

Abstract
Purpose
Supplementation with the n3 polyunsaturated fatty acid docosahexaenoic acid (DHA) is beneficial in heart failure patients, however the mechanisms are unclear. DHA is incorporated into membrane phospholipids, which may prevent mitochondrial dysfunction. Thus we assessed the effects of DHA supplementation on cardiac mitochondria and the development of heart failure caused by aortic pressure overload.
Methods
Pathological cardiac hypertrophy was generated in rats by thoracic aortic constriction. Animals were fed either a standard diet or were supplemented with DHA (2.3 % of energy intake).
Results
After 14 weeks, heart failure was evident by left ventricular hypertrophy and chamber enlargement compared to shams. Left ventricle fractional shortening was unaffected by DHA treatment in sham animals (44.1±1.6 % vs. 43.5±2.2 % for standard diet and DHA, respectively), and decreased with heart failure in both treatment groups, but to a lesser extent in DHA treated animals (34.9±1.7 %) than with the standard diet (29.7±1.5 %, P <0.03). DHA supplementation increased DHA content in mitochondrial phospholipids and decreased membrane viscosity. Myocardial mitochondrial oxidative capacity was decreased by heart failure and unaffected by DHA. DHA treatment enhanced Ca2+ uptake by subsarcolemmal mitochondria in both sham and heart failure groups. Further, DHA lessened Ca2+-induced mitochondria swelling, an index of permeability transition, in heart failure animals. Heart failure increased hydrogen peroxide-induced mitochondrial permeability transition compared to sham, which was partially attenuated in interfibrillar mitochondria by treatment with DHA.
Conclusions
DHA decreased mitochondrial membrane viscosity and accelerated Ca2+ uptake, and attenuated susceptibility to mitochondrial permeability transition and development of left ventricular dysfunction.
Keywords: Cardiac failure, Metabolism, Polyunsaturated fatty acids, Reactive oxygen species
Introduction
Chronic heart failure remains a major cause of hospitalization and death despite aggressive treatment with current therapies, and new approaches are needed. Recent studies found that pharmacological doses of marine n3-polyunsaturated fatty acids (PUFA) comprised of docosahexaenoic acid (DHA; 22:6n3) or eicosapentaenoic acid (EPA: 20:5n3) can improve left ventricular function and reduce clinical events in heart failure patients [1–4]. While the underlying mechanisms for clinical benefit are likely multifactorial [5, 6], recent evidence suggest that favorable effects of n3-PUFA in heart failure may be partially due to changes in mitochondrial structure and function [7–11].
We recently found that dietary supplementation with DHA at a clinically relevant dose increases DHA incorporation into mitochondrial membrane phospholipids and decreases the susceptibility of isolated cardiac mitochondria to undergo mitochondrial permeability transition (MPT) induced by Ca2+ stress [7–10, 12]. MPT is a catastrophic event that is triggered by exposure to high Ca2+ or reactive oxygen species (ROS), resulting in the collapse of the mitochondrial inner membrane potential, matrix swelling, loss of ATP production, and release of matrix proteins that trigger cell death [13–15]. While it is now well established that DHA supplementation decreases susceptibility to Ca2+-induced MPT, the ability of DHA supplementation to enhance resistance to ROS-induced MPT has not been reported. The mechanism(s) for greater resistance to Ca2+-induced MPT following DHA supplementation is not clear, but could be due to decreased membrane viscosity and greater ease of movement of membrane proteins [9, 16]. Pepe et al. found that increasing DHA in mitochondrial phospholipids with dietary fish oil did not affect mitochondrial Ca2+ content, but attenuated the acute increase in mitochondrial Ca2+ following norepinephrine stimulation in isolated perfused hearts from normal rats [17]. This suggests that an increase in mitochondrial membrane DHA content may slow Ca2+ uptake in response to an extra-mitochondrial Ca2+ load, though this has not been directly assessed in isolated mitochondria.
Interventions that enhance resistance to stress-induced MPT in the heart do not necessarily translate into resistance to the development of heart failure in response to cardiac injury or pressure overload [8, 12, 18, 19]. While our previous studies showed that dietary supplementation with purified DHA at levels similar to those use in human studies improved resistance to Ca2+ induced MPT, we did not observe improved left ventricular function and/or survival in rodents with genetic or infarct-induced heart failure [10, 12] or with mild LV hypertrophy induced by constriction of the abdominal aorta [8]. On the other hand, DHA supplementation improved cardiac function in response to moderate pressure overload in studies by us and others [20–25]. Severe hypertensioninduced LVH results in mitochondrial pathology, as seen in an impaired capacity for oxidative phosphorylation [11, 26, 27]. While studies in genetic and infarct-induced models found that heart failure increased susceptibility to MPT induced by cell stress [12, 28, 29], to our knowledge this has not been assessed in models of pressure overload-induced heart failure. Further, while we have demonstrated that DHA supplementation delays Ca2+-induced MPT, the effects on ROS-induced MPT have not been reported.
Based on this background, the present study addressed the following unanswered questions: 1) does heart failure induced by aortic pressure overload and cardiac hypertrophy increase susceptibility to ROS-induced MPT? 2) Does DHA supplementation prevent this effect? 3) Does the increase in DHA in mitochondrial phospholipids following DHA supplementation decrease membrane viscosity? 4) Does DHA alter the rate of Ca2+ uptake by mitochondria in the normal and failing heart? and 5) Does DHA prevent LV remodeling and contractile dysfunction in response to severe aortic pressure overload? We hypothesized that heart failure induced by pressure overload would increase susceptibility to MPT, and that dietary supplementation with DHA would decrease membrane viscosity and accelerate mitochondrial Ca2+ uptake, which would be associated with greater resistance to stress-induced MPT and attenuation of LV dysfunction. Studies were performed in a well characterized rat model of heart failure caused by constriction of the transverse aorta to generate in LVH, LV chamber expansion, and mitochondrial and contractile dysfunction [11, 27, 30].
Material and Methods
Experimental Design
The effects of DHA were assessed in rats subjected to either sham or transverse aortic constriction in a 2×2 design comparing surgical and dietary treatments. Surgery was performed in young rats and followed by assignment to treatment with either a standard diet (15 sham rats and 22 with aortic constriction) or with DHA supplementation (14 sham rats and 22 with aortic constriction). The animal protocol was approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee and conducted according to the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health publication 85–23). The investigators were blinded to treatment when measurements were performed.
Surgery was performed in 6–8 week old (70–100 g) male Sprague Dawley rats (Harlan, Indianapolis, IN) to constrict the transverse thoracic aorta and cause cardiac hypertrophy and heart failure. Rats were placed in a chamber with 5 % isoflurane to induce anesthesia, intubated, mechanically ventilated and maintained on isoflurane (1.5–2.5 % to effect). The aortic arch was exposed and a tantalum clip (0.50 mm internal diameter; Pilling-Weck, Germany) was positioned around the transverse aorta between the brachiocephalic trunk and the left common carotid artery, as described previously [11, 31, 32]. Sham animals underwent the same procedure but without clip application. The incision was sutured closed and the animal was maintained for 14 weeks.
Three days following surgery, rats were assigned to one of two custom manufactured diets made with purified ingredients (Research Diets, New Brunswick, NJ, USA). They each contained 20 % of energy from protein (casein + L-cystine), 68 % of energy from carbohydrate sources (maltodextrin, 13 % of total energy and corn starch, 55 %) and 12 % of energy from fat. They were matched for the content of cellulose (50 g/kg), and vitamins and minerals. The fat source in the standard diet was a mixture of lard, cocoa butter and soybean oil that was free of DHA, and the DHA diet had a similar fat mixture at 9.7 % of total fat plus the addition of DHA ethylester oil (90 % purity, KD Pharma, Bexbach, Germany) at 2.3 % of energy intake. This dose of DHA corresponds to a human does of approximately 5 g/day (assuming 9 kcal/g DHA oil and a human energy intake of 2,000 kcals/day).
Echocardiography
LV function was evaluated by echocardiography using a high-resolution small animal imaging system (Vevo 770 with transducer model RMV 716, VisualSonics Inc., Toronto, Canada). Rats were anesthetized with isoflurane (induced in a chamber with 1.5 % isoflurane and maintained on by mask), the chest shaved, and the animal situated in the supine position on a warming platform. M-mode and two-dimensional echocardiographic studies were performed from short axis and end systolic and diastolic diameters where assessed, and end systolic and end diastolic volumes calculated as previously described [21]. Percent fractional shortening was calculated 100 × ((end diastolic diameter − end systolic diameter)/ end diastolic diameter).
Tissue Harvest
The rats were anesthetized with isoflurane (induced on 5 % in a chamber and maintained on 5 % by mask). The chest was opened, blood rapidly was collected by cardiac puncture, and the heart removed, dissected, and weighed. Mitochondria were freshly isolated from LV myocardium. A piece of LV tissue for measurement of the activity of mitochondrial enzymes was frozen in liquid nitrogen and stored at −80 °C until analyzed.
Mitochondria Isolation
The two spatially distinct subpopulations of cardiac mitochondria, subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM), were isolated as described previously by Palmer et al. [33] with modifications [8, 10]. LV tissue was minced and homogenized in 1:10 cold modified Chappel-Perry buffer [100 mM KCl, 50 mM MOPS, 5 mM MgSO4, 1 mM EGTA, 1 mM ATP, 0.2 mg/ml bovine serum albumin (BSA)]. Homogenates were centrifuged at 500 x g and supernatant yielded SSM. IFM were extracted through tryptic digestion (5 mg/g wet weight) for 10 min on ice, and further purified through a series of centrifugation steps. Mitochondrial protein was assessed by the Lowry method using BSA as a standard.
Mitochondrial Respiration
Mitochondrial oxygen consumption was measured in SSM and IFM as described previously [8, 10]. Isolated mitochondria (0.5 mg mitochondrial protein/ml) were respired in buffer containing 100 mM KCl, 50 mM MOPS, 5 mM KH2PO4, 1 mM EGTA, and 1 mg/ml BSA. State 3 and 4 respiration was measured utilizing glutamate + malate (10 mM and 5 mM), palmitoylcarnitine (10 mM), and succinate with rotenone (10 mM and 7.5 μM). The respiratory exchange ratio (RCR) was calculated as state 3/state 4.
Ca2+ Uptake and ROS-induced MPT
Ca2+ uptake and tert-butyl hydrogen peroxide (tBH) induced Ca2+ release were assessed in isolated SSM and IFM as previously described [34]. In short, mitochondria (1.5 mg mitochondrial protein) were resuspended in 1.5 mL of Ca2+-free buffer containing 100 mM KCl, 50 mM MOPS, 5 mM KH2PO4, 5 μM EGTA, 1 mM MgCl2, 5 mM glutamate, and 5 mM malate. Mitochondrial Ca2+ uptake was measured in a fluorescence spectrophotometer at 37 °C from the fluorescence of the Ca2+ indicator calcium green-5N (CaGN-5N; Molecular Probes) with an excitation and emission of 488 and 530 nm. Ca2+ uptake was taken from the fall in extramitochondrial Ca2+ following a bolus injection of 3 μL of 15 mM Ca2+ (30 nmoles Ca2+/mg mitochondrial protein). After stabilization of Ca2+ uptake (6 to 8 min), a continuous infusion of tBH (400 mM) was initiated into the cuvette at a rate of 0.2 μL/min (53 nmols tBH·mg mitochondrial protein 1·min 1), and the concentration of free Ca2+ in the medium was monitored. Mitochondria were also monitored without infusion of tBH or Ca2+, which established that MPT did not occur without addition of tBH or Ca2+ (data not shown).
Ca2+ Induced Mitochondrial Swelling
Ca2+-induced mitochondrial swelling, an established measure of MPT, was monitored at 540 nm using a 96 well spectrophotometic plate reader (SpectraMax, Molecular Devices, USA) at 37 °C as previously described [7]. Briefly, 50 μg of mitochondrial protein in 200 μL of Ca2+ free buffer was monitored for 2 min to obtain a baseline, then 100 nmoles Ca2+/mg mitochondrial protein was added and the absorbance monitored for 15 min.
Membrane Viscosity
Mitochondrial membrane viscosity was assessed by measuring anisotropy using fluorescence polarization of 1,6diphenyl-1,3,5-hexatriene (DPH, Invitrogen), which decreases with increased rotation of the dye (i.e. decreased membrane viscosity) [35]. 200μg mitochondrial protein was incubated with 10 μM DPH for 30 min at 37 °C in 3 mL of 46 mM KH2PO4. Anisotropy was measured in black opaque 96-well polystyrene plates at 37 °C with an excitation and emission of 355 and 430 nm, respectively. Results were corrected for background by subtracting measurements from wells containing only buffer. Anisotropy was calculated as:
where r is the anisotropy, I VV and I VH are the fluorescent intensities measured in both the vertical and horizontal channels, and G (gating) was assigned a value of 1.00.
Flow Cytometry Analyses
Mitochondrial size and membrane potential were assessed in SSM and IFM by flow cytometry (BD Biosciences) as previously described [34, 36]. Briefly, isolated SSM and IFM were stained with MitoTracker Green FM (Molecular Probes) and the arithmetic mean output from the forward scatter detector was used as an index of mitochondrial size. Mitochondrial membrane potential was assessed using 5,5′,6,6′-tetrachloro1,1′,3,3′-tetraethylbenzimidazol carbocyanine iodide (JC-1) (Molecular Probes, Carlsbad, CA). Briefly, mitochondria were incubated with 0.3 μM JC-1 for 30 min at 37 °C. Membrane potential was measured as a ratio of orange to green fluorescence. Increased membrane potential causes JC-1 aggregates to form, shifting the fluorescence of emitted light from 530 nm (green) to 590 nm (orange).
Mitochondrial Enzyme Activities
To determine if heart failure and/or DHA affected the total mitochondrial oxidative capacity, the activities of citrate synthase, aconitase, and medium-chain acyl coenzyme A dehydrogenase were measured from myocardial homogenates from the frozen LV using spectrophotometric assays as previously described [10, 37].
Mitochondrial Phospholipid Fatty Acid Composition
The phospholipid fatty acid composition of SSM and IFM were measured using gas chromatography–mass spectrometry using the transesterification method as previously described [9, 12, 38].
Statistical Analysis
Data are presented as mean ± SEM. The effect of TACinduced heart failure was assessed with a 2-way ANOVA, with a Bonferroni post hoc test to determine potential difference among groups. A P <0.05 was taken as significant.
Results
Survival and Cardiac Function
After 14 weeks of treatment there was a similar rate of mortality in both untreated and DHA treated groups for sham animals (15/15 (0 %) and 14/14(0 %) for sham standard diet and DHA, respectively, and 20/22 (10 %) and 20/22 (10 %) for heart failure standard diet and DHA, respectively). There were no significant differences in body mass (Table 1). Transverse aortic constriction caused cardiac hypertrophy as evident by a significant increase in the mass of the LV, right ventricle and atria compared to sham animals (Table 1, Fig. 1). Treatment with DHA had no effect on cardiac mass in either sham or aortic constricted animals. Heart failure was evident, as seen in LV chamber enlargement at both end systole and end diastole, and a significant fall in LV fractional shortening and ejection fraction in both treatment groups (Table 1, Fig. 1). LV fractional shortening and ejection fraction were unaffected by DHA treatment in sham animals. In heart failure treatment with DHA significantly attenuated the decline in left ventricular fractional shortening and ejection fraction compared to the standard diet, suggesting that DHA has a modest but significant protective effect (Table 1, Fig. 1).
Table 1.
Body and cardiac masses, and echocardiographic results 14 weeks after surgery
| Untreated |
DHA treated |
|||
|---|---|---|---|---|
| Sham | Heart failure | Sham | Heart failure | |
| Body mass (g) | 411±8 | 390±7 | 437±5 | 403±7 |
| Atria mass (mg) | 57±5 | 210±13* | 61±4 | 189±14* |
| RV mass (mg) | 198±7 | 413±12* | 225±7 | 374±19* |
| LV mass (mg) | 876±18 | 1,282±34* | 943±19 | 1,298±39* |
| Echocardiography: | ||||
| Heart rate (bpm) | 359±7 | 342±5 | 349±12 | 337±5 |
| End Diastolic Diameter (mm) | 7.5±0.2 | 8.3±0.2# | 7.7±0.2 | 8.4±0.2# |
| End Systolic Diameter (mm) | 4.3±0.2 | 5.9±0.2* | 4.3±0.2 | 5.4±0.2* |
| End Diastolic Volume (μL) | 494±47 | 641±42* | 470±55 | 625±47# |
| End Systolic Volume (μL) | 90±20 | 228±17* | 96±22 | 183±19# |
| Ejection Fraction (%) | 81.9±2.1 | 64.2±1.8* | 81.0±2.3 | 71.5±1.8#† |
P <0.001
P <0.05 compared to sham within the same dietary group,
P <0.01 vs. the untreated heart failure group
Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Fig. 1.
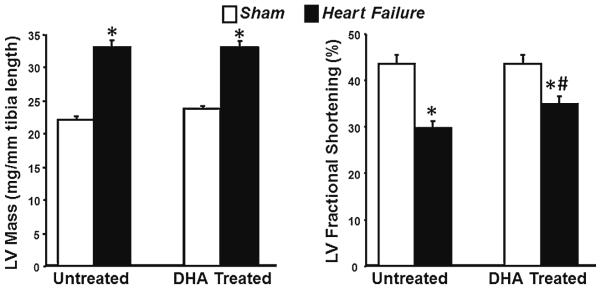
Left ventricular mass and fractional shortening after 14 weeks of treatment. * P <0.05 compared to respective sham. # P <0.05 compared to untreated heart failure group. Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Mitochondrial Function
Heart failure decreased myocardial oxidative capacity, as seen in lower activity of the citric acid cycle enzymes citrate synthase and aconitase, and the fatty acid β-oxidation enzyme medium chain acyl-CoA dehydrogenase in myocardial homogenates from the LV (Fig. 2). Further, mitochondrial yield was lower with heart failure in the standard diet group in SSM and in both dietary groups in IFM (Fig. 2). Thus, heart failure decreased total mitochondrial oxidative capacity in LV myocardium in both treatment groups. Treatment with DHA had no significant effect on any of these parameters (Fig. 2).
Fig. 2.

Top panels : activity of mitochondrial enzymes in whole LV tissue homogenates. Bottom panels: mitochondrial yields for SSM and IFM. *P <0.05 compared to sham animals within the dietary treatment
Maximal respiratory function in isolated mitochondria (state 3) was not impaired by heart failure in either SSM or IFM with any of the substrates tested (Table 2). On the other hand, DHA treatment increased state 3 respiration of SSM in sham rats with palmitoylcarnitine as a substrate, and showed a similar nonsignificant trend in IFM. This response was not observed in heart failure animals treated with DHA, as the state 3 rate in IFM with palmitoylcarnitine was similar to levels observed in the standard diet group (Table 2). DHA had no effect on state 3 respiration with glutamate + malate or succinate as substrates.
Table 2.
Mitochondrial respiratory function after 14 weeks of treatment
| Untreated |
DHA treated |
|||
|---|---|---|---|---|
| Sham | Heart failure | Sham | Heart failure | |
| SSM | ||||
| Glutamate + Malate | ||||
| State 3 | 179±14 | 183±12 | 201±10 | 183±8 |
| State 4 | 30.1±1.7 | 30.5±2.0 | 32.3±2.6 | 29.9±2.9 |
| Respiratory control ratio | 5.9±0.3 | 6.2±0.4 | 6.7±0.6 | 6.7±0.4 |
| ADP:O | 2.26 ±0.09 | 2.15±0.07 | 2.08±0.13 | 2.20±0.08 |
| Palmitoylcarnitine | ||||
| State 3 | 184±13 | 181±8 | 251±11^ | 193±7* |
| State 4 | 50.7±3.7 | 51.5±2.8 | 57.1±4.7 | 49.3±3.9 |
| Respiratory control ratio | 3.8±0.3 | 3.6±0.1 | 5.0±0.7 | 4.2±0.3 |
| ADP:O | 2.12±0.05 | 2.19±0.04 | 2.13±0.05 | 2.28±0.05 |
| Succinate + Rotenone | ||||
| State 3 | 286±15 | 267±15 | 327±13 | 304±10 |
| State 4 | 114±4 | 103±7 | 120±5 | 114±5 |
| Respiratory control ratio | 2.5±0.1 | 2.7±0.1 | 2.7±0.1 | 2.7±0.1 |
| ADP:O | 1.33±0.04 | 1.40±0.11 | 1.28±0.04 | 1.47±0.06 |
| IFM | ||||
| Glutamate + Malate | ||||
| State 3 | 198±15 | 194±12 | 215±9 | 199±10 |
| State 4 | 36.2±2.9 | 42.2±2.3 | 42.1±5.1 | 32.7±2.1# |
| Respiratory control ratio | 5.7±0.4 | 4.7±0.3 | 5.9±0.6 | 6.4±0.4# |
| ADP:O | 2.71±0.21 | 2.57±0.16 | 2.25±0.11 | 2.52±0.08 |
| Palmitoylcarnitine | ||||
| State 3 | 225±17 | 224±9 | 261±10 | 231±10 |
| State 4 | 70.5±5.8 | 76.4±4.3 | 68.2±4.6 | 61.8±3.4# |
| Respiratory control ratio | 3.4±0.3 | 3.0±0.1 | 4.1±0.3^ | 3.8±0.2# |
| ADP:O | 2.27±0.09 | 2.30±0.06 | 2.48±0.12 | 2.47±0.07 |
| Succinate + Rotenone | ||||
| State 3 | 397±15 | 426±20 | 440±17 | 438±13 |
| State 4 | 153±5 | 154±7 | 167±14 | 157±5 |
| Respiratory control ratio | 2.6±0.1 | 2.8±0.1 | 2.7±0.2 | 2.8±0.1 |
| ADP:O | 1.44±0.07 | 1.27±0.04 | 1.43±0.05 | 1.31±0.05 |
P <0.05 Heart failure vs. respective sham.
P <0.05 DHA treatment vs. standard diet in heart failure.
P <0.05 DHA treatment vs. standard diet in shams
Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
State 4 respiration was not different among groups in SSM, but in IFM it was decreased by DHA treatment compared to the standard diet with heart failure when glutamate + malate or palmitoylcarnitine were used as substrates. This corresponded to an increase in the respiratory control ratio (RCR; state 3/state4) in these groups. There were no effects of heart failure or DHA on the ADP:O ratio (Table 2). Combined with the lack of elevation in state 4 respiration, this demonstrates that heart failure did not result in mitochondrial uncoupling.
Mitochondrial Phospholipid Fatty Acid Composition
Analysis of phospholipids in isolated SSM and IFM revealed that heart failure induced changes in mitochondrial phospho-lipid fatty acyl side chain composition, decreasing linoleic acid (18:2n6) in SSM in both treatment groups, and in the standard diet group in IFM compared to their respective shams (Table 3). There was an increase in arachidonic acid in IFM with heart failure in untreated animals (Fig. 3). In SSM, heart failure significantly increased palmitate (16:0) in the standard diet group (Table 3). Further, heart failure caused a decrease in total n6-PUFA and n3-PUFA in the DHA heart failure group compared to the DHA treated sham group in SSM (Table 3).
Table 3.
Fatty acid composition of mitochondrial phospholipids expressed as a percent of total phospholipid fatty acids
| Fatty acid | Untreated |
DHA treated |
||
|---|---|---|---|---|
| Sham | Heart failure | Sham | Heart failure | |
| SSM | ||||
| 14:0 | 0.58±0.11 | 0.65±0.10 | 0.60±0.10 | 0.68±0.07 |
| 16:0 | 25.9±1.7 | 31.9±2.8^ | 27.4±2.1 | 30.8±1.1 |
| 16:1 | 0.32±0.05 | 0.39±0.04 | 1.3±0.7 | 0.31±0.05 |
| 18:0 | 39.8±3.1 | 37.4±2.2 | 31.3±3.7 | 37.2±1.1 |
| 18:1n9 | 5.5±0.3 | 4.5±0.5 | 5.1±0.3 | 6.1±0.4 |
| 18:1n7 | 2.7±0.3 | 3.6±0.4 | 2.4±0.2 | 3.1±0.2 |
| 18:2n6 | 15.5±2.0 | 9.0±1.2& | 17.8±1.1 | 11.3±1.0& |
| 18:3n3 | 0.14±0.01 | 0.12±0.01 | 0.40±0.20 | 0.11±0.01 |
| 20:3n6 | 0.04±0.00 | 0.04±0.01 | 0.24±0.13 | 0.06±0.00 |
| 20:5n3 | 0.01±0.00 | 0.01±0.00 | 0.15±0.02† | 0.13±0.01† |
| 22:5n3 | 0.17±0.02 | 0.16±0.02 | 0.12±0.02† | 0.08±0.01† |
| Σ n6-PUFA | 24.2±3.1 | 18.8±2.2 | 21.4±1.4 | 14.9±1.1^ |
| Σ n3-PUFA | 1.8±0.3 | 1.8±0.2 | 10.3±1.6† | 6.9±0.7†^ |
| Σ MUFA | 8.8±0.5 | 9.4±0.8 | 8.9±0.7 | 9.5±0.4 |
| Σ SFA | 65.3±3.3 | 69.9±2.8 | 59.3±3.4 | 68.6±1.8 |
| IFM | ||||
| 14:0 | 0.52±0.06 | 0.56±0.11 | 0.55±0.06 | 0.45±0.04 |
| 16:0 | 26.3±1.6 | 29.1±1.2^ | 31.1±1.5 | 30.1±1.1 |
| 16:1 | 0.39±0.02 | 0.44±0.04 | 0.33±0.05 | 0.39±00.3 |
| 18:0 | 29.1±2.7 | 30.4±2.1 | 29.1±2.1 | 29.5±1.8 |
| 18:1n9 | 8.5±0.6 | 6.2±0.7 | 6.6±0.7 | 6.8±0.3 |
| 18:1n7 | 3.8±0.6 | 7.0±0.4 | 3.3±0.4 | 4.5±0.3 |
| 18:2n6 | 21.8±3.1 | 12.4±1.4& | 18.1±12.0 | 15.7±1.7 |
| 18:3n3 | 0.04±0.03 | 0.07±0.03 | 0.07±0.02 | 0.07±0.02 |
| 20:3n6 | 0.04±0.01 | 0.05±0.01 | 0.07±0.03 | 0.08±0.01 |
| 20:5n3 | 0.01±0.00 | 0.01±0.02 | 0.13±0.02† | 0.17±0.02† |
| 22:5n3 | 0.20±0.03 | 0.28±0.03 | 0.15±0.03 | 0.11±0.01† |
| Σ n6-PUFA | 29.4±2.9 | 23.5±2.1 | 21.9±2.2* | 20.1±1.8 |
| Σ n3-PUFA | 1.8±0.4 | 2.7±0.3 | 7.0±0.9† | 8.3±0.8† |
| Σ MUFA | 12.7±1.1 | 13.7±1.0 | 10.3±0.8 | 11.7±0.5 |
| Σ SFA | 55.9±4.1 | 60.1±3.3 | 60.7±3.4 | 60.0±2.7 |
P <0.05,
P <0.001 compared to the standard diet within the respective surgical treatment group.
P <0.05,
P <0.005 compared to respective sham within diet group
Groups sizes were 10 and 12 for the untreated sham and heart failure groups, respectfully, and 10 and 12 for the DHA treated sham and heart failure groups, respectfully
Fig. 3.

Phospholipid content of DHA (top panels) and arachidonic acid (bottom panels ) in SSM and IFM expressed as a percent of total fatty acids. Data are mean ± SEM. * P <0.001 compared to the respective untreated group. † P <0.05 compared to untreated sham group
Treatment with DHA increased DHA, EPA and the sum of total n3-PUFA in both IFM and SSM in both sham and heart failure rats (Fig. 3, Table 3). Supplementation with DHA also decreased arachidonic acid (20:4n6) in sham and heart failure animals in both mitochondrial populations (Fig. 3) and the sum of n6-PUFA in IFM (Table 3). There were no differences in the sum of either monounsaturated or saturated fatty acids among groups (Table 3).
Mitochondrial Structure
Mitochondrial size, internal complexity and membrane potential were examined in isolated mitochondria using flow cytometry. Neither heart failure nor DHA significantly altered mitochondrial size, internal complexity or membrane potential in SSM or IFM (Table 4).
Table 4.
Mitochondrial morphology and membrane potential (Δψm) determined by flow cytometry
| Untreated |
DHA treated |
|||
|---|---|---|---|---|
| Sham | Heart failure | Sham | Heart failure | |
| SSM | ||||
| Mitochondrial Diameter (AU) | 506±13 | 511±15 | 493±32 | 512±16 |
| Mitochondrial Internal Complexity (AU) | 108.9±2.2 | 108.7±1.8 | 96.1±5.7 | 106.2±3.5 |
| Δψm (JC-1 aggregate/monomer) | 3.55±0.33 | 4.05±0.37 | 5.06±0.78 | 4.54±0.40 |
| IFM | ||||
| Mitochondrial Diameter (AU) | 328±19 | 324±16 | 323±45 | 317±25 |
| Mitochondrial Internal Complexity (AU) | 80.8±0.4 | 82.0±0.8 | 72.5±5.3 | 79.6±2.4 |
| Δψm (JC-1 aggregate/monomer) | 4.20±0.36 | 3.76±0.35 | 4.31±0.56 | 4.45±0.34 |
Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Mitochondrial Membrane Viscosity
DHA supplementation and heart failure differentially effected membrane viscosity in the two mitochondrial subpopulations. In SSM, there was no significant effect of DHA treatment, however heart failure decreased membrane viscosity (P <0.02 as a main effect vs. sham), as seen in a decrease in anisotropy (Fig. 4). On the other hand, in IFM there was no effect of heart failure, but DHA treatment significantly decreased membrane viscosity compared to the standard diet.
Fig. 4.
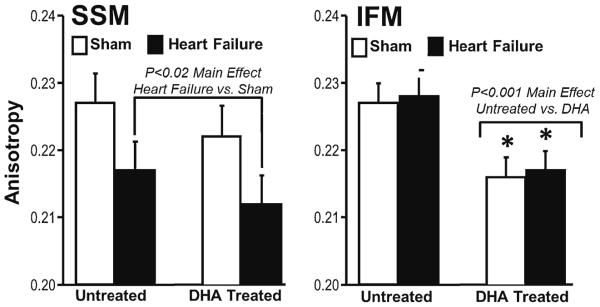
Fluorescence polarization of 1,6 diphenyl 1,3,5 hexatriene in SSM and IFM, which reflects mitochondrial membrane viscosity. Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Ca2+ Induced Mitochondrial Swelling
The ability of isolated mitochondria to resist Ca2+-induced MPT was assessed from the fall in absorbance at 540 nm following a bolus injection of Ca2+, which is an established index of mitochondrial swelling as a result of permeability transition. In SSM, heart failure resulted in a significantly greater decrease in absorbance compared to sham rats fed the standard diet, while treatment with DHA prevented this effect (Fig. 5). In IFM, heart failure did not accelerate the fall in absorbance compared to sham animals on the standard diet, however DHA treatment slowed the fall in absorbance compared to the standard diet heart failure group.
Fig. 5.

Change in absorbance at 540 nm, an index of LV mitochondrial swelling, following addition of 100 nmol Ca2+/mg mitochondrial protein. *P <0.05 for the DHA treated heart failure group compared to untreated heart failure; ^ P <0.05 for untreated sham compared to untreated heart failure. Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Mitochondrial Ca2+ Uptake and tBH-induced Ca2+ Release
Mitochondrial Ca2+ uptake was assessed by measuring the fall in extramitochondrial Ca2+ fluorescence after subjecting iso-lated mitochondria to a bolus of Ca2+. Heart failure significantly delayed Ca2+ uptake in both SSM and IFM (Fig. 6). Supplementation with DHA increased the rate of Ca2+ uptake in both heart failure and sham animals in SSM but not in IFM. Further, the total amount of Ca2+ buffered by the mitochondria was decreased with heart failure both with and without DHA treatment compared to the sham animals on standard diet.
Fig. 6.

Extramitochondrial Ca2+ fluorescence. * P <0.05 heart failure compared to respective sham, # P <0.05 DHA treatment compared to standard diet in heart failure, ^ P <0.05 DHA treatment compared to standard diet in the sham groups. Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Ca2+ was released from the mitochondria during a continuous infusion of tBH with monitoring of extramitochondrial Ca2+ fluorescence. Mitochondria required significantly less tBH to release Ca2+ in animals with heart failure compared to shams in both untreated and DHA treated groups in SSM and IFM, demonstrating greater susceptibility to hydrogen peroxide-induced permeability transition in heart failure (Fig. 7). DHA partially attenuated this response in IFM but not SSM.
Fig. 7.

Time course of extramitochondrial Ca2+ fluorescence during a progressive increase in tBH. Values are normalized to total Ca2+ buffered during the loading phase (Fig. 5). * P <0.05 heart failure compared to respective sham, #P <0.05 DHA treatment compared to standard diet in heart failure, ^ P <0.05 DHA treatment compared to standard diet in the sham groups. Groups sizes were 15 and 20 for the untreated sham and heart failure groups, respectfully, and 14 and 20 for the DHA treated sham and heart failure groups, respectfully
Discussion
The present investigation provides important new information regarding the effects of dietary supplementation with purified DHA on the development of heart failure and the susceptibility of mitochondria to undergo permeability transition. First, we observed that cardiac mitochondria from animals with heart failure induced by aortic pressure overload were more susceptible to ROS-induced MPT in both untreated and DHA treated animals. Second, treatment with DHA resulted in modest but significant improvement in ROS-induced MPT in IFM in rats with heart failure. Third, the increase in DHA in mitochondrial phospholipids was associated with a decrease in membrane viscosity in IFM, but not in SSM. Further, viscosity was decreased by heart failure in SSM in both untreated and DHA treated animals. Together these findings suggest that the changes in viscosity with heart failure and DHA did not contribute to the differences in mitochondrial function among groups. Lastly, in a model of pressure overload induced heart failure that was more severe than those used in previous studies with marine n3-PUFA [8, 21–23, 25], we observed that DHA did not prevent loss of myocardial mitochondrial oxidative capacity in heart failure, and only modestly attenuated the development of LV dysfunction. Taken together, these findings suggest that while dietary supplementation with DHA significantly changed mitochondrial phospholipids and resistance to MPT, it provided only modest protection against the development of heart failure.
The effects of pressure overload-induced heart failure on MPT have not been extensively studied. We previously assessed MPT in isolated mitochondria from rats with mild left ventricular hypertrophy due to abdominal aortic constriction, and observed no differences from sham animals [8]. We recently showed that 15 weeks of aldosterone-induced hypertension with resultant modest LV hypertrophy in old female beagles resulted in a modest decrease in Ca2+ in cardiac SSM, but not IFM [39]. These findings are in contrast to our recent finding in cardiomyopathic hamsters, where IFM had greater susceptibility to Ca2+-induced MPT than normal healthy hamster, while SSM were similar [40]. The present investigation extends these previous findings by showing that with more severe LV hypertrophy with clear LV dilation there is greater susceptibility to Ca2+-induced MPT only in SSM, not IFM. Further, we show that in this model, ROS-induced MPT is clearly enhanced in heart failure (Fig. 7). The role of MPT in the development and progression of heart failure is controversial and unresolved, and thus the implications of these findings are not clear and remain under extensive investigation.
Several issues arise that require additional investigation. First, the present investigation assessed the effects of DHA on prevention of heart failure induced by pressure overload and cardiac hypertrophy, and did not evaluate the ability of DHA to treat established heart failure. Further, we did not assess the impact of early pre-treatment with DHA prior to initiation of pressure overload, as we did in a previous study with less severe aortic constriction [21]. This approach may have provided superior protection and is worthy of further study. Second, we assessed relatively short term treatment in young rats. Since human heart failure is largely a disease in the elderly, it is important to assess the effects of DHA in a similar model in animals with advanced age. That said, we recently observed minimal beneficial effect of DHA on LV function and no improvement in survival in cardiomyopathic hamsters with treatment lasting up to 78 weeks and with treatment initiated in adulthood [12]. This remains to be tested in a non-genetic hypertrophic pressure overload model of heart failure. Third, the diets we used were low in total fat and high in carbohydrate (12 % and 68 % of energy intake) which likely results in development of worse LV and mitochondrial dysfunction than higher fat intake (see reference [41] for review). On the other hand, we have previous observed that very high intake of mainly saturated fat (60 % of energy intake) blocked the beneficial effect of dietary supplementation of a mixture of DHA and EPA. It is possible that this effect would not be seen with a more modest high fat intake using mainly monounsaturated fatty acids, though this remains to be seen. It is also important to consider that most human heart failure develops after several years of hypertension that is often combined with ischemic heart disease and metabolic abnormalities such as obesity, diabetes and physical inactivity. The aortic banding model is highly reproducible and recapitulates many of the key components of heart failure due to aortic stenosis, but has distinct differences from most human heart failure. Future studies should consider evaluating the long term effects of DHA in a model of myocardial infarction in combination with more modest hypertension and/or metabolic disease. Lastly, treatment with DHA could affect arterial function and vascular resistance, thus in future studies it is import to assess systemic and proximal aortic blood pressure and the gradient across the aortic constriction.
In summary, the present investigation established that heart failure induced by pressure overload increases susceptibility to ROS-induced MPT in both untreated and DHA treated animals, but that this effect is modestly attenuated by treatment with DHA. Elevated DHA in mitochondrial membranes was associated with a decrease in membrane viscosity in the mitochondria found among the myofibrils, but not those adjacent to the sarcolemmal membrane. On the other hand, DHA was ineffective at preventing loss of myocardial mitochondrial oxidative capacity and only modestly attenuated of the development of LV dysfunction.
Acknowledgments
Funding support
This work was supported by the National Institutes of Health, National Heart Lung and Blood Institute [Grant numbers HL074237, HL110731 and HL101434].
Footnotes
Conflict of Interest
William Stanley is the inventor on a US patent application filed by the University of Maryland for the use of DHA for the treatment of heart failure. All other authors have no conflicts.
References
- 1.Ghio S, Scelsi L, Latini R, Masson S, Eleuteri E, Palvarini M, et al. Effects of n 3 polyunsaturated fatty acids and of rosuvastatin on left ventricular function in chronic heart failure: a substudy of GISSI HF trial. Eur J Heart Fail. 2010;12:1345–53. doi: 10.1093/eurjhf/hfq172. [DOI] [PubMed] [Google Scholar]
- 2.Gissi Hf I. Effect of n 3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI HF trial): a randomised, double blind, placebo controlled trial. Lancet. 2008;372:1223–30. doi: 10.1016/S0140-6736(08)61239-8. [DOI] [PubMed] [Google Scholar]
- 3.Nodari S, Triggiani M, Campia U, Manerba A, Milesi G, Cesana BM, et al. Effects of n 3 polyunsaturated fatty acids on left ventric ular function and functional capacity in patients with dilated cardio myopathy. J Am Coll Cardiol. 2011;57:870–9. doi: 10.1016/j.jacc.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 4.Moertl D, Hammer A, Steiner S, Hutuleac R, Vonbank K, Berger R. Dose dependent effects of omega 3 polyunsaturated fatty acids on systolic left ventricular function, endothelial function, and markers of inflammation in chronic heart failure of nonischemic origin: a double blind, placebo controlled, 3 arm study. Am Heart J. 2011;161:915–9. doi: 10.1016/j.ahj.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Mozaffarian D, Wu JH. Omega 3 fatty acids and cardiovascular disease: effects on risk factors, molecular pathways, and clinical events. J Am Coll Cardiol. 2011;58:2047–67. doi: 10.1016/j.jacc.2011.06.063. [DOI] [PubMed] [Google Scholar]
- 6.Duda MK, O’shea KM, Stanley WC. omega 3 polyunsaturated fatty acid supplementation for the treatment of heart failure: mechanisms and clinical potential. Cardiovasc Res. 2009;84:33–41. doi: 10.1093/cvr/cvp169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khairallah RJ, Sparagna GC, Khanna N, O’shea KM, Hecker PA, Kristian T, et al. Dietary supplementation with docosahexaenoic acid, but not eicosapentaenoic acid, dramatically alters cardiac mitochondrial phospholipid fatty acid composition and prevents permeability transition. Biochim Biophys Acta. 2010;1797:1555–62. doi: 10.1016/j.bbabio.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khairallah RJ, O’shea KM, Brown BH, Khanna N, des Rosiers C, Stanley WC. Treatment with docosahexaenoic acid, but not eicosapentaenoic acid, delays Ca2+ induced mitochondria perme ability transition in normal and hypertrophied myocardium. J Pharmacol Exp Ther. 2010;335:155–62. doi: 10.1124/jpet.110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khairallah RJ, Kim J, O’Shea KM, O’Connell KA, Brown BH, Galvao TDRC, et al. Improved mitochondrial function with diet induced increase in either docosahexaenoic acid or arachidonic acid in membrane phospholipids. PLoS One. 2012;7:e34402. doi: 10.1371/journal.pone.0034402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’shea KM, Khairallah RJ, Sparagna GC, Xu W, Hecker PA, Robillard Frayne I, et al. Dietary omega 3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+ induced permeability transition. J Mol Cell Cardiol. 2009;47:819–27. doi: 10.1016/j.yjmcc.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload induced heart failure. Cardiovasc Res. 2010;85:376–84. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 12.Galvao TF, Khairallah RJ, Dabkowski ER, Brown BH, Hecker PA, O’Connell KA, et al. Marine n3 polyunsaturated fatty acids enhance resistance to mitochondrial permeability transition in heart failure, but do not improve survival. Am J Physiol Heart Circ Physiol. 2013;73:H12–21. doi: 10.1152/ajpheart.00657.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharov VG, Todor A, Khanal S, Imai M, Sabbah HN. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol. 2007;42:150–8. doi: 10.1016/j.yjmcc.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halestrap AP. A pore way to die: the role of mitochondria in reper fusion injury and cardioprotection. Biochem Soc Trans. 2010;38:841–60. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 15.Sharov VG, Todor AV, Imai M, Sabbah HN. Inhibition of mitochon drial permeability transition pores by cyclosporine A improves cyto chrome C oxidase function and increases rate of ATP synthesis in failing cardiomyocytes. Heart Fail Rev. 2005;10:305–10. doi: 10.1007/s10741-005-7545-1. [DOI] [PubMed] [Google Scholar]
- 16.Stanley WC, Khairallah RJ, Dabkowski ER. Update on lipids and mitochondrial function: impact of dietary n 3 polyunsaturated fatty acids. Curr Opin Clin Nutr Metab Care. 2012;15:122–6. doi: 10.1097/MCO.0b013e32834fdaf7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pepe S, Tsuchiya N, Lakatta EG, Hansford RG. PUFA and aging modulate cardiac mitochondrial membrane lipid composition and Ca2+ activation of PDH. Am J Physiol. 1999;276:H149–58. doi: 10.1152/ajpheart.1999.276.1.H149. [DOI] [PubMed] [Google Scholar]
- 18.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–7. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin d and the mitochondrial permeability transition pore. Circ J. 2013;77:1111–22. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’shea KM, Chess DJ, Khairallah RJ, Hecker PA, Lei B, Walsh K, et al. omega 3 Polyunsaturated fatty acids prevent pressure overload induced ventricular dilation and decrease in mitochondrial enzymes despite no change in adiponectin. Lipids Health Dis. 2010;9:95. doi: 10.1186/1476-511X-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duda MK, O’shea KM, Tintinu A, Xu W, Khairallah RJ, Barrows BR, et al. Fish oil, but not flaxseed oil, decreases inflammation and prevents pressure overload induced cardiac dysfunction. Cardiovasc Res. 2009;81:319–27. doi: 10.1093/cvr/cvn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duda MK, O’shea KM, Lei B, Barrows BR, Azimzadeh AM, McElfresh TE, et al. Dietary supplementation with omega 3 PUFA increases adiponectin and attenuates ventricular remod eling and dysfunction with pressure overload. Cardiovasc Res. 2007;76:303–10. doi: 10.1016/j.cardiores.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah KB, Duda MK, O’shea KM, Sparagna GC, Chess DJ, Khairallah RJ, et al. The cardioprotective effects of fish oil during pressure overload are blocked by high fat intake: role of cardiac phospholipid remodeling. Hypertension. 2009;54:605–11. doi: 10.1161/HYPERTENSIONAHA.109.135806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Shearer GC, Chen Q, Healy CL, Beyer AJ, Nareddy VB, et al. Omega 3 fatty acids prevent pressure overload induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation. 2011;123:584–93. doi: 10.1161/CIRCULATIONAHA.110.971853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLennan PL, Abeywardena MY, Dallimore JA, Raederstorff D. Dietary fish oil preserves cardiac function in the hypertrophied rat heart. Br J Nutr. 2011;108:645–54. doi: 10.1017/S0007114511005915. [DOI] [PubMed] [Google Scholar]
- 26.Gong G, Liu J, Liang P, Guo T, Hu Q, Ochiai K, et al. Oxidative capacity in failing hearts. Am J Physiol Heart Circ Physiol. 2003;285:H541–8. doi: 10.1152/ajpheart.01142.2002. [DOI] [PubMed] [Google Scholar]
- 27.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura Clapier R. Depressed mitochondrial transcription factors and oxida tive capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551:491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Javadov S, Choi A, Rajapurohitam V, Zeidan A, Basnakian AG, Karmazyn M. NHE 1 inhibition induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochon drial permeability transition. Cardiovasc Res. 2008;77:416–24. doi: 10.1093/cvr/cvm039. [DOI] [PubMed] [Google Scholar]
- 29.Javadov S, Huang C, Kirshenbaum L, Karmazyn M. NHE 1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J Mol Cell Cardiol. 2005;38:135–43. doi: 10.1016/j.yjmcc.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Faerber G, Barreto Perreia F, Schoepe M, Gilsbach R, Schrepper A, Schwarzer M, et al. Induction of heart failure by minimally invasive aortic constriction in mice: reduced peroxisome proliferator activated receptor gamma coactivator levels and mitochondrial dysfunction. J Thorac Cardiovasc Surg. 2011;141(492-500):500. doi: 10.1016/j.jtcvs.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 31.Zaha V, Grohmann J, Gobel H, Geibel A, Beyersdorf F, Doenst T. Experimental model for heart failure in rats induction and diagnosis. Thorac Cardiovasc Surg. 2003;51:211–5. doi: 10.1055/s-2003-42264. [DOI] [PubMed] [Google Scholar]
- 32.Doenst T, Pytel G, Schrepper A, Amorim P, Farber G, Shingu Y, et al. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res. 2010;86:461–70. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- 33.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–9. [PubMed] [Google Scholar]
- 34.Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF, Stanley WC, et al. Cardiomyocyte deletion of mitofusin 1 leads to mitochondrial fragmentation and improves tol erance to ROS induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–79. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J, Yu BP, Herlihy JT. Modulation of cardiac mitochondrial membrane fluidity by age and calorie intake. Free Radic Biol Med. 1999;26:260–5. doi: 10.1016/s0891-5849(98)00195-6. [DOI] [PubMed] [Google Scholar]
- 36.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, et al. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–40. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chess DJ, Xu W, Khairallah R, O’shea KM, Kop WJ, Azimzadeh AM, et al. The antioxidant tempol attenuates pressure overload Induced cardiac hypertrophy and contractile dysfunction in mice fed a high fructose diet. Am J Physiol Heart Circ Physiol. 2008;295:H2223–30. doi: 10.1152/ajpheart.00563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gelinas R, Thompson Legault J, Bouchard B, Daneault C, Mansour A, Gillis MA, et al. Prolonged QT interval and lipid alterations beyond {beta} oxidation in very long chain acyl CoA dehydrogenase null mouse hearts. Am J Physiol Heart Circ Physiol. 2011;301:H813–23. doi: 10.1152/ajpheart.01275.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asemu G, O’Connell KA, Cox JW, Dabkowski ER, Xu W, Ribeiro RF, Jr, et al. Enhanced resistance to permeability transition in interfibrillar cardiac mitochondria in dogs: effects of aging and long term aldoste rone infusion. Am J Physiol Heart Circ Physiol. 2013;304:H514–28. doi: 10.1152/ajpheart.00674.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galvao TF, Brown BH, Hecker PA, O’Connell KA, O’shea KM, Sabbah HN, et al. High intake of saturated fat, but not polyunsatu rated fat, improves survival in heart failure despite persistent mito chondrial defects. Cardiovasc Res. 2012;93:24–32. doi: 10.1093/cvr/cvr258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stanley WC, Dabkowski ER, Ribeiro RF, Jr, O’Connell KA. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circ Res. 2012;110:764–76. doi: 10.1161/CIRCRESAHA.111.253104. [DOI] [PMC free article] [PubMed] [Google Scholar]