

Over the past 40 years, a number of seemingly unrelated mechanisms have been implicated in the early stages of glucose-mediated damage responsible for diabetic complications. Starting in 2000, a series of publications showed that each of these mechanisms reflects a single hyperglycemia-induced process: overproduction of superoxide by the mitochondrial electron transport chain. This discovery created a paradigm shift in the field1,2.
In the kidney, hyperglycemia causes increased reactive oxygen species (ROS) in both glomerular mesangial cells and proximal tubular cells. The central pathogenic role of hyperglycemia-induced superoxide in initating diabetic glomerular injury is directly supported by the observation that overexpression of superoxide dismutase as a transgene protects 8-month-old diabetic mice from developing increased fractional mesangial volume, increased glomerular transforming growth factor-β, increased collagen IV and increased plasma creatinine3. Transgenic expression of superoxide dismutase (SOD) similarly prevents diabetic retinopathy and cardiomyopathy.
This paradigm has been challenged in a recent paper by Dugan et al.4, who claim that in the diabetic kidney, ROS are decreased rather than increased. The authors also report that adenine monophosphate (AMP)-activated protein kinase (AMPK) activity, proliferator-activated receptor γ coactivator 1α (PGC1α) protein level and mitochondrial density are decreased in the diabetic kidney, and suggest that these reflect a feed-forward cycle of decreased AMPK activity, decreased PGC1α, and decreased mitochondrial biogenesis, initiated and maintained by decreased mitochondrial ROS production.
A Closer Look at the New ROS Data
Over the past decade, it has become clear that both physiological and pathological states are determined by complex interactions among different molecular and cellular populations. In addition, temporal and spatial factors are critically important. In early experimental and human diabetic nephropathy, pathognomonic changes occur exclusively in the glomeruli, where increased mesangial fractional area and glomerular basement membrane thickness are observed. Importantly, evidence of increased ROS production in the diabetic kidney is found almost exclusively in the glomeruli and proximal tubules. However, glomeruli and proximal tubules represent a very small percentage of the total kidney volume. Dugan et al.4 used electron paramagnetic resonance (EPR) and the cyclic nitrone spin trap, 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO), to measure ROS in whole kidney homogenate, but not in isolated glomeruli. Thus, these data only reflect diabetes-associated changes in the entire kidney. For live animal imaging of ROS, Dugan et al.4 used dihydroethidium (DHE) fluorescence to show superoxide production in whole kidney. They also used DHE fluorescence to detect DHE oxidation in kidney slices with confocal microscopy. However, DHE is readily spontaneously oxidized and often gives a high background. It can also undergo direct metabolic oxidation without any free radical reactions. Without HPLC confirmation of superoxide-specific DHE-derived products, plus demonstration that fluorescence is inhibited by SOD, interpretation of changes in DHE red fluorescence is problematic5. ROS need to be more specifically measured in renal glomeruli and proximal tubules isolated from diabetic kidneys to know the effect of diabetes on the renal structures whose morphology and function are affected by experimental diabetes.
Alternative Model of a Feed-Forward Cycle Involving Decreased AMPK activity and Decreased Mitochondrial Biogenesis, Caused by Increased ROS
Dugan et al.4 found that AMPK activity, PGC1α protein level and mitochondrial density are decreased in the diabetic kidney. They suggest that these reflect a feed-forward cycle initiated and maintained by decreased mitochondrial ROS, based on the observation that increased ROS can activate AMPK without affecting AMP concentrations. Dugan et al.4 propose that decreased ROS production causes decreased AMPK activity. This decrease in AMPK activity leads to a decrease in PGC1α protein, resulting in decreased mitochondrial density and a consequent decrease in mitochondrial ROS. Restoration of AMPK activity by the AMP-analog, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), normalized AMPK activity, PGC1α levels and mitochondrial density. Their model predicts that increased AMPK activity would increase mitochondrial ROS. However, when AMPKα is silenced in human endothelial cells, there is an increase, not a decrease, in ROS, and increased AMPK activity achieved either by AICAR or by overexpression of constitutively activated AMPK inhibits, rather than enhances, superoxide production induced by high glucose exposure in human umbilical vein endothelial cells (HUVECs)6. The AICAR-induced decrease in glomerular levels of the oxidative marker, 8-hydroxydeoxyguanosine (8-OHdG), observed by Dugan et al.4 is consistent with these data.
An alternative model of a feed-forward cycle involving decreased AMPK activity and decreased mitochondrial biogenesis caused by increased mitochondrial ROS is suggested by our discovery that high glucose-induced ROS causes deoxyribonucleic acid (DNA) strand breaks in the nucleus (Figure1). This activates the latent DNA repair enzyme poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP)2, which degrades nicotinamide adenine dinucleotide+ (NAD+) in the process of synthesizing ADP-ribose. High glucose reduces NAD+ content by 50% in wild-type endothelial cells, and PARP deficiency or inhibition prevents this7. This is important, because reduced NAD+ concentration inhibits the activity of the NAD+-dependent protein deacetylase sirtuin 1 (SIRT1)8. SIRT1 deacetylates and activates both PGC1α and liver kinase B1 (LKB1), the kinase that activates AMPK. This model provides an alternate explanation for the decreased AMPK activity, PGC1α expression and consequent decrease in mitochondrial mass observed by Dugan et al.4, which is caused by increased, rather than decreased mitochondrial ROS in response to high glucose. Experimental support for this comes from the recent demonstration that SIRT1 is downregulated in proximal tubules of diabetic mice before albuminuria occurs. Proximal tubule-specific SIRT1 transgenic and knockout mice showed prevention and aggravation of the glomerular changes that occur in diabetes, respectively, whereas non-diabetic knockout mice developed diabetic-like albuminuria. Proximal tubule SIRT1 overexpression was shown to epigenetically suppress expression of the tight junction protein, claudin-1, in podocytes of diabetic mice9.
Figure 1.
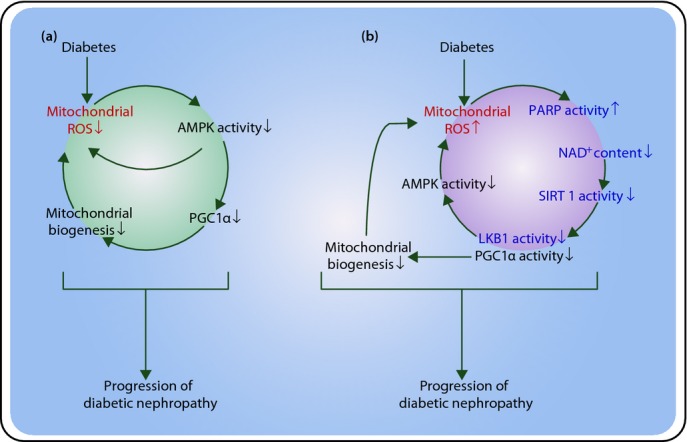
Proposed mechanisms involved in the pathogenesis of diabetic nephropathy. (a) Model of a feed-forward cycle involving decreased adenine monophosphate (AMP)-activated protein kinase (AMPK) activity and decreased mitochondrial biogenesis caused by decreased reactive oxygen species (ROS; proposed by Dugan et al.4). Dugan et al.4 reported that AMPK activity, proliferator-activated receptor γ coactivator 1α (PGC1α) protein level and mitochondrial density are decreased in the diabetic kidney. They suggested that these reflect a feed-forward cycle initiated and maintained by decreased mitochondrial ROS. They also showed that decreased ROS production was associated with decreased AMPK activity. (b) An alternate model of a feed-forward cycle involving decreased AMPK activity and decreased mitochondrial biogenesis caused by increased mitochondrial ROS. We previously reported that diabetes increased mitochondrial ROS. ROS causes deoxyribonucleic acid (DNA) strand breaks in the nucleus, resulting in activation of poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP), which degrades nicotinamide adenine dinucleotide+ (NAD+) in the process of synthesizing ADP-ribose. Reduced NAD+ concentration inhibits the activity of the NAD+-dependent protein deacetylase sirtuin 1 (SIRT1). SIRT1 deacetylates and activates both PGC1α and liver kinase B1 (LKB1), the kinase which activates AMPK.
The study by Dugan et al.4 is important, because science progresses through critical examination of current hypotheses in the light of new data and claims. Although their feed-forward loop data can be better explained by a model consistent with a diabetes-induced increase in ROS production, this study will stimulate new ideas and new experiments. The recent finding by Hodgin et al.10 that glomerular transcription networks in diabetic mice most closely resemble transcriptional changes that occur quite early in human diabetic nephropathy vs those that occur later reminds us that dominant pathogenic mechanisms likely differ for different stages of diabetic nephropathy. Discovering what the dominant multilevel molecular patterns are at different clinical/pathological stages of diabetic nephropathy will require imaginative new approaches using human tissue. Only with such knowledge will it be possible to develop therapeutic approaches that prevent the progression of very early diabetic nephropathy to end-stage renal failure.
References
- Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven PA, Melhem MF, Phillips SL, et al. Overexpression of Cu2+/Zn2+ superoxide dismutase protects against early diabetic glomerular injury in transgenic mice. Diabetes. 2001;50:2114–2125. doi: 10.2337/diabetes.50.9.2114. [DOI] [PubMed] [Google Scholar]
- Dugan LL, You YH, Ali SS, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol. 2004;142:231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Song P, Zou MH. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin Sci (Lond) 2012;122:555–573. doi: 10.1042/CS20110625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Soriano F, Virag L, Jagtap P, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- Yu J, Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2010;62:35–41. doi: 10.1016/j.phrs.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Wakino S, Simic P, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19:1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgin JB, Nair V, Zhang H, et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes. 2013;62:299–308. doi: 10.2337/db11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]