required [18].
In conclusion, free Hb is a major plasma contaminate in SCD and Hb depletion may be necessary for successful detection and identification of low-abundance biomarkers. While Ni-NTA effectively depletes Hb, it must be used cautiously as it also binds low and high abundance proteins that may have clinical and biological relevance for biomarker discovery. This can add complexity to assigning importance to differentially detected proteins for validation by spectral counting. Despite these limitations, this method is a practical approach for depleting Hb from plasma of children with hemolytic processes. Ni-NTA is commercially available, can be used in very small volumes, and affords flexibility with the amount of Hb that is depleted by changing the ratio of beads to solution; furthermore, this method may avoid the protein dilution that results from affinity chromatography techniques, which is important for the volume restrictions imposed by partitioning systems for protein enrichment. There are other metals with affinity for Hb, such as cobalt, but the risk for nonselective protein binding is still relevant since a number of proteins have metal-binding capabilities [9]. Our study demonstrates important considerations for experimental design and implementation of clinical proteomic studies involving patient samples with increased free Hb. Further studies to explore additional depletion techniques in this patient population should be considered.
Supplementary Material
Figure S1. Location of Ni-NTA bound proteins 1D-GEL bands excised for MS/MS identification. Twenty-one gel bands were excised, trypsin digested and analyzed using LTQ-Orbitrap MS.
Figure S2. Coomassie-stained 1D SDS-PAGE of plasma proteins bound to NiNTA agarose and NTA agarose. I = proteins bound to NiNTA agarose. II = proteins bound to NTA agarose. III= NTA agarose non-bound proteins.
Figure S3. Coomassie-stained 1D SDS-PAGE of Ni-NTA non-bound plasma proteins from individuals with sickle cell disease and hemolysis or normal control. High= increased hemolysis (average reticulocyte count 16%; average bilirubin 7.6 mg/dL); low = low hemolysis (avg. reticulocyte count 4.5%; average bilirubin 2.2 mg/dL); N= no hemolysis. Molecular height markers are indicated on left side of gel (KDa).
Statement of clinical relevance.
The high abundant free hemoglobin found in the plasma of patients with sickle cell and other diseases, as well as with in vitro hemolysis, represents a pre-analytic problem that can potentially limit the effectiveness of biomarker discovery. While hemoglobin depletion has been shown to increase the detection and identification of low abundance proteins, the consequences of using nickel beads for this depletion technique has not yet been explored in clinical proteomic studies. We report on the depletion of heme and non-heme proteins by nickel beads, including potential candidate biomarkers, and demonstrate the potential limitations in using this technique as determined by ELISA or mass spectrometry based quantitation (e.g. spectral counting) of differentially expressed proteins. This finding provides new considerations for experimental design of future proteomic studies and biomarker discovery in hemolytic disorders.
Acknowledgments
The authors would like to thank Michael Debaun, MD, MPH for his thoughtful comments during the inception and preparation of this report. This study was supported by award numbers U54HL090515 and 5R01HL091759 (AE and JFC) from the National Heart, Lung and Blood Institute (NHLBI) and the Johns Hopkins ITCR/CTSA Biomarker Development Center funded in part by National Institutes of Health (NIH) grant U54RR023561 (JVE). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
Abbreviations
- Hb
hemoglobin
- IPI
International Protein Index
- MS
mass spectrometry
- NHLBI
National Heart, Lung and Blood Institute
- NIH
National Institutes of Health
- Ni-NTA
nickel-nitrilotriacetic acid
- SC
spectral count(s)
- SCD
sickle cell disease
- SCI
silent cerebral infarcts
- TSP1
thrombospondin 1
Footnotes
The authors have declared no conflict of interest.
References
- 1.Lezcano NE, Odo N, Kutlar A, Brambilla D, et al. Regular transfusion lowers plasma free hemoglobin in children with sickle-cell disease at risk for stroke. Stroke. 2006;37:1424–6. doi: 10.1161/01.STR.0000221173.97108.01. [DOI] [PubMed] [Google Scholar]
- 2.Levine J, Weickert M, Pagratis M, Etter J, et al. Identification of a nickel(II) binding site on hemoglobin which confers susceptibility to oxidative deamination and intramolecular cross-linking. J Biol Chem. 1998;273:13037–13046. doi: 10.1074/jbc.273.21.13037. [DOI] [PubMed] [Google Scholar]
- 3.Ringrose JH, van Solinge WW, Mohammed S, O’Flaherty MC, et al. Highly Efficient Depletion Strategy for the Two Most Abundant Erythrocyte Soluble Proteins Improves Proteome Coverage Dramatically. Journal of Proteome Research. 2008;7:3060–3063. doi: 10.1021/pr8001029. [DOI] [PubMed] [Google Scholar]
- 4.Casella JF, King AA, Barton B, White DA, et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatr Hematol Oncol. 2010;27:69–89. doi: 10.3109/08880010903360367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebastiani P, Nolan VG, Baldwin CT, Abad-Grau MM, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110:2727–35. doi: 10.1182/blood-2007-04-084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–6855. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 7.Arrell DK, Elliott ST, Kane LA, Guo Y, et al. Proteomic analysis of pharmacological preconditioning: novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706–714. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 8.Vizcaíno JA, Côté R, Reisinger F, Foster JM, et al. A guide to the Proteomics Identifications Database proteomics data repository. Proteomics. 2009;9:4276–83. doi: 10.1002/pmic.200900402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porath J, Olin B. Immobilized metal ion affinity adsorption and immobilized metal ion affinity chromatography of biomaterials. Serum protein affinities for gel-immobilized iron and nickel ions. Biochemistry. 1983;22:1621–30. doi: 10.1021/bi00276a015. [DOI] [PubMed] [Google Scholar]
- 10.Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signa. 2009 Oct 2; doi: 10.1007/s12079-009-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isenberg JS, Hyodo F, Matsumoto K, Romeo MJ, et al. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide-mediated vascular smooth muscle relaxation. Blood. 2007;109:1945–1952. doi: 10.1182/blood-2006-08-041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugihara K, Sugihara T, Mohandas N, Hebbel RP. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood. 1992;80:2634–2642. [PubMed] [Google Scholar]
- 13.Bargatze RF, Kurk S, Butcher EC, Jutila MA. Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J Exp Med. 1994;180:1785–1792. doi: 10.1084/jem.180.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian WJ, Kaleta DT, Petritis BO, Jiang H, et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol Cell Proteomics. 2008;7:1963–1973. doi: 10.1074/mcp.M800008-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics. 2008;7:2373–2385. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics. 2010;7:39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 17.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Wu WW, Wang G, Baek SJ, Shen RF. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel- or LC-MALDI TOF/TOF. J Proteome Res. 2006;5:651–658. doi: 10.1021/pr050405o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Location of Ni-NTA bound proteins 1D-GEL bands excised for MS/MS identification. Twenty-one gel bands were excised, trypsin digested and analyzed using LTQ-Orbitrap MS.
Figure S2. Coomassie-stained 1D SDS-PAGE of plasma proteins bound to NiNTA agarose and NTA agarose. I = proteins bound to NiNTA agarose. II = proteins bound to NTA agarose. III= NTA agarose non-bound proteins.
Figure S3. Coomassie-stained 1D SDS-PAGE of Ni-NTA non-bound plasma proteins from individuals with sickle cell disease and hemolysis or normal control. High= increased hemolysis (average reticulocyte count 16%; average bilirubin 7.6 mg/dL); low = low hemolysis (avg. reticulocyte count 4.5%; average bilirubin 2.2 mg/dL); N= no hemolysis. Molecular height markers are indicated on left side of gel (KDa).