

Abstract
Context
Fine particulate matter (PM) air pollution has been associated with alterations in circulating endothelial progenitor cell (EPC) levels, which may be one mechanism whereby exposures promote cardiovascular diseases. However, the impact of coarse PM on EPCs is unknown.
Objective
We aimed to determine the effect of acute exposure to coarse concentrated ambient particles (CAP) on circulating EPC levels.
Methods
Thirty-two adults (25.9±6.6 years) were exposed to coarse CAP (76.2±51.5 μgm−3) in a rural location and filtered air (FA) for 2 h in a randomized double-blind crossover study. Peripheral venous blood was collected 2 and 20 h post-exposures for circulating EPC (n=21), white blood cell (n=24) and vascular endothelial growth factor (VEGF) (n=16–19) levels. The changes between exposures were compared by matched Wilcoxon signed-rank tests.
Results
Circulating EPC levels were elevated 2 [108.29 (6.24–249.71) EPC mL−1; median (25th–75th percentiles), p=0.052] and 20 h [106.86 (52.91–278.35) EPC mL−1, p=0.008] post-CAP exposure compared to the same time points following FA [38.47 (0.00–84.83) and 50.16 (0.00–104.79) EPC mL−1]. VEGF and white blood cell (WBC) levels did not differ between exposures.
Conclusions
Brief inhalation of coarse PM from a rural location elicited an increase in EPCs that persisted for at least 20 h. The underlying mechanism responsible may reflect a systemic reaction to an acute “endothelial injury” and/or a circulating EPC response to sympathetic nervous system activation.
Keywords: Air pollution, endothelial function, endothelial progenitor cells, inflammation
Introduction
Fine particulate matter <2.5 μm (PM2.5) air pollution, typically derived from combustion processes (e.g. coal-fired power plants), is a leading cause of worldwide mortality (Lim et al., 2012). The majority of the deaths following exposures have been shown to be due to cardiovascular (CV) causes. In support of these epidemiological associations, numerous studies have confirmed that PM2.5 can elicit an array of adverse biological responses potentially capable of promoting acute CV events among susceptible individuals (Brook et al., 2010).
Conversely, the health impacts of exposure to larger “coarse PM” (2.5–10 μm in aerodynamic diameter; PM2.5–10) are less conclusive (Brunekreef & Forsberg, 2005; Chang et al., 2011; Peng et al., 2008; Puett et al., 2009; Zanobetti & Schwartz, 2009). In addition to differing sources and components from PM2.5, the composition of PM2.5–10 itself often varies to a greater extent across seasons and locations. PM2.5–10 represents a mixture of particles generated from mechanical processes (e.g. crushing, re-suspension of ground material) with sources ranging from farming, roadway dust, to construction. The constituents also differ according to nearby activities as well as landscape features and include metals, crustal material (e.g. silicon, calcium, natural elements) and bio-aerosols (e.g. endotoxin) (Brook et al., 2010; Brunekreef & Forsberg, 2005).
PM2.5 has been shown to instigate a variety of adverse CV responses such as vascular dysfunction, reduced heart rate variability (HRV) and enhanced coagulation-thrombosis potential (Brook et al., 2009, 2010). Recently, a few studies have also shown that exposure over a period of 24 h to several weeks can cause reductions in circulating endothelial progenitor cell (EPC) levels (Haberzetti et al., 2012; Liberda et al., 2010; O’Toole et al., 2010). On the contrary, a brief 30-min exposure to secondhand smoke (SHS) was shown to cause acute elevations in circulating EPCs, albeit of a dysfunctional nature (Heiss et al., 2008). EPCs play an important role in vascular homeostasis, endothelial reparative capacity and modulating vasomotor functionality. EPCs normally comprise a small fraction (<1%) of all circulating WBCs. They are derived from the bone marrow and are known to home to areas of endothelial injury where they both directly (e.g. incorporate into vessels) and indirectly (e.g. help orchestrate local cells to) repair damaged vessel tissue. Hence, any alteration in their circulating numbers (e.g. a reduction likely leading to impaired endothelial regenerative capacity or an increase often reflecting a response to acute endothelial injury) and/or their proper functioning (even if their numbers remain unchanged) is known to convey an adverse CV prognosis (Mobius-Winkler et al., 2009; Povsic & Goldschmidt-Clermont, 2008; Shantsila et al., 2007).
To date, no experiment has evaluated the impact of PM2.5–10 on EPC levels. Due to differences in size, chemistry and fate upon inhalation (Brook et al., 2010; Brunekreef & Forsberg, 2005), there is substantial reason to believe that coarse particles might elicit dissimilar responses when compared with PM2.5. Despite its important contribution to global air pollution, the paucity of existing experimental studies demonstrating harmful actions induced by PM2.5–10 exposure is one critical barrier against adding regulations of this specific size fraction to existing air quality standards, particularly given the mixed epidemiological findings (Brunekreef & Forsberg, 2005). Mechanistic studies help to inform on our present limitations in knowledge regarding the plausibility that coarse PM could prompt CV events. As such, we aimed to evaluate if controlled exposure to coarse concentrated ambient particle (CAP) alters circulating EPC levels – an important determinant of CV health. To gain insights into the potential biological reasons for any changes in EPCs, we also evaluated vascular endothelial growth factor (VEGF) levels and HRV metrics (i.e. autonomic balance), both known modulators of circulating EPC concentrations.
Methods
The study was approved by the Institutional Review Board of the University of Michigan. Subjects signed an informed consent document during a screening visit when blood laboratory tests were drawn (fasting lipids, glucose) and a history and physical exam was performed. To be enrolled, patients were healthy non-smoking adults (no active smoking within the past year) who live in non-smoking households and were from 18 to 50 years of age without any established CV disease or traditional CV risk factors. Enrolled subjects had screening blood pressures <140/90mmHg and fasting glucose levels <126 mg dL−1. No subject was taking medications (e.g. statins) or over-the-counter pills (e.g. antioxidants) that might alter vascular function.
Qualifying subjects entered into a randomized double-blind crossover study comparing the health effects of 2 h long exposures to coarse CAP versus filtered air (FA). Exposures began during May 2011 and were all completed by the end of June 2012. There was a 1–3 week washout period between exposures. Subjects came to the facility on each day fasting for >8 h. Randomized exposures occurred from 10 am to noon. Peripheral venous blood was drawn 2 and 20 h post-exposures.
In this article, we report only the findings regarding changes in EPC levels (a pre-defined secondary study outcome) induced by exposures. The results of the primary study outcomes [blood pressure, brachial flow-mediated dilatation (FMD), HRV], determined as previously described (Brook et al., 2009), are reported elsewhere (Brook et al., in review).
Exposure facility and air pollution measurements
We selected Dexter, a village in Michigan as the location for coarse PM exposures as it is heavily influenced by rural sources (http://www.epa.gov/castnet/javaweb/site_pages/ANA115.html). This location is >10 km from major freeways and >60 km west of the Detroit metropolitan area. Coarse CAP was generated using a Two-Stage Virtual Impactor System (Demokritou et al., 2002), which concentrates ambient coarse PM (predominantly from 2.5 to 10 μm) without substantively altering their composition or chemistry. The AirCARE Mobile Air Research Laboratory is described elsewhere in more detail (Brook et al., 2009; Harkema et al., 2004). Exposures were delivered to subjects seated within an air-tight chamber via a facemask with an air flow rate between 25 and 28 L min−1. All gaseous pollutants (e.g. ozone) remained at or below ambient levels. During FA exposures a high-efficiency PM filter was inserted at the inlet of the concentrator (i.e. “upstream” to the series of virtual impactors). Coarse CAP mass levels were monitored during exposures downstream of the concentrator using a personal DataRAM Monitor (Thermo Scientific, Waltham, MA). Outdoor and within chamber temperatures and relative humidity were also monitored. Chamber temperature was maintained at ~24 °C. Filter samples were collected immediately upstream of the exposure chamber on 47-mm Teflon Filters (Pall Life Sciences, Ann Arbor, MI) at a flow rate of 6 L min−1. The samples were analyzed gravimetrically using a Microbalance (MT-5 Mettler Toledo, Columbus, OH) in a temperature/humidity-controlled clean laboratory as described in the Federal Reference Method (US Environmental Protection Agency, 1997. Reference method for the determination of fine PM as PM2.5 in the atmosphere. EPA 40 CFR Part 50, Washington, DC).
WBC and EPC levels
Peripheral blood mononuclear cells (PBMCs) were obtained from blood by Ficoll–Hypaque gradient and stained with anti-CD34–fluorescein isothiocyanate (Ancell Co., Bayport, MN) and anti-CD133–phycoerythrin (PE) (Miltenyi Biotec, Auburn, CA), in combination with a cocktail of PE/ Cyanine5-conjugated antibodies recognizing CD3, CD79b and CD56 (BD Biosciences, San Jose, CA). Immunofluorescence was measured using a Coulter Epics XL Flow Cytometer (Coulter Corporation, Hialeah, FL). EPCs were identified in the lymphocyte population as CD34+/ CD133+ cells in the CD3−/CD79b−/CD56− gate. The number of positive events detected by fluorescence-activated cell sorter was divided by the sum of lymphocytes plus monocyte events obtained during the acquisition. The percentage of CD34+/CD133+ cells in the PBMC pool (CD34/CD133 divided by lymphocytes plus monocytes) was used to calculate the total number of CD34+/CD133+ cells recovered from the PBMC isolation and then divided by the initial blood volume to determine CD34+/CD133+ cellsmL−1 (Denny et al., 2007; Rajagopalan et al., 2004). Total WBC levels and the complete WBC differential counts were performed by the University of Michigan Hospital Main Laboratory.
VEGF levels
The Michigan Diabetes Research Training Center Chemistry Laboratory, University of Michigan Health System analyzed blood samples for VEGF-A using a Luminex Multiplex System (analyzer: LX10010091401; platform: LXY10099103) (Luminex Corp., Austin, TX). Data collection and interpretation were performed by Luminex xPONENT Software for LX100/LX200 Systems [version 3.1.871.0 (2009)] (Luminex Corp., Austin, TX) and VigeneTech Milliplex Analyst [version 3.5.5.0 standard (2005)] (VigeneTech Inc., Carlisle, MA), respectively. The intra-assay coefficient of variation is 3.7% with a minimal detectable concentration of 26.3 pgmL−1.
Statistical methods
Summary statistics were computed for continuous measures as mean ± standard deviation (SD) or median (interquartile range, IQR) if the distribution was skewed. Due to the factorial design or pre-after longitudinal study design, the matched pairs of measurements were compared using the matched Wilcoxon signed-rank tests given the limited sample size. To analyze the dose–response relationship, we evaluated the association between the coarse PM level during exposures and the EPCs (both time points) using linear mixed models. We used similar models to evaluate the associations between HRV metrics (as well as brachial FMD) with EPC levels during all exposures.
Results
The characteristics of all the 32 subjects that completed the study are presented in Table 1. PM2.5–10 levels were significantly higher during coarse CAP compared to FA exposures (Table 2). There were no differences between exposures in ambient temperature or humidity (results not shown).
Table 1.
Subject characteristics (n=32, 16 female subjects).
| Variable | Mean (SD) | Minimum | Maximum |
|---|---|---|---|
| Age (years) | 25.9 (6.6) | 18.0 | 46.0 |
| Weight (kg) | 78.4 (16.3) | 55.9 | 111.4 |
| Height (m) | 1.7 (0.1) | 1.6 | 2.0 |
| Body mass index (kgm−2) | 26.3 (5.7) | 18.3 | 43.5 |
| Fasting glucose (mg dL−1)a | 86.9 (6.9) | 70.0 | 103.0 |
| Total cholesterol (mg dL−1) | 163.9 (31.4) | 104.0 | 244.0 |
| HDL-C (mg dL−1) | 55.4 (15.9) | 25.0 | 91.0 |
| LDL-C (mg dL−1) | 88.8 (26.1) | 49.0 | 135.0 |
| Triglycerides (mg dL−1) | 106.0 (80.9) | 40.0 | 401.0 |
HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.
Missing one subject data point.
Table 2.
PM2.5–10 concentrations during exposures (n=32).
| Exposure | Mean (SD) | Minimum | Maximum |
|---|---|---|---|
| FA | 10.1 (7.1) | 2.6 | 27.4 |
| Coarse CAP | 76.2* (51.5) | 10.3 | 246.5 |
PM2.5–10 concentrations are in microgram per cubic meter and determined by Teflon filter-based gravimetric mass measurements for the 2 h period of exposures. Mass levels below the detection limit (6.8 μgm3) were recorded at this value for analyses (n=15, FA exposures only).
p<0.01 for differences of mean or median levels between exposure types.
Circulating EPC levels were elevated both 2 h [108.29 (6.24, 249.71) EPCmL−1; median (25th, 75th percentiles), p=0.052) and 20 h [106.86 (52.91, 278.35) EPCmL−1, p=0.008] post-CAP exposure compared to the same time points following FA [38.47 (0, 84.83) and 50.16 (0.00, 104.79) EPCmL−1] (Figure 1). During the 20 h time point when differences were statistically significant, 16 of 21 available subjects had higher EPC levels VEGF and total WBC levels, including individual sub-type of WBCs in the differential count evaluated (i.e. neutrophils and lymphocytes), did not differ between exposures (Table 3).
Figure 1.
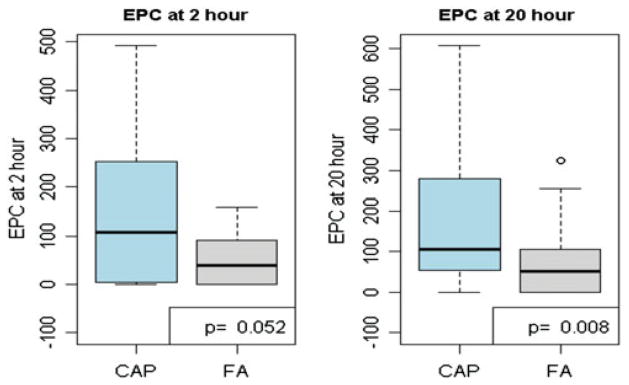
EPC levels 2 and 20 h after exposures (n=21 subjects with results on EPC levels).
Table 3.
VEGF levels and complete blood count results.
| Post-CAP
|
Post-FA
|
p Value | |||
|---|---|---|---|---|---|
| n | Median (IQR) | n | Median (IQR) | ||
| VEGF at 2 h (pgmL−1) | 16 | 159 (36.8–431.5) | 16 | 245 (63.2–516.8) | 0.99 |
| VEGF at 20 h (pgmL−1) | 19 | 149 (25.6–463) | 19 | 131 (48.9–555) | 0.37 |
| WBCs (1000mm−3)* | 24 | 6.85 (5.38–7.63) | 24 | 6.30 (5.58–7.05) | 0.73 |
| Neutrophils (1000mm−3)* | 24 | 3.65 (2.88–4.93) | 24 | 3.45 (2.80–4.13) | 0.46 |
| Lymphocytes (1000mm−3)* | 24 | 2.10 (1.70–2.70) | 24 | 2.10 (1.78–2.50) | 0.95 |
Laboratory values were collected and analyzed only 2 h post-exposures. p Values are calculated by paired Wilcoxin signed-rank test. n, number of subjects with results for each outcome.
We found no significant linear dose–response association between PM2.5–10 levels during exposures and the changes in EPCs at either time point. As part of the main study (Brook et al., in review), we also assessed HRV during exposures and brachial FMD 2 h following exposures. There was no evidence for significant associations between alterations in any HRV metric or endothelial-dependent vasodilatation (brachial FMD) with the changes in EPCs at either time point.
Discussion
Brief exposure to coarse PM derived from a rural setting elicited an increase in circulating EPC levels that persisted for at least 20 h. Changes in VEGF levels (a primary stimulator of EPC release) or all WBC fractions (representing a generalized cellular inflammation) did not account for this response. We speculate that the rapid increase in ECPs may be consistent with a broadly conserved systemic circulating response to an underlying “endothelial injury” and/or with acute autonomic imbalance favoring sympathetic nervous system (SNS) activation (Mobius-Winkler et al., 2009; Povsic & Goldschmidt-Clermont, 2008; Shantsila et al., 2007).
Prior studies relating air pollutants to EPCs
To date, published studies in the literature that have evaluated the effects of air pollution or SHS on EPCs have been mixed, with some showing increased while others showing decreased levels – likely depending on several study-related factors. A few studies have shown that PM2.5 (Haberzetti et al., 2012; O’Toole et al., 2010) and certain pollution components such as nickel (Liberda et al., 2010) and acrolein (Wheat et al., 2011) can reduce circulating EPC levels. Mechanistic experiments suggested that the EPC reductions were likely caused by a blunting of VEGF-mediated signaling that impairs the mobilization of EPCs from the bone marrow (Haberzetti et al., 2012). However, these studies were performed in animals and largely evaluated the chronic effects following several days to weeks of exposures.
Only two studies have been published regarding the effects of PM2.5 (O’Toole et al., 2010) and SHS (Heiss et al., 2008) on EPCs in humans over brief time periods. O’Toole et al. (2010) demonstrated that higher concentrations of ambient PM2.5 averaged over the prior 24 h were associated with reductions in circulating EPCs in young adults living in Utah. As in our study, alterations in VEGF levels (along with other cytokines) did not explain the EPC changes. On the contrary, Heiss et al. (2008) showed that a 30-min long exposure to SHS actually caused a rapid elevation in both EPC and VEGF levels. Sub-populations of more mature EPCs remained elevated for 24 h; however, they displayed several dysfunctional features (e.g. impaired chemotaxis to VEGF). The investigators provided evidence that reduced nitric oxide bioavailability played a role in facilitating the acute increase in EPC mobilization as well as contributed to their dysfunction.
Potential mechanisms
Numerous cardio-metabolic disease states (e.g. hypertension) as well as chronic smoking (Povsic & Goldschmidt-Clermont, 2008; Shantsila et al., 2007) and longer-term pollution exposures (Haberzetti et al., 2012; Liberda et al., 2010) have typically been associated with directionally opposite changes (i.e. lower) in EPC levels. We therefore speculate that the observed circulating EPC responses as well as the responsible mechanistic pathways may differ between acute (i.e. increased levels) and more chronic (i.e. reduced levels) time frames of pollution exposures. Lower EPC concentrations previously observed with longer-term air pollution exposures (≥24 h in duration) may reflect cumulative damage to the bone marrow or circulating EPCs, an impairment in EPC release, and/or a depletion of bioavailable stem cells due to their continuous mobilization and subsequent exhaustion.
On the other hand, the biological responses following brief high-dose air pollution exposures more likely resemble those induced by other sudden insults associated with acute elevations in EPCs (e.g. myocardial ischemia, exercise, 30 min of SHS exposure) (Dimmeler, 2010; Heiss et al., 2008). This response may reflect their normal mobilization in an effort to aid in the repair of endothelial damage, in this case caused by coarse PM exposure. Numerous factors (cytokines, colony-stimulating factors, VEGF, erythropoietin, stromal cell-derived factor-1, hypoxemia) play roles in recruiting EPCs as part of a biologically conserved response following vascular injury (Dimmeler, 2010). We did not observe a concomitant elevation in VEGF levels as was seen with SHS exposure (Heiss et al., 2008); thus, if this is the underlying mechanism the precise danger signals responsible for mobilizing EPCs remain to be clarified.
It is also plausible that augmented SNS activity induced by coarse PM inhalation could have played a role. PM2.5 is well-known to blunt HRV in a manner reflecting SNS activation (Brook et al., 2009, 2010). Results from this current study (Brook et al., in review) demonstrated that coarse CAP exposure likely also acutely promotes heightened SNS activity (reflected in elevated heart rates and reducted HRV). In this context it is important to note that prior studies have shown that enhanced β-adrenergic tone is a relevant mediator of acute EPC release from the bone marrow (Dimmeler, 2010; Galasso et al., 2013). We therefore hypothesize that PM2.5–10 induced SNS activation may also be a relevant pathway whereby acute exposures could trigger a rapid mobilization of EPCs.
Clinical implications
We believe that the circulating EPC elevation is indicative of (or serves as a marker for) harmful biological changes regardless of the underlying mechanism – endothelial injury and/or SNS activation. EPC mobilization most often represents a response to injurious factors (e.g. cardiac ischemia) upon the endothelium. In such a scenario, the observed elevation is evidence that coarse PM can cause a biologically relevant injury to the systemic vasculature. On the other hand, if the findings represent a response to an acute imbalance in autonomic tone (as with post-exercise), the changes are still of health importance. SNS activation is a well-established trigger of sudden CV events in vulnerable patients (Mittleman & Mostofsky, 2011). While the precise health implications of our results remain speculative, overall they support the notion that PM2.5–10 may be capable of contributing to the triggering of ischemic CV events – particularly among susceptible individuals with underlying vulnerable atherosclerotic lesions.
Limitations
These results represent analyses of pre-defined secondary study outcomes, but nonetheless they should be considered exploratory. However, the magnitude of EPC changes and statistical significance (particularly 20 h post-exposure) suggest that this was not a chance finding alone. The similar responses previously shown to occur following SHS exposure also support the veracity of our results. We acknowledge that future experiments are required to determine if the circulating EPCs, although increased in numbers, were dysfunctional in nature. There were no significant associations between the EPC elevations and reductions in FMD (endothelial dysfunction) or alterations in HRV (autonomic imbalance). Thus, the mechanisms (as we posited) underlying the EPC elevation must remain speculative. In addition, follow-up analyses are planned to explore the particle constituents (e.g. elements, organic carbon) responsible for the changes and whether or not coarse PM derived from urban sources causes similar (or potentially) greater EPC changes. Once the analyses of PM constituents are completed, we also plan to evaluate the effect on health outcomes of other important determinants of particle composition (e.g. sources of pollutants, seasons of exposure).
“While mean coarse PM levels were significantly lower during FA compared to CAP exposures, we recognize that the levels were above ambient outdoor concentrations during some of the FA exposures. The position of our HEPA filter in the coarse exposure facility prior to the series of concentrators may have reduced its efficiency as compared to some of our prior studies using a fine CAP system (Brook et al., 2009). Nonetheless, on every occasion each individual subject was exposed to higher PM level during their CAP compared to their own respective FA exposure scenario. Given the nature of the main statistical analyses comparing within subject changes in biological outcomes, we believe our findings therefore remain valid despite this limitation”. Finally, we recognize that the precise characterization and optimal methods to identify EPCs remains a subject of active debate in the scientific literature (Fadini et al., 2012; Mobius-Winkler et al., 2009); nevertheless, the techniques and markers used have been successfully employed and corroborated in our prior studies (Denny et al., 2007).
Conclusions
Brief inhalation of rural coarse PM prompted an acute increase in EPCs persisting for at least 20 h. This response likely reflects a circulating reaction to acute endothelial injury and/or heightened SNS activity. Our findings therefore support the plausibility that acute exposures to high levels of coarse PM could contribute to the risk for acute CV events among susceptible individuals.
Acknowledgments
This study was funded by grants from the National Institutes of Health’s Clinical and Translational Science Awards (NIH CTSA) (UL1RR024986) and from the US Environmental Protection Agency (RD83479701 and R833740).
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Brook RD, Rajagopalan S, Pope CA, III, et al. Particulate matter air pollution and cardiovascular disease. An update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–78. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- Brook RD, Urch B, Dvonch JT, et al. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009;54:659–67. doi: 10.1161/HYPERTENSIONAHA.109.130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunekreef B, Forsberg B. Epidemiological evidence on effects of coarse airborne particles on health. Eur Respir J. 2005;26:309–18. doi: 10.1183/09031936.05.00001805. [DOI] [PubMed] [Google Scholar]
- Chang HH, Peng RD, Dominici F. Estimating the acute health effects of coarse particulate matter accounting for exposure measurement error. Biostatistics. 2011;12:637–52. doi: 10.1093/biostatistics/kxr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demokritou P, Gupta T, Ferguson S, et al. Development and laboratory characterization of a prototype coarse particle concentrator for inhalation toxicology studies. Aerosol Sci. 2002;33:111–23. [Google Scholar]
- Denny MF, Thacker S, Mehta H, et al. Interferon-α promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood. 2007;110:2907–15. doi: 10.1182/blood-2007-05-089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S. Regulation of bone marrow-derived vascular progenitor cell mobilization and maintenance. Arteriolscler Thromb Vasc Biol. 2010;30:1088–93. doi: 10.1161/ATVBAHA.109.191668. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res. 2012;110:624–37. doi: 10.1161/CIRCRESAHA.111.243386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasso G, De Rosa R, Ciccarelli M, et al. β2-Adrenergic receptor stimulation improves endothelial progenitor cells mediated ischemic neoangiogenesis. Circ Res. 2013;112:1026–34. doi: 10.1161/CIRCRESAHA.111.300152. [DOI] [PubMed] [Google Scholar]
- Haberzetti P, Lee J, Duggineni D, et al. Exposure to ambient air fine particulate matter prevents VEGF-induced mobilization of endothelial progenitor cells from the bone marrow. Environ Health Perspect. 2012;120:848–56. doi: 10.1289/ehp.1104206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkema JR, Keeler GJ, Wagner JG, et al. Health Effects Institute Research Report 120. Boston (MA): Health Effects Institute; 2004. Effects of inhaled urban air particulates on normal and hypersecretory airways in rats. [PubMed] [Google Scholar]
- Heiss C, Amabile N, Lee AC, et al. Brief secondhand smoke exposure depresses endothelial progenitor cell activity and endothelial function. J Am Coll Cardiol. 2008;51:1760–71. doi: 10.1016/j.jacc.2008.01.040. [DOI] [PubMed] [Google Scholar]
- Liberda EN, Cuevas AK, Gillespie PA, et al. Exposure to inhaled nickel nanparticles causes a reduction in number and function of bone marrow endothelial progenitor cells. Inhal Toxicol. 2010;22:95–9. doi: 10.3109/08958378.2010.515269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of the burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study. Lancet. 2012;380:2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittleman MA, Mostofsky E. Physical, psychological and chemical triggers of acute cardiovascular events: preventive strategies. Circulation. 2011;124:346–54. doi: 10.1161/CIRCULATIONAHA.110.968776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobius-Winkler S, Hollriegel R, Schuler G, Adams V. Endothelial progenitor cells: implications for cardiovascular disease. Cytometry. 2009;75A:25–37. doi: 10.1002/cyto.a.20669. [DOI] [PubMed] [Google Scholar]
- O’Toole TE, Hellmann J, Wheat L, et al. Episodic exposure to fine particulate air pollution decreases circulating levels of endothelial progenitor cells. Circ Res. 2010;107:200–3. doi: 10.1161/CIRCRESAHA.110.222679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng PD, Chang HH, Bell ML, et al. Coarse particulate matter air pollution and hospital admissions for cardiovascular and respiratory diseases among Medicare patients. JAMA. 2008;299:2172–9. doi: 10.1001/jama.299.18.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povsic TJ, Goldschmidt-Clermont PJ. Endothelial progenitor cells: markers of vascular reparative capacity. Therapeut Advanc Cardiovasc Dis. 2008;2:199–213. doi: 10.1177/1753944708093412. [DOI] [PubMed] [Google Scholar]
- Puett RC, Hart JE, Yanosky JD, et al. Chronic fine and coarse particulate exposure, mortality, and coronary heart disease in the Nurses’ Health Study. Environ Health Perspect. 2009;117:1697–701. doi: 10.1289/ehp.0900572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Somers EC, Brook RD, et al. Endothelial cell apoptosis in systemic lupus erythematosis: a common pathway for abnormal vascular function and thrombosis propensity. Blood. 2004;103:3677–83. doi: 10.1182/blood-2003-09-3198. [DOI] [PubMed] [Google Scholar]
- Shantsila E, Watson T, Lip GYH. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol. 2007;49:741–52. doi: 10.1016/j.jacc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- Wheat LA, Haberzetti P, Hellmann J, et al. Acroelin inhalation prevents VEGF-induced mobilization of Llk-1+/Sca-1+ cells in mice. Arterioscler Thromb Vasc Biol. 2011;31:1598–606. doi: 10.1161/ATVBAHA.111.227124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanobetti A, Schwartz J. The effect of fine and coarse particulate matter air pollution on mortality: a national analysis. Environ Health Perspect. 2009;117:898–903. doi: 10.1289/ehp.0800108. [DOI] [PMC free article] [PubMed] [Google Scholar]