

Abstract
Stroke in general, and ischemic stroke in particular, are routinely defined using clinical criteria. Incorporating brain imaging and neuropathological findings into an expanded conceptual definition of stroke will result in a vastly increased prevalence of the disease. The resultant category of mixed cerebrovascular disease thus may include subclinical infarct, cerebral white matter disease, and cerebral microbleeds. Subclinical brain infarcts occur five times more frequently than does clinical ischemic stroke. Abnormalities of cerebral white matter are present in more than 95% of the population over the age of 65 years, and magnetic resonance imaging evidence of cerebral microbleeds is found in at least 18% of the population, beginning at the age of 60 years. Pathologic evidence supports at least a partial microvascular origin for cerebral white matter disease and cerebral microbleeds. Emphasizing mixed cerebrovascular disease as a conceptual framework allows for a focus on common underlying mechanisms and new therapeutic strategies.
Keywords: stroke, blood–brain barrier, ischemia, hemorrhage
Introduction
Stroke is typically viewed as a disease entity with obvious clinical consequences, and therapeutic efforts directed at stroke focus on clinical end points. This paper will attempt to address stroke from a broader perspective, focusing on a larger spectrum of cerebrovascular syndromes. It will be argued that expansion of stroke as a disease entity will have substantial therapeutic and mechanistic implications.
A conceptual expansion of stroke can be achieved by incorporation of imaging and to a lesser extent neuropathological analyses, with resultant attention to potential common pathways of the disease. This is in contradistinction to the traditional view of stroke as a relatively heterogeneous disorder with commonality substantially determined by risk factors rather than the disease itself. Note that expansion of a definition of stroke, beyond clinical symptomatology, is entirely consistent with recent attempts to redefine “transient ischemic attack” from a combined clinical and imaging perspective.1
Clinical and subclinical stroke
Extent of stroke prevalence is largely determined by age of population and whether one relies on clinical or radiological criteria. For example, most recent data from American Heart Association show prevalence of less than 1% for clinical stroke in age group of 20–39 years, 1–3% for ages 40–59 years, 7–8% for ages 60–79 years, and 13–15% for ages 80 years and older.2 These numbers exhibit the usual and widely accepted age-dependent prevalence, culminating in stroke prevalence that is substantial but not necessarily overwhelming.
However, when one extends criteria for stroke prevalence to include subclinical or silent stroke, prevalence becomes far more extensive. Using brain magnetic resonance imaging (MRI) in a population-based investigation of more than 1,000 subjects between the age of 60 and 90 years, silent brain infarcts were found to be five times more frequent compared to symptomatic infarcts.3 Prevalence of silent infarcts ranged from 8% in the age range of 60–64 years to 35% in the 85–90-year-old group (Fig. 1).
Figure 1.
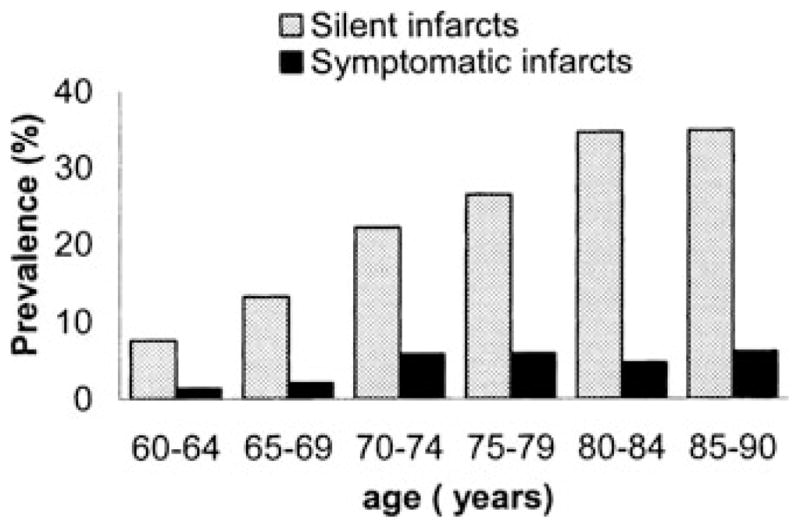
Prevalence (%) of silent and symptomatic infarcts visible on magnetic resonance imaging (MRI) per 5-year age category (from Vermeer et al.3 with permission).
Cerebral white matter disease
Radiology reports for brain MRI performed on older individuals will regularly describe “chronic microvascular ischemia” or variations thereof. These findings reflect what is commonly referred to as “cerebral white matter disease” or “leukoaraiosis.”4 The MRI findings are ubiquitous in an older population, with more than 95% of individuals age 65 years and older exhibiting at least some white matter changes5 (Fig. 2). This prevalence, it must be stressed, is for individuals with no clinical history of cerebrovascular disease. It would seem that the dividing line between “disease” (as in cerebral white matter disease) and “normal brain aging” is uncertain. Most of the white matter changes are minor, but approximately one third appear substantial and more extensive findings are associated with deterioration in cognitive function and gait.5 The underlying pathological substrate of cerebral white matter disease has been surprisingly underinvestigated, but the consensus view has been that it represents a variant of cerebrovascular disease.
Figure 2.

Distribution of white matter grades for 3,301 subjects without history of stroke or transient ischemic attack (TIA), undergoing magnetic resonance imaging (MRI). Grade 0 represented studies with no white matter changes, whereas Grade 9 represented the most extensive findings (from Longstreth et al.5 with permission).
Cerebral microbleeds
Cerebral microbleeds are another entity demonstrable by MRI and understood to represent small or microscopic subclinical intracerebral hemorrhage. The microbleeds are hemosiderin deposits typically located in cortex and in the deep subcortical hemispheres and in a population-based study have been found in 18% of individuals age 60–69 years and 38% of individuals over the age of 80 years.6 If one goes beyond MRI imaging to neuropathological analysis, microscopic microbleeds in the deep subcortical hemispheres are ubiquitous in postmortem brain specimens of individuals over the age of 70 years.7 Cerebral microbleeds on MRI are clearly associated with hypertension and cerebral amyloid angiopathy and are most frequently located at lobar sites.6 The current consensus view is that the cortical microbleeds of MRI represent cerebral amyloid angiopathy, whereas subcortical microbleeds represent the consequence of chronic hypertension.6
Mechanistic considerations
Commonality of stroke has traditionally focused on similarity of risk factors and, of course, the common pathological features of ischemic infarction for large-vessel, cardiogenic, and small-vessel stroke. There is considerable evidence, however, that commonality goes well beyond these observations. Specific relationships may focus on small vessel stroke–subclinical stroke–cerebral white matter disease and cerebral white matter disease–cerebral microbleeds. Basic pathological features of cerebral white matter disease have been worked out in a less-than-definitive fashion. Important questions remain: Is cerebral white matter disease a stroke syndrome? Or perhaps more precisely, is cerebral white matter disease a cerebrovascular syndrome? As it turns out, there is now evidence for both.
Cerebral white matter disease is typically defined by MRI and has traditionally been viewed as distinct from small-vessel infarction (both clinical and subclinical). That distinction has been based on the presumption that infarction from small-vessel disease (sometimes referred to as “lacunar infarction”) creates cavitary lesions that are routinely distinguishable from cerebral white matter disease. Recent evidence demonstrates that this distinction is largely incorrect.8 Careful analysis of 90 patients presenting with acute small-vessel infarction showed that definite cavitary lesions developed in only 20% of patients. For the remaining patients, it appeared likely that the infarcts were incorporated into cerebral white matter disease, and it has been suggested that prevalence of small-vessel stroke may be vastly underestimated by brain imaging.8 In other words, small deep infarcts and cerebral white matter disease may be substantially indistinguishable by brain imaging.
Pathological studies of cerebral white matter disease have been relatively infrequent, surprisingly so given the prevalence of the disorder. Recent detailed examination of 43 postmortem brain samples has yielded some new insights.9 There was no significant correlation between degradation of white matter fibers and extent of in vivo MRI white matter changes. The only pathological predictor of the MRI findings was vascular integrity (measured by CD31-positive staining), with a significant inverse relationship.9 For postmortem MRI, there was no significant association between myelin pallor and MRI lesions. However, vascular integrity was again the only pathological variable with a significant predictive value, with an inverse relationship.9 Moreover, staining for blood–brain barrier efflux transporter P-glycoprotein showed reduced expression of P-glycoprotein in white matter with lesions.9 These finding emphasize the vascular nature of cerebral white matter disease and imply that enhanced permeability at the blood–brain barrier is an important underlying mechanism.
Another line of investigation of cerebral white matter disease has emphasized an inflammatory component. Initial focus has been on Binswanger’s disease, which may be considered a severe and progressive form of white matter disease incorporating lacunar infarcts and presenting clinically as vascular dementia.10 An inflammatory component of Binswanger’s disease was demonstrated by activated microglia, increased perivascular lymphocytes, and macrophage clusters.10 Later work showed increased expression of a variety of matrix metalloproteinases linked to microglial activation,11 with increased levels of matrix metalloproteinase-9 found in cerebrospinal fluid.12,13 It remains to be seen how these findings linking inflammation, matrix metalloproteinase activation, and blood–brain barrier alteration in Binswanger’s disease and vascular dementia relate to the more common entity of cerebral white matter disease.
Cerebral white matter diseases and cerebral microbleeds are probably the most common form of subclinical cerebrovascular disease. An important question is whether and how these two entities may be related. This question has been most extensively studied in a population of patients with cerebral amyloid angiopathy.14 In this group of 26 patients studied with sequential brain MRI over the course of approximately 1 year, progression of cerebral white matter disease and cerebral microbleeds was compared. Not surprisingly, baseline white matter disease was highly correlated with progressive white matter disease. Interestingly, the correlation between baseline white matter disease and progression of cerebral microbleeds was nearly as high.14 The obvious implication is that in the presence of cerebral amyloid angiopathy, there are likely common mechanisms for both cerebral white matter disease and cerebral microbleeds. Whether this reflects common mechanisms of the two entities in the absence of cerebral amyloid angiopathy is a question that has not yet been addressed.
Recent pathological evidence indicates that microscopic cerebral microbleeds frequently occur at the capillary level,7 suggesting changes at the level of the blood–brain barrier. At the pathological level, there is also evidence that cerebral microbleeds can occur in the aging brain in the absence of both hypertension and deposition of beta-amyloid at site of microbleeds.7 These findings are consistent with a conceptual model in which the aging vasculature is the primary substrate for cerebral microbleeds, with the presence of amyloid angiopathy and/or hypertension acting to potentiate a common underlying pathophysiological process. Additional events such as transient blood–brain barrier injury may allow for brief extravasation of red blood cells from vascular lumen into the vessel wall or into parenchyma; the latter would constitute microscopic microbleeds.
Mixed cerebrovascular disease
We can define “mixed cerebrovascular disease” to encompass stroke clinical and subclinical, ischemic, and hemorrhagic. The current target of stroke prevention trials is almost invariably clinical ischemic stroke. But there is an alternative approach, one that integrates clinical, subclinical, ischemic, and hemorrhagic elements. A minimal definition of mixed cerebrovascular disease will include clinical ischemic stroke and cerebral microbleeds, thus combining clinical and subclinical disease with both ischemic and hemorrhagic elements. A more expansive definition will include subclinical ischemic stroke and cerebral white matter disease. It would appear logical that future stroke trials expand their endpoints, integrating the clinical and subclinical. Of the various elements of mixed cerebrovascular disease, cerebral microbleeds and cerebral white matter disease appear particularly relevant and compelling.
Given that approximately 20–40% of all patients aged 60 years and older will have cerebral microbleeds demonstrable by MRI,6 efforts at ischemic stroke prevention require attention to prevention of subclinical hemorrhagic stroke. The question becomes the following: How to prevent ischemic stroke while simultaneously avoiding hemorrhagic stroke clinically (i.e., intracerebral hemorrhage) or subclinically (i.e., microbleeds)? Although intracerebral hemorrhage is a typical adverse clinical outcome for stroke trials, microbleed progression has been largely ignored.
One can also make a good argument that cerebral white matter disease be included as an endpoint in stroke trials. This is based on evidence that there is a strong vascular element in the pathology of cerebral white matter disease,9 presence of stroke risk factors accentuate cerebral white matter disease,4,5 and focal infarcts may be seamlessly incorporated into MRI findings of apparent cerebral white matter disease.8 The prevalence of cerebral white matter disease in the stroke-prone age group and its variegated relationship to ischemic stroke provide, it would seem, sufficient reason to include it in mixed cerebrovascular disease.
A shift from the usual dimensions of stroke to the broader definition of mixed cerebrovascular disease leads to a strikingly different set of investigative priorities. Even a minimal definition of mixed cerebrovascular disease, focusing on clinical ischemic stroke and subclinical cerebral microbleeds, results in a more nuanced therapeutic focus. For example, an agent’s platelet effects may need to be combined with vessel wall protecting properties. Development of more potent platelet agents will not be advantageous unless these agents also have vascular protective effects that mitigate any proclivity to enhance cerebral microbleeds. A therapeutic focus such as this goes well beyond viewing intracerebral hemorrhage as simply an adverse event. Instead, a vascular protective element in treatment of mixed cerebrovascular disease addresses those processes that make intracerebral hemorrhage a future likelihood.
Mixed cerebrovascular disease as a conceptual definition of stroke thus has substantial implications. This definition implicitly encourages a focus on common underlying mechanisms. Emerging evidence indicates microvascular and blood–brain barrier involvement in both cerebral microbleeds and cerebral white matter disease. Moreover, this expansive definition invokes the necessity for therapeutic interventions that have a broad focus, a focus that encompasses both the clinical and subclinical, and the ischemic and hemorrhagic.
Acknowledgments
This work was supported in part by National Institutes of Health grant number RO1 NS20989.
Footnotes
Conflicts of interest
Boehringer–Ingelheim: research grant, speakers’ bureau, and honoraria; Otsuka Pharmaceutical Co.: research grant and honoraria.
References
- 1.Easton JD, Saver JL, Albers GW, et al. Definition and evaluation of transient ischemic attack. Stroke. 2009;40:2276–2293. doi: 10.1161/STROKEAHA.108.192218. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics-2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 3.Vermeer SE, Koudstaal PJ, Oudkerek M, et al. Prevalence and risk factors of silent brain infarcts in the population-based Rotterdam scan study. Stroke. 2002;33:21–25. doi: 10.1161/hs0102.101629. [DOI] [PubMed] [Google Scholar]
- 4.Pantoni L, Basile AM, Pracucci G, et al. Impact of age-related cerebral white matter changes on the transition to disability–the LADIS study: rationale, design, and methodology. Neuroepidemiology. 2005;24:51–62. doi: 10.1159/000081050. [DOI] [PubMed] [Google Scholar]
- 5.Longstreth WT, Manolio TA, Arnold A, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. Stroke. 1996;27:1274–1282. doi: 10.1161/01.str.27.8.1274. [DOI] [PubMed] [Google Scholar]
- 6.Vernooij MW, Van Der Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70:1208–1214. doi: 10.1212/01.wnl.0000307750.41970.d9. [DOI] [PubMed] [Google Scholar]
- 7.Fisher M, French S, Ji P, Kim RC. Cerebral microbleeds in the elderly: a pathological analysis. Stroke. 2010;41:e377. doi: 10.1161/STROKEAHA.110.593657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potter GM, Doubal FN, Jackson CA, et al. Counting cavitating lacunes underestimates the burden of lacunar infarction. Stroke. 2010;41:267–272. doi: 10.1161/STROKEAHA.109.566307. [DOI] [PubMed] [Google Scholar]
- 9.Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804–811. doi: 10.1212/01.wnl.0000319691.50117.54. [DOI] [PubMed] [Google Scholar]
- 10.Akiguchi I, Tomimoto H, Suenaga T, et al. Alterations in glia and axons in the brains of Binswanger’s disease patients. Stroke. 1997;28:1423–1429. doi: 10.1161/01.str.28.7.1423. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg GA, Sullivan N, Esiri MM, Sobel RA. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–1168. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- 12.Adair JC, Charlie J, Dencoff JE, et al. Measurement of gelatinase B (MMP-9) in the cerebrospinal fluid of patients with vascular dementia and Alzheimer’s disease. Stroke. 2004;35:e159–e162. doi: 10.1161/01.STR.0000127420.10990.76. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg GA. Inflammation and white matter damage in vascular cognitive impairment. Stroke. 2009;40(Suppl 1):S20–S23. doi: 10.1161/STROKEAHA.108.533133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YW, Gurol ME, Rosand J, et al. Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology. 2006;67:83–87. doi: 10.1212/01.wnl.0000223613.57229.24. [DOI] [PMC free article] [PubMed] [Google Scholar]