

Several themes motivate the synthetic modeling of the active sites of the hydrogenase enzymes: interest in complexes of dihydrogen and hydride ligands,1 novel biochemistry implied by structurally unusual active sites,2–4 and the potential for utilizing hydrogen as an energy carrier.5 Through recent intense efforts, synthetic models for the active site of the [FeFe]-H2ases are yielding useful mechanistic insights.6,7 Modeling the more prevalent [NiFe]-H2ases, however, has proven more challenging.8 The functional core of this enzyme features a nickel tetrathiolate wherein two thiolates are bridging to an Fe(CN)2(CO) center. Several states of the [NiFe]-H2ases have been detected, although only two oxidation states are catalytically significant, Fe(II)Ni(III) (Ni–C, with a hydride) and Fe(II)Ni(II) (Ni–R, with a hydride, and Ni–SI, without; see Figure 1).9 Despite intensive efforts, compounds featuring the NiFeS2(μ-H) core have resisted synthetic modeling, although progress with related RuNiS2(μ-H) has recently been described.10 We now report successful and potentially generalizable routes to the long-sought nickel–iron dithiolato hydrides.
Figure 1.
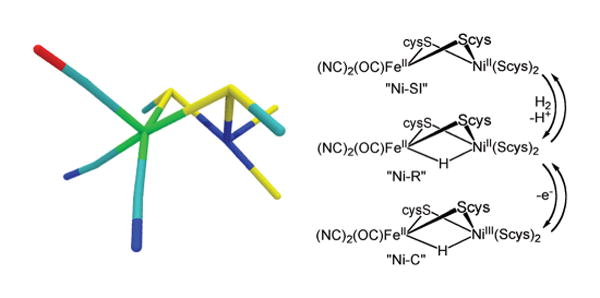
Structure of the active site in [NiFe]-hydrogenase (left, PDB #1WUL) and its key conversions (right).9
In our initial approach to biomimetic models, we treated the iron dithiolato complex Fe(pdt)(CO)2(dppe)11 (1) with NiCl2(dppe) in hot acetone solution to give the μ-chloro bimetallic cation [(CO)-(dppe)Fe(pdt)(μ-Cl)Ni(dppe)]+ ([2Cl]+), isolated as its BF4− salt (pdt= 1,3-propanedithiolate; dppe = 1,2-C2H4(PPh2)2; OTs− = CH3C6H4-4-SO3−) (Scheme 1). The brown-red colored complex is diamagnetic, consistent with low spin Fe(II) and Ni(II) centers. Attempts to replace the chloride ligand with hydrides proved unfruitful. Nonetheless, with three bridging ligands, the structure of the bimetallic complex verified that the ligand set is geometrically compatible with the corresponding targeted μ-hydrido complexes.
Scheme 1. Synthetic Approaches to Nickel–Iron Dithiolato Hydrides.
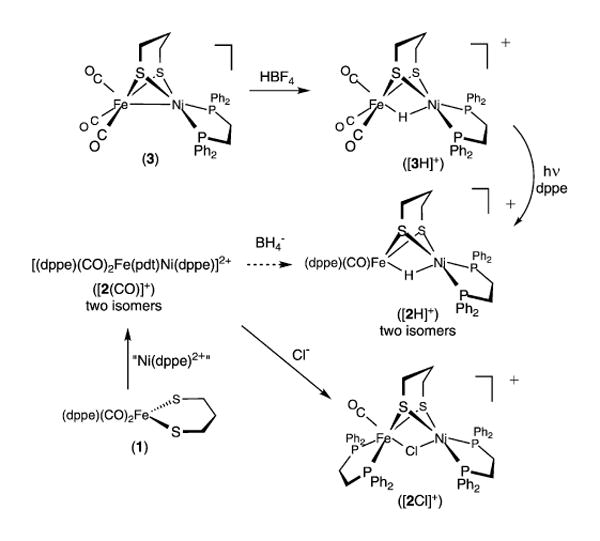
Seeking chloride-free precursors, we next examined the related condensation of 1 with “Ni(OTs)2(dppe),” generated from [Ni(H2O)6](OTs)212 and dppe. Treatment of a CH2Cl2 solution of Ni(OTs)2(dppe) with 1 rapidly gave the salt of the dicationic dicarbonyl [(CO)2(dppe)Fe(pdt)Ni(dppe)]2+ ([2(CO)]2+), obtained as a mixture of symmetric and unsymmetric stereoisomers. A pair of isomers (in a similar ratio) are also seen in the iron precursor. Thus, it appears that the Ni(dppe)2+ module binds to the dithiolate without perturbing the coordination sphere of the Fe(II) center. The dicationic nickel–iron complex [2(CO)]2+ is observed as an intermediate in the reaction of 1 and NiCl2(dppe). Solutions of [2(CO)]2+ in CH2Cl2 were found to react with NBu4BH4 to give a complex mixture. Analysis of this mixture revealed the presence of the targeted cationic hydride [(CO)(dppe)Fe(pdt)(μ-H)Ni(dppe)]+ [2H]+: a molecular ion was observed in the ESI-MS, and the 1H NMR spectrum revealed two high-field triplets consistent with two isomers depending on the stereochemistry of the Fe(dppe) center. Solutions of crude [2H]+ are more robust than the dicationic precursor consistent with a complex that has three bridging ligands (like the μ-chloride [2Cl]+).
Because of the inefficiency of the hydride routes but encouraged by the stability of crude samples of [2H]+, we investigated an alternative route to [2H]+ starting from more reduced reagents. The Fe(I) complex (CO)3Fe(pdt)Ni(dppe) (3)13 readily protonated with HBF4·Et2O in CH2Cl2 solution to give the corresponding hydride [3H]+ (Scheme 1). When monitored by FT-IR spectroscopy, protonation induces a shift in νCO of 54 cm−1. The retention of the νCO pattern suggests that the protonation occurs at the Ni–Fe bond. Consistent with this assignment, the hydride region of the 1H NMR spectrum for[3H]BF4 displays a triplet (δ –3.53, JPH ∼6 Hz), and the 31P NMR spectrum shows a singlet. Excess acid is not deleterious; i.e. [3H]+ does not protonate further, even with a large excess of HBF4·Et2O. Solutions of [3H]BF4 are stable in air for days.
Representing the first example of a nickel–iron thiolato hydride,14 [3H]BF4 was characterized crystallographically (Figure 2). Including the bridging hydride ligand, the Fe center is quasi-octahedral and Ni is approximately square pyramidal. The hydride connectivity, the metal coordination numbers, and the presence of three diatomic terminal ligands on Fe match features of the active site of the enzyme in the Ni–R state.2,9 Completing the synthetic cycle outlined in the scheme, [3H]+ reacts with dppe under photochemical conditions to give the hydride [2H]+, which exhibits spectroscopic properties seen in the crude samples (high field 1H NMR, ESI/MS, FT-IR). This substitution method should allow the synthesis of related complexes with a range of ligands on Fe.
Figure 2.
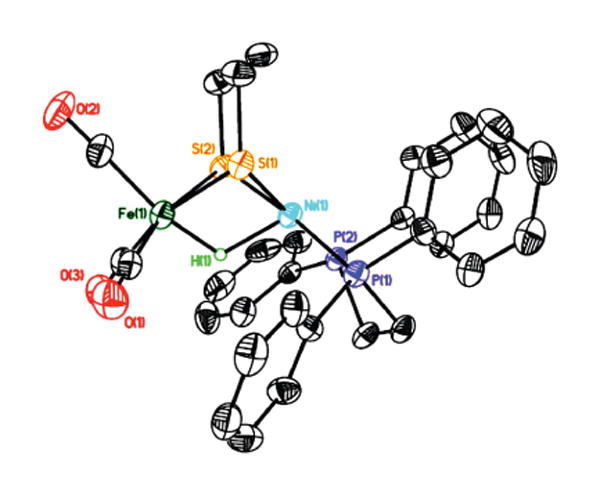
Structure of the cation in [(dppe)NiFe(pdt)(H)(CO)3]BF4 ([3H]BF4). Key distances (Å): Ni–Fe, 2.6131(14); Ni–S(1), 2.210(2); Ni–S(2), 2.219(2); Fe–S(1), 2.321(2); Fe–S(2), 2.322(2); Ni–H, 1.64(6); Fe–H, 1.46(6); S(⋯)S, 3.101. The Ni–Fe distance in [(CO)(dppe)Fe(pdt)(μ-Cl)Ni(dppe)]BF4 is 3.076 Å.
The complex [3H]+ resembles the 34e diiron dithiolato hydrides [Fe2(pdt)(μ-H)(CO)6−x(PR3)x]+, which are considered to be useful models for the [FeFe]-hydrogenases.6 Deprotonation of methylene chloride solutions of [3H]+ with NEt3 ([HNEt3]BF4, pKa = 18) were found to rapidly give 3. Complex [3H]+ is an active catalyst for the reduction of protons to H2, as indicated by electrochemical measurements. For a CH2Cl2 solution of CF3CO2H (pKa in CH3CN = 12.65; E° ∼ −0.90 V vs Fc/Fc+),15 a catalytic current is observed near −1.37 V vs Fc/Fc+, which is milder than that seen for the diiron complexes (Figure 3). The proposed catalytic cycle begins with protonation of 3 and probably follows the mechanistic pattern described for the substituted diiron catalysts, i.e., a cycle that involves reduction of the hydride followed by protonation.6,16
Figure 3.
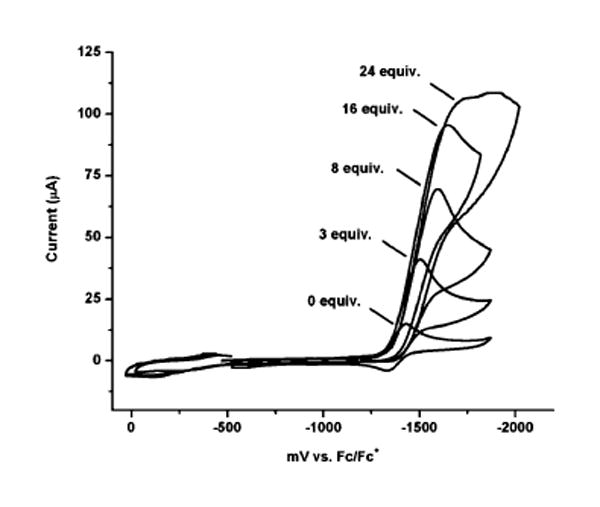
Cyclic voltammetry of [3H]BF4 in the presence of varying equivalents of CF3CO2H. Conditions: 0.001 M [3H]BF4 in CH2Cl2 solution, 0.1 M NBu4PF6, scan rate of 100 mV/s.
Complex 3 is also easily oxidized to the monocation, E1/2∼ −520 V (vs Fc/Fc+). Using ferrocenium as the oxidant (FcBF4), we generated the salt [3]BF4. This cation exists in the same oxidation state (FeNi)3+ assigned to the Ni–L state of the enzyme.4 The IR spectrum shows that oxidation shifts νCO by ∼29 cm−1. Although the argument is qualitative, the values for νCO indicate that one-electron oxidation affects the Fe(CO)3 center less than protonation, which formally corresponds to a Fe(II) ion. A frozen 8:2 CH2Cl2/THF solution of [3]BF4 exhibits an intense nearly axial EPR spectrum lacking phosphine hyperfine coupling. The EPR and FT-IR properties are similar to those for square pyramidal Fe(I) species.17 It is likely that replacement of CO ligands in 3 will enable access to higher oxidation states of the NiFe(SR)2 system beyond the (FeNi)3+ state.
The methods described above provide access to functional models for the [NiFe]-hydrogenases. The models are amenable to extensive modifications by variations of the ligands. Our results should encourage the development of a host of biomimetic hydrides, leading to new mechanistic insights relevant to Nature's most pervasive catalysts for processing hydrogen.
Acknowledgments
This research was supported by NIH. We thank Dr. Mark Nilges for assistance with the EPR spectroscopy. We thank Dr. Jinzhu Chen for the assistance with the preparation of the Fe precursors.
Footnotes
Supporting Information Available: Preparative, spectroscopic, and crystallographic details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kubas GJ. Proc Natl Acad Sci USA. 2007;104:6901–6907. doi: 10.1073/pnas.0609707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 3.Vincent KA, Parkin A, Armstrong FA. Chem Rev. 2007;107:4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- 4.De Lacey AL, Fernández VM, Rousset M, Cammack R. Chem Rev. 2007;107:4304–4330. doi: 10.1021/cr0501947. [DOI] [PubMed] [Google Scholar]
- 5.Esswein AJ, Nocera DG. Chem Rev. 2007;107:4022–4047. doi: 10.1021/cr050193e. [DOI] [PubMed] [Google Scholar]; Lewis NS, Nocera DG. Proc Natl Acad Sci USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gloaguen F, Rauchfuss TB. Chem Soc Rev. 2009;38:100–108. doi: 10.1039/b801796b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheah MH, Tard C, Borg SJ, Liu X, Ibrahim SK, Pickett CJ, Best SP. J Am Chem Soc. 2007;129:11085–11092. doi: 10.1021/ja071331f. [DOI] [PubMed] [Google Scholar]; Thomas CM, Liu T, Hall MB, Darensbourg MY. Inorg Chem. 2008;47:7009–7024. doi: 10.1021/ic800654a. [DOI] [PubMed] [Google Scholar]
- 8.Ohki Y, Yasumura K, Kuge K, Tanino S, Ando M, Li Z, Tatsumi K. Proc Natl Acad Sci USA. 2008;105:7652–7657. doi: 10.1073/pnas.0800538105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Linck RC, Rauchfuss TB. In: Bioorganometallics: Biomolecules, Labeling, Medicine. Jaouen G, editor. Wiley-VCH; Weinheim: 2005. [Google Scholar]; Evans DJ, Pickett CJ. Chem Soc Rev. 2003;32:268–275. doi: 10.1039/b201317g. [DOI] [PubMed] [Google Scholar]; Li Z, Ohki Y, Tatsumi K. J Am Chem Soc. 2005;127:8950–8951. doi: 10.1021/ja051590+. [DOI] [PubMed] [Google Scholar]; Riordan CG. In: Comprehensive Coordination Chemistry II. Meyer TJ, McCleverty JA, editors. Pergamon Press; London: 2004. pp. 677–713. [Google Scholar]
- 9.Cammack R, Frey M, Robson R. Hydrogen as a Fuel: Learning from Nature. Taylor & Francis; London: 2001. [Google Scholar]
- 10.Ogo S, Kabe T, Uehara K, Kure B, Nishimura T, Menon SC, Harada R, Fukuzumi S. Science. 2007;316:585–588. doi: 10.1126/science.1138751. [DOI] [PubMed] [Google Scholar]; (a) Oudart Y, Artero V, Pécaut J, Fontecave M. Inorg Chem. 2006;45:4334–4336. doi: 10.1021/ic060510f. [DOI] [PubMed] [Google Scholar]
- 11.Takács J, Markó L, Párkányi L. J Organomet Chem. 1989;361:109–116. [Google Scholar]
- 12.Holmes SM, McKinley SG, Girolami GS. Inorg Synth. 2002;33:91–103. [Google Scholar]
- 13.Zhu W, Marr AC, Wang Q, Neese F, Spencer DJE, Blake AJ, Cooke PA, Wilson C, Schröder M. Proc Natl Acad Sci USA. 2005;102:18280–18285. doi: 10.1073/pnas.0505779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nickel-iron-hydrides are known in the form of [HFe5NiN(CO)14]2−:; Pergola RD, Fumagalli A, Garlaschelli L, Manassero C, Manassero M, Sansoni M, Sironi A. Inorg Chim Acta. 2008;361:1763–1769. [Google Scholar]
- 15.Felton GAN, Glass RS, Lichtenberger DL, Evans DH. Inorg Chem. 2006;45:9181–9184. doi: 10.1021/ic060984e. [DOI] [PubMed] [Google Scholar]; Felton GAN, Vannucci AK, Chen J, Lockett LT, Okumura N, Petro BJ, Zakai UI, Evans DH, Glass RS, Lichtenberger DL. J Am Chem Soc. 2007;129:12521–12530. doi: 10.1021/ja073886g. [DOI] [PubMed] [Google Scholar]
- 16.Gloaguen F, Lawrence JD, Rauchfuss TB. J Am Chem Soc. 2001;123:9476–9477. doi: 10.1021/ja016516f. [DOI] [PubMed] [Google Scholar]
- 17.MacNeil JH, Chiverton AC, Fortier S, Baird MC, Hynes RC, Williams AJ, Preston KF, Ziegler T. J Am Chem Soc. 1991;113:9834–9842. [Google Scholar]