

Abstract
A key goal of cancer therapeutics is to selectively target the genetic lesions that initiate and maintain cancer cell proliferation and survival. While most cancers harbor multiple oncogenic mutations, a wealth of preclinical and clinical data supports that many cancers are sensitive to inhibition of single oncogenes, a concept referred to as ‘oncogene addiction’. Herein, we describe the clinical evidence supporting oncogene addiction and discuss common mechanistic themes emerging from the response and acquired resistance to oncogene-targeted therapies. Finally, we suggest several opportunities toward exploiting oncogene addiction to achieve curative cancer therapies.
Keywords: feedback, oncogene addiction, oncogenic shock, targeted therapy
Introduction
Cancer is a disease resulting from the acquisition of somatic genetic alterations. The results of extensive cancer genome sequencing and myriad preclinical in vitro and in vivo functional studies have underscored that cancers are initiated and maintained by recurrent ‘driver’ oncogene and/or tumor suppressor gene mutations. Established cancers in humans harbor, on average, approximately 30–60 mutations capable of altering protein function, with cancers such as melanoma bearing roughly 200 protein function-altering mutations per tumor 1. A key goal of cancer therapeutics development is to selectively target somatic cancer mutations—however, targeting all of these alterations in any one cancer seems a daunting task. Although cancer develops through progressive gene mutations that activate a variety of oncogenic functions, compelling evidence from preclinical studies, and most importantly from cancer patients treated with oncogene-targeted therapeutics, suggests that cancer cell survival relies on relatively few key genetic driver events. The term ‘oncogene addiction’ was coined to describe this phenomenon of exquisite cancer cell dependence on individual oncogenes to sustain the malignant phenotype 2.
Clinical evidence for oncogene addiction
As we focus herein on the clinical evidence for oncogene addiction, we direct the reader to excellent reviews of the preclinical data supporting oncogene and non-oncogene addiction 3, 4. A prime clinical example of oncogene addiction is in CML. CML is driven by the BCR-ABL mutant oncogene, produced as a result of chromosome 9:22 translocation, otherwise known as the ‘Philadelphia’ chromosome 5, 6. While preclinical studies provided evidence that BCR-ABL was a bona fide oncogene both in vitro and in vivo 7, 8, addiction of CML to BCR-ABL was demonstrated in patients through the profound clinical responses attained with the kinase inhibitor imatinib, which targets BCR-ABL. This addiction was further reinforced by the description of genetic mechanisms of resistance that largely led to reactivation of BCR-ABL kinase activity (described in more detail below). These observations in aggregate provided a transformative proof-of-concept for oncogene-targeted cancer therapy 9. As summarized in Table1 (and referenced therein), the strategy of targeting mutant oncogenic kinases has now been repeated many times over in a variety of cancer types. The common theme from these studies is a marked improvement in initial patient responses when oncogene-targeted therapies, tested in the correct oncogene-mutated patient population, are compared head-to-head with prior standard of care therapeutics.
Table 1.
Examples of approved oncogene-targeted therapies and observed resistance mechanisms in patients
| Target/indication | Inhibitor(s) | Observed clinical responses | Resistance mechanisms | ||
|---|---|---|---|---|---|
| Secondary oncogene mutation | Pathway mutations | Bypass | |||
| BCR-ABL mutant CML | Imatinib, nilotinib, dasatinib, ponatinib | Complete cytogenetic responses: 65–80% 9, 112, 113, 166 | T315I and other mutations, BCR-ABL amplification 26, 167, 168 | SRC family upregulation FGF2/FGFR3 activation 168, 169 | |
| KIT mutant GIST | Imatinib | 53.7% partial response in patients with refractory disease 170 | KIT mutations (e.g., V654A, T670I) or amplification 171, 172 | PDGFRA mutation 172 Rhabdomyosarcomatous differentiation 109 | |
| BRAF mutant melanoma | Vemurafenib, dabrafenib | 45–51% response rate; benefits observed versus prior standard of care 58, 59, 173 | P61-BRAF splice variant 28; BRAF amplification 174 | NRAS, NF1, MAP2K1, MAP2K2 mutation MITF amplification 31, 32, 33, 175, 176 | PI3K pathway mutations; CRAF, RTK, COT, AXL upregulation 29, 31, 33, 110, 175, 176, 177 |
| EGFR mutant NSCLC | Gefitinib, erlotinib, afatinib | 9–13 months progression-free survival; 73.7% response rate for gefitinib; benefit versus standard chemotherapy 178, 179, 180, 181, 182 | T970M mutation (+/− gene amplification ∽40–65%); other EGFR point mutations (∽1–2%) 105, 183, 184, 185 | PIK3CA mutation, BRAF mutation (∽1%) 105, 185 | MET or HER2 amplification, histologic transformation (EMT, SCLC ∽12–14%) 37, 38, 105, 185, 186 |
| EGFR-amplified colorectal cancer | Cetuximab, panitumumab | Improvements in progression-free survival versus best supportive care 187 | KRAS, BRAF, PIK3CA, PTEN mutation 187 | ||
| ALK-translocated NSCLC | Crizotinib, ceritinib, alectinib | 55–65% response rate; improved response rate versus standard chemotherapy 116, 117, 188 | L1196M, I1171T, V1180L, and other mutations, with or without amplification (∽28–65%) 185, 189, 190, 191 | KRAS mutation 191 | KIT amplification, EGFR upregulation or mutation, IGF-1R upregulation 190, 191, 192 |
| HER2/ERBB2-amplified breast cancer | Trastuzumab, lapatinib, pertuzumab | Trastuzumab: 33% combined complete and partial response rate 193; Lapatinib: 39% partial response rate 194 | Trastuzumab epitope mutations: p95-HER2, D16 27, 195 | PIK3CA/PTEN mutation 196 | EGFR, HER3, HER4, IGF-1R, MET upregulation and heterodimerization 195, 196 |
| ROS1-translocated NSCLC | Crizotinib | 72% objective response rate 197 | G2032R mutation 198 | ||
| RET mutant medullary thyroid carcinoma (MTC) | Vandetanib | 46% objective response rate in patients with hereditary MTC harboring RET mutation 199 | |||
| Retinoic acid receptor (RARA)-translocated APL | ATRA | Complete response rates of > 90%; superior to prior chemotherapy regimens 200 | Ligand-binding domain mutation (∽40%) 11, 201 | ||
| AR-positive castration-resistant prostate cancer | Enzalutamide | 18.4-month overall survival, 54% PSA reduction 202 | F876L mutation 99, 100, 101, AR-V7 ligand-binding domain truncation 203 | GR upregulation 203 | |
| ER-positive metastatic breast cancer | Tamoxifen, toremifene, fulvestrant, letrozole, anastrozole, exemestane | Tamoxifen: approximately 50% drop in mortality with 10 years of treatment 204 | Ligand-binding domain mutation 30, 205 | ||
The oncogene-addicted phenotype is not unique to mutated kinases. One of the earliest examples of targeted therapy (albeit one where clinical efficacy was established prior to molecular cloning of the causative oncogene) was the use of ATRA in APL. APL bears characteristic translocations affecting the retinoic acid receptor, generating fusion proteins such as PML-RARA that interfere with normal cell differentiation 10. ATRA binds to the ligand-binding domain of PML-RARA, which inhibits its oncogenic function 11. An additional example is the use of antiandrogens for the treatment of prostate cancers, which are ‘lineage-addicted’ 12 to AR and bear recurrent AR amplifications or mutations upon resistance to first-line therapies 13, 14. Finally, recent cancer genome sequencing has revealed a prevalent novel class of mutated oncogenes involved in the regulation of epigenetic states 15. Examples include oncogenic point mutations or chromosomal translocations affecting EZH2,NSD2,BRD4,IDH1, and IDH2. Given the aforementioned history of cancer addiction to mutated driver oncogenes, as well as emerging preclinical studies demonstrating the dependence on these oncogenes for tumor maintenance, drugs targeting these lesions have been rapidly developed 16, 17, 18, 19, 20. Several of these have already entered early clinical investigation, with encouraging initial responses 21, 22, 23, 24. Together, these striking results demonstrate that the concept of oncogene addiction indeed translates into clinical responses.
Therapeutic resistance reveals oncogenic pathway addiction
Despite the robust initial clinical responses described above, chronic exposure to most targeted therapeutics often gives way to relapse, and cures remain elusive. Does this argue against oncogene addiction? Answers lie in the observed clinical mechanisms of resistance to oncogene-targeted therapeutics. As detailed below, three common themes emerge upon resistance to many oncogene-targeted therapies; these themes demonstrate that most cancers retain an underlying addiction to oncogene-induced signaling pathways, if not a monolithic addiction to the originally mutated oncogene.
Secondary alterations of the oncogene drug target
Single-agent BCR-ABL inhibition often results in cancer cell apoptosis and profound long-term responses 9, 25; however, a significant fraction of patients show resistance to existing therapies. The main observed mechanism of resistance to BCR-ABL inhibition is the acquisition of second-site mutations in BCR-ABL itself 26. Predominant among these is mutation of the ATP-binding pocket at the ‘gatekeeper’ residue threonine 315. Mutation at this site prevents optimal binding of imatinib and other inhibitors, while still allowing ATP hydrolysis, and hence restoring BCR-ABL signaling in the presence of inhibitors (Fig1A). Treatment of lung cancers with drugs targeting mutant EGFR, ALK, and ROS1 also results in a significant fraction of resistant disease bearing second-site oncogene mutations that restore oncogene function in the presence of drug. These acquired mutations often occur within the highly conserved gatekeeper residue (Table1). The HER2 oncogene commonly develops resistance to the humanized HER2 antibody trastuzumab in a slightly different fashion; in this case, the trastuzumab-binding epitope is lost, while oncogene function is retained 27. BRAF obtains resistance to kinase inhibitors, at least in part, through either kinase amplification or truncations that further activate kinase activity 28, 29. Outside of kinases, AR, PML-RAR, and ER (a lineage driver for many breast cancers) acquire mutations in their ligand-binding domains that reduce or abrogate drug efficacy (Table1, 30) while restoring oncogene function. The common theme of treatment-acquired secondary oncogene alterations is that they provide resistance to therapy while reinstating oncogene function—this clinically observed resistance mechanism makes the most compelling argument for oncogene addiction.
Figure 1. Common mechanisms of resistance to oncogene-targeted therapeutics.

(A) Second-site mutations can reinstate oncogene function while abrogating inhibitor activity, as exemplified by BCR-ABL gatekeeper mutations as an inhibitor resistance mechanism. (B) Mutations in oncogene pathway components can reinstate pathway signaling despite continued oncogene inhibition, as exemplified by MAP2K1 mutations as a resistance mechanism for BRAF inhibitors. (C) Mutational or non-mutational activation of bypass signaling pathways can render cancer cells independent of the original oncogene, as exemplified by MET activation as a resistance mechanism for EGFR inhibition.
Activating mutations in oncogenic pathway components
Acquired resistance to oncogene-targeted drugs also occurs via mutation of alternate components of oncogene-induced signaling pathways. For example, mutant BRAF signals through the MAPK signaling pathway to promote melanoma growth. As such, one key resistance mechanism to BRAF inhibitors such as vemurafenib is the acquisition of activating mutations in other known MAPK signaling pathway components such as NRAS 31, or more rarely MAP2K1, and MAP2K2 31, 32; loss of function mutations in the negative MAPK pathway regulator NF1 31, 33; or amplification and activation of the MAPK pathway target gene MITF, a lineage driver of melanoma 31. All of these mutations restore MAPK oncogenic pathway signals despite continued pharmacological inhibition of mutant BRAF (Fig1B). This theme recurs in NSCLC, where activation of RTKs such as EGFR and ALK are key driver events. RTKs signal through several intracellular pathways, including the MAPK and PI3K pathways. As such, acquired resistance to HER2-, EGFR-, and ALK-targeted therapies includes selection for activating mutations in the MAPK or PI3K pathways (Table1 and references therein). As with second-site mutation of the oncogene itself, resistance mutations in key members of an oncogenic signaling pathway highlights that many cancers retain dependence upon specific oncogenic pathways, if not always the oncogene itself.
Induction of bypass pathway signaling
The third common theme in acquired resistance is the induction of bypass signaling pathways. A key example is observed in resistance to BRAF inhibitors. RAF family proteins normally function as dimers; common oncogenic mutations in BRAF (e.g., V600E) allow monomeric BRAF proteins to activate downstream signaling pathways in the absence of upstream activating signals 28. This activity is blocked by selective BRAF kinase inhibitors. However, CRAF, another key RAF isoform, can still activate the MAPK pathway in the presence of upstream pathway signals. This commonly occurs through the induction of growth factor-dependent and/or RAS-dependent signals 34. Such upstream activating signals are often paradoxically induced by oncogene inhibition, as discussed in more detail below. Despite mutant BRAF inhibition, upstream pathway activation can still signal through CRAF to reinstate MAPK pathway signaling. This is facilitated through the activation of CRAF homodimers, or through CRAF:BRAF heterodimers that are stabilized in the presence of some BRAF inhibitors 34, 35, 36.
Induction of oncogene bypass signaling is not unique to BRAF inhibitors. As discussed in more detail below, acquired resistance to EGFR-, HER2-, and ALK-targeted therapeutics commonly occurs through the upregulation or amplification of alternate RTK's (Table1). This bypasses the need for the mutated oncogene, but often reinstates the original downstream signaling pathways. A key example is the selection for activated MET signaling as a resistance mechanism to EGFR-targeted therapies 37, 38 (Fig1C). As another recent example, resistance to the AR inhibitor enzalutamide can be caused by glucocorticoid receptor (GR)-dependent bypass of AR signaling 39. While many bypass resistance mechanisms reinstate the same signaling pathways originally activated by the oncogene, this is not always the case. For acquired resistance to BRAF inhibitors in melanoma, mutational activation of the PI3K-PTEN-AKT pathway has been identified as a prevalent mechanism in bypassing tumor dependence on MAPK signaling 29, 40.
In summary, despite the apparent mechanistic diversity of acquired resistance to oncogene-targeted therapies, the three major themes of resistance outlined above demonstrate that most cancers retain addiction to specific oncogene-activated pathway signals. This suggests that the key dependencies of cancer cells remain tractable despite acquired resistance and that a better knowledge of resistance mechanisms can lead to rational therapeutic strategies that reduce or prevent resistance in the clinic.
‘Oncogenic shock’ as a model to understand response versus resistance to therapy
While many oncogene-addicted cancers show striking initial responses to targeted therapies, the heterogeneity of response within and across cancers must be noted. Why is the proportion of response and resistance so different between different oncogenes—for example, why are durable single-agent responses often seen with BCR-ABL inhibition in CML, while inhibitors of FLT3-ITD in AML appear to only provide transient benefits 41, 42? Similarly, why does inhibition of the same oncogene have divergent responses in different cancer types—as exemplified by a ∽50% response rate to BRAF inhibitors in BRAF mutant melanoma, but a less than 5% response rate in BRAF mutant colorectal cancers 43? Such inconsistencies could be explained by an inability to achieve complete and sustained target inhibition in different tumor types, due to pharmacological limitations across different drugs and among different patient populations 44. However, emerging data demonstrate that intrinsic biological differences across oncogenes and tumor types also exist.
A useful paradigm to understand the biological diversity of responses to oncogene inhibition is that of ‘oncogenic shock’ 45, 46. This hypothesis builds on the knowledge that activated oncogenes promote proliferation and survival, but at the same time paradoxically activate signals that promote arrest or apoptosis 47, 48, 49, 50, 51. Upon acute inactivation of oncogene signaling, the timing of how these two pathways respond may differ for different oncogenes, or in different contexts. If the oncogenic pathway is quickly blocked by a drug, while the paradoxical oncogene-activated growth inhibitory pathway is slow to turn off, then apoptosis, or oncogenic shock, prevails. Conversely, if the paradoxical growth inhibitory signals from the oncogene can quickly reset, this provides a scenario where cells may survive to become resistant to oncogene inhibition. This differential in pathway response, presumably due to differences in the turnover of signaling proteins such as phosphatases that negatively regulate discreet prosurvival or proapoptotic pathways 46, may explain why some oncogenes show more profound responses than others upon acute inhibition. What are the mechanisms that allow oncogenic shock phenotypes to occur—or to be bypassed—and are they common among cancers?
An early view into the mechanism of paradoxical oncogene-induced growth inhibitory pathways in cancer cells was afforded by a study of the differential sensitivity of BRAF mutant versus RTK-activated cancer cells to MAPK pathway inhibition 52. This study suggested that mutant BRAF activates a unique ERK-dependent transcriptional output, including the upregulation of DUSP phosphatases and the SPRY family of secreted RTK inhibitory proteins, both of which negatively regulate MAPK pathway signaling 53, 54. BRAF inhibition blocks MAPK-dependent growth signaling, but also shuts off MAPK-dependent SPRY expression, which relieves SPRY-dependent inhibition of HER-family-, FGFR-, and/or IGF-1R-dependent responsiveness to exogenous growth factors 55, 56. Such feedback appears to be particularly active in BRAF mutant colorectal cancers via the rapid activation of EGFR upon BRAF inhibition 57. While this explains the limited efficacy of BRAF inhibitors in this indication, it also provides the rationale for dual inhibition of BRAF and EGFR in BRAF mutant colorectal cancers 43, 57. In melanoma, paradoxical feedback pathway activation (as well as most other clinically observed resistance mechanisms) reinstates MAPK signaling, providing the rationale for dual BRAF/MEK inhibition 58, 59.
While BRAF signaling inhibition has become a paradigm for paradoxical feedback pathway activation, oncogenic alterations in EGFR, HER2, ALK, and MET also function through a MAPK-dependent feedback pathway to block IL-6-facilitated activation of STAT3 and PI3K pathway-mediated survival signals 60. Also like BRAF, BCR-ABL and FLT3-ITD oncogenes can block the expression of growth factor receptors via a MAPK pathway-dependent feedback mechanism 61. The contrasting behavior of BCR-ABL- and FLT3-ITD-dependent feedback responses noted in an isogenic cell background 61 is of particular interest for the oncogenic shock hypothesis: pulsed inhibition of BCR-ABL rapidly shuts down BCR-ABL-dependent downstream survival signaling (including MAPK pathway signaling), but BCR-ABL- and MAPK pathway-dependent inhibition of normal growth factor-dependent signaling is slow to revert back to its basal state. This creates a window of time where no prosurvival signals are present, resulting in apoptosis. While pulsed FLT3 inhibition in FLT3-ITD mutant expressing cells similarly inhibits oncogenic signaling, MAPK pathway-dependent negative feedback is rapidly lost and growth factor-dependent signaling pathway signaling is quickly restored. In this setting, there is not enough time for apoptosis to be induced before the FLT3-ITD-inhibited cells restore functional growth factor receptor-dependent signaling.
Together, these data provide evidence that oncogene-dependent feedback inhibition of growth factor-dependent signaling may be pervasive across many cancers (even BCR-ABL mutant CML). These studies furthermore suggest that the turnover rate of these feedback mechanisms in different cancers dictates the fine line between oncogenic shock versus the activation of bypass resistance mechanisms (e.g., BCR-ABL inhibition in CML versus BRAF inhibition in colorectal cancer). Finally, the data implicate the MAPK pathway as a key node regulating the oncogene-induced feedback inactivation of growth factor receptor signaling (Fig2).
Figure 2. Oncogenic shock versus feedback reactivation of growth factor receptor signaling.
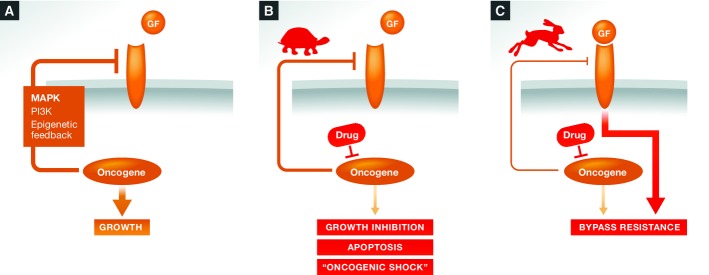
(A) Many oncogenes actively suppress growth factor receptor (GF-R)-dependent signaling in addition to activating oncogenic pathway signaling. (B) Oncogenic shock may predominate when GF-R survival signaling is slow to reinstate after oncogene inhibition (tortoise), creating a window where cells have no prosurvival signals. (C) Bypass resistance may predominate when GF-R responsiveness is quickly reactivated upon oncogene inhibition (hare).
While MAPK pathway-dependent feedback may commonly attenuate oncogenic shock responses across cancers, alternate mechanisms have been reported. First, altered epigenetic regulation can generate ‘drug-tolerant’ states that allow for the survival of small subpopulations within otherwise treatment-sensitive cancer cells; despite this alternate mechanism of resistance, survival is still generated through the upregulation of growth factor receptors such as IGF-1R 62. Second, PI3K-AKT-dependent feedback pathways have also been reported 63, 64—once again, resistance is driven through relief of oncogene-induced negative feedback regulation of growth factor receptors. A particularly interesting case is in prostate cancer—oncogenic PI3K and AR activation exists in a vicious cycle where inhibition of either pathway results in feedback upregulation of the other, via a mechanism that induces EGFR family RTK signaling 65. As above, knowledge of these feedback pathways sheds light onto rationally designed therapeutic combinations to prevent resistance—in this case, dual inhibition of AR and PI3K signaling pathways shuts down paradoxical bypass signaling and achieves remarkable efficacy in preclinical models.
A Road map for targeting oncogene addiction
The clinical benefits observed with agents targeting mutated oncogenes provide hope that the ‘one-step remedy’ to oncogene addiction initially proposed by Weinstein 2 may be attainable for some cancers. However, the common patterns of resistance to oncogene-targeted therapies must be anticipated and intercepted in order to achieve deep and sustained clinical benefit, and ultimately cures. The road map to curative therapy will require a rationally designed ‘one-two punch’ with combinations of targeted agents, rather than a one-step remedy. Below, we offer some suggestions to reach this goal.
Genetically define all cancers
Classical characterization of cancer subtypes include distinctions based on tissue type, histology, pathology, and level of differentiation, characteristics that are open to biased interpretation 66. Targeted therapies require a genetic definition for the patient populations most likely to respond. The most straightforward example is that of CML, where the BCR-ABL fusion defines the disease better than any histologic distinction. This is precisely because the genetic lesion directly impacts the therapeutic strategy (i.e., sensitivity to BCR-ABL inhibitors). This paradigm extends to other tumor types with more heterogeneous genetic etiology. Clinical trials of the EGFR inhibitor gefitinib in otherwise non-selected NSCLC patients originally failed to show strong differences in overall survival. It was not until the ‘outlier’ patients who responded to therapy 67 were retrospectively analyzed for EGFR mutations that the patterns of response in NSCLC could be rationalized 68, 69, 70. EGFR mutant NSCLC is therefore a key clinical subtype. This initial finding now extends to other mutations in NSCLC that are paired with targeted therapeutics (e.g., ALK translocations and ceritinib). An initially homogeneous histological subtype of lung cancer is therefore redefined into a mutation-stratified spectrum of diseases that dictates different therapeutic options 71. Many other tumor types such as glioma and glioblastoma are being similarly redefined based on molecular subtypes 72.
An extreme version of the mutation-driven definition of cancer is apparent in so-called basket trials (e.g., NCI-MATCH and the Novartis Signature trial), where both the histological and even lineage definitions of tumors break down. Patients with the mutation that predicts sensitivity to the disease are entered into a trial regardless of tumor type or lineage 73. The utility of such studies requires the sequencing of large panels of actionable oncogenes across all tumor types, with the affordability and rapid turnaround to routinely impact clinical decision-making.
To best pair the mutant oncogenes with targeted therapies, we need to complete our understanding of the genetic underpinnings of all cancers. Our knowledge of the key driver mutations across many cancers is approaching saturation for genes mutated at a frequency > 10%. However, our knowledge of important driver mutations is rapidly expanding for oncogenes mutated at lower frequency 74. Elucidating lower frequency oncogenes will uncover new molecular driver events and could highlight common signaling pathways that can be exploited therapeutically. In some cancers however, it has been challenging to define key oncogenic driver mutations. An extreme example of this is in pediatric ependymomas of the posterior fossa, where zero recurrent driver mutations in either oncogenes or tumor suppressor genes have been identified 75. Identifying driver mutations could be aided by improving our ability to sequence difficult regions of the genome, or by improving the analysis of existing cancer genome sequences 76, 77. In addition, improvements can be made in the analysis of genetic or epigenetic alterations in the ‘dark matter’ of noncoding DNA sequence 1. Regulatory sequence mutations can drive oncogenic function, as has been recently shown for TERT promoter mutations 78, 79, as well as somatic enhancer mutations that activate MYB oncogene binding 80. Similar mutations or epigenetic alterations that exist in other DNA regulatory sequences remain to be uncovered, as the characterization of these dark regions of the genome is only starting to come to light 81, 82.
Address tumor heterogeneity
A key consideration for proper molecular diagnosis of cancers is to account for inter- and intratumor heterogeneity. Issues with correctly diagnosing oncogene alterations from patient to patient could be influenced by the particular test used, as has been noted for expression-based tests for HER2 amplification 83—diagnostics focused on the specific detection of somatic cancer mutations will likely prove superior. Tumor biopsies can be biased by analyses of a small portion of the tumor that does not appropriately reflect intratumor heterogeneity, as has been documented in glioblastoma 84, 85, 86, or that does not capture the mutational heterogeneity of disseminated disease 1, 31. More systematic methods of identifying oncogene mutations and acquired resistance mutations, such as with the analysis of circulating tumor DNA 87, may provide a more holistic view of actionable mutations in a patient's disease, particularly in the case of treatment resistance.
Understanding intratumor heterogeneity is critical not just for diagnosis, but also for choosing the best targets for durable antitumor responses. The acquisition of mutations upon the progression of individual tumors resembles a phylogenetic tree, where ‘trunk’ mutations are initiating mutations present in every cancer cell, ‘branch’ mutations occur later during tumor progression and are present in distinct subregions, and ‘private’ mutations occur only in small individual regions of the disease. Patterns of trunk versus branch mutations are seen across a wide variety of cancer types 88, 89, 90, 91. Targeting trunk mutations, while unfortunately reducing the universe of possible drug targets, may provide key benefit in that it reduces the contribution of intratumor mutational heterogeneity in engendering resistance to treatment. Understanding branch mutations in parallel may allow one to recognize independently arising but convergent resistance mechanisms.
Address therapeutic resistance early
Due to the widespread heterogeneity of most cancers, the alterations promoting ‘acquired’ therapeutic resistance are almost certainly preexisting in most cancers 1. Therefore, resistance should be expected to arise as a by-product of any single-agent-targeted therapy, and as such should be addressed up-front. Resistance may be minimized by using multiple drugs targeting the same oncogene or oncogenic pathway in combination, as the chance any one tumor cell bears resistance mutations to multiple agents is small (discussed in more detail below). Resistance may also be minimized by expediting the use of novel targeted therapies in treating earlier stage cancers versus advanced metastatic disease. The chance of earlier stage tumors bearing resistance mutations is likely reduced, and therefore, responses may be more durable. To this point, the movement of trastuzumab into the adjuvant and neoadjuvant settings for early-stage HER2-positive breast cancer has demonstrated significant benefits 92, 93. On the other hand, the durable responses and long-term improvements in overall survival seen in chronic-phase CML upon BCR-ABL inhibition 94 are much less apparent in CML that has progressed to the accelerated phase or blast crisis 95, 96.
Two key areas of research are essential. First, it is critical that wherever feasible, serial biopsies from untreated tumors and resulting treatment-refractory tumors are analyzed for mutations and other alterations in oncogenic signaling pathways. Such studies have already revealed a broader picture of resistance mechanisms 31. As mentioned above for initial cancer diagnoses, systemic analyses of resistance in patients through methods such as circulating tumor DNA analyses 97 may aid in rapidly identifying clinically relevant resistance mutations. Second, prospective studies to identify acquired drug resistance mechanisms should be undertaken in the laboratory 98, even before resistance is seen in the clinic. Where performed comprehensively, such studies can guide subsequent analyses in clinical samples, as has been seen for enzalutamide-resistant prostate cancers 99, 100, 101 and BRAF and MEK inhibitor-resistant melanoma 102. Similarly, comprehensive functional genomic screens to identify novel combination targets with existing therapeutics, or screens to identify novel nodes to target treatment-resistant disease, should be actively explored 103, 104.
While, as described above, most cancers acquire therapeutic resistance through common themes that retain addiction to core oncogene pathways, an insidious emerging exception must be noted. Upon treatment with EGFR inhibitors, a substantial proportion of EGFR mutant lung cancers makes histological transformations to a small cell/neuroendocrine or mesenchymal phenotypes 105, 106, 107. This is not unique to NSCLC; prostate cancers transform to small cell/neuroendocrine phenotypes in order to evade androgen deprivation therapies, and there is growing concern that this may be an increasing issue for antiandrogen therapies such as enzalutamide and abiraterone 108. GISTs can also transdifferentiate, in this case to a rhabdomyoblastic state, upon imatinib resistance 109. Recently, a change in melanoma cell state has also been suggested as a novel resistance mechanism for MAPK pathway inhibition 110. Changes in cell state can completely abrogate dependencies on the signaling pathways present in the original tumor, negating most previously known therapeutic options. As targeted inhibitors (and combinations thereof) become more potent and selective, the transformation of cancers to a different cell type may become a more commonly observed phenomenon to bypass oncogene addiction. A better understanding of this phenomenon, and how to prevent it, is highly warranted.
Make better inhibitors
For many oncogenes, incomplete antitumor response and/or resistance can be facilitated simply by incomplete target inhibition. Incomplete inhibition could be due to insufficient drug potency, suboptimal pharmacokinetics, or poorly tolerated side effects that either preclude dosing to maximum efficacy or limit patient compliance. There are several clinical examples of more potent and selective ‘next-generation’ inhibitors showing responses superior to those of their predecessors. These include the efficacy of nilotinib and other next-generation BCR-ABL inhibitors in imatinib-pretreated CML 111, 112, 113, and the benefit of vemurafenib and dabrafenib in BRAF mutant melanoma, where the RAF/multikinase inhibitor sorafenib had previously failed 58, 114. Enzalutamide, a more potent androgen receptor (AR) antagonist, has shown clinical efficacy in castration-resistant prostate cancer, further validating continued addiction of these cancers to AR-dependent signaling 115. The more potent and selective ALK inhibitors ceritinib and alectinib show efficacy in crizotinib-resistant NSCLC 116, 117. It is anticipated that next-generation PI3K inhibitors with an optimized selectivity profile will show benefit in PIK3CA mutant cancers, where many earlier generation drugs have not yet shown durable responses 118, 119. Well-optimized next-generation inhibitors therefore are not merely ‘me too’ drugs, but an essential part of our cancer therapeutic armamentarium.
As inhibitors become more potent and selective for their intended targets, an upper limit can be reached where the dose-limiting toxicity is due to inhibition of the normal physiological function of the wild-type proto-oncogene. For example, earlier generation EGFR inhibitors are associated with diarrhea, skin rash, and other likely ‘on-target’ toxicities due to inhibition of physiological EGFR-dependent functions in epithelia 120, 121. Acquired resistance mutations such as those at the T790M gatekeeper residue increase the affinity of the mutant enzyme for ATP and further tip the balance of early generation EGFR inhibitors in favor of blocking wild-type over mutant oncogenic EGFR function 122. To address this issue, third-generation EGFR inhibitors have been designed to enhance the targeting of mutant EGFR isoforms, including the T790M gatekeeper mutation. These inhibitors show remarkable selectivity for mutant EGFR versus wild-type and therefore can be dosed more optimally to fully inhibit mutant EGFR signaling with a reduced liability of affecting physiological EGFR function in normal tissues 123, 124. This reduction in on-target side effects is a landmark step toward improving the efficacy of oncogene-selective drugs. Mutant-selective inhibition, wherever possible, should be a goal. Evidence suggests that this can be feasible for other oncogenes, as has been reported with the discovery of mutant-selective inhibitors of the IDH1 and IDH2 oncogenes 18, 19.
Develop different modes (and nodes) of target inhibition
For many oncogenes, it is likely that there is more than one way to pharmacologically inhibit protein function—all potential therapeutic options should be explored, as there is no a priori notion of which mode of target inhibition is best. For example, most BCR-ABL inhibitors work through similar mechanisms, and therefore, all eventually engender resistance through kinase domain mutations such as the gatekeeper T315I alteration. Recently, allosteric BCR-ABL inhibitors have been discovered, which show a different spectrum of secondary resistance mutation versus ATP-binding site inhibitors 125. Therefore, the combination of two different modes of inhibition targeting the same protein in parallel could dramatically reduce the occurrence of any one type of resistance mechanism. Allosteric kinase inhibition is not unique to BCR-ABL, as allosteric inhibition of other kinases such as MEK and AKT is attainable 126, 127. Indeed, allosteric MEK inhibitors that lock RAF:MEK protein complexes in an inactive state may provide a novel opportunity for more effective inhibition of BRAF- and MEK-dependent MAPK pathway signaling 128.
An alternate example of combining different therapeutic strategies against the same target is with the HER2 oncogene in breast cancer. Combination of HER2 targeting using both trastuzumab (targeting the extracellular domain) and lapatinib (targeting kinase activity) has shown significant improvements in pathological complete responses versus either agent alone 129, presumably because each drug targets a different part of the HER2 oncogene. Similarly, the HER2 antibody pertuzumab, which works in a mechanism complementary to trastuzumab, also shows combination benefit 130. In addition, the observation that HER2 expression is maintained in trastuzumab-resistant breast cancer has spurred the use of T-DM1, an ADC that delivers the cytotoxic microtubule inhibitor DM1 to cancers via HER2-dependent internalization. This modality has shown remarkable efficacy in patients previously treated with trastuzumab and taxanes 131—therefore, a combination of functional antibody and ADC approaches could reduce resistance.
Insight into oncogene-addicted signaling pathways affords the opportunity to discover key signaling nodes that can be exploited to more potently kill tumor cells. As mentioned above, most of the treatment-acquired resistance mechanisms to BRAF inhibitors in melanoma occur via restoration of MAPK pathway signaling. Thus, ‘vertical’ combination therapies simultaneously targeting different signaling nodes of the MAPK pathway (e.g., BRAF-, MEK-, ERK-, RTK-dependent signals) could overcome mutational or feedback-mediated resistance mechanisms and increase response to therapies. Indeed, results from clinical studies comparing single-agent RAF inhibitors versus RAF plus MEK inhibitor combinations demonstrate an increased rate of progression-free survival with the combination treatments 58, 59. Attacking multiple nodes in the HER2 signaling pathway may also provide benefit 132, 133. Some cancers may require ‘parallel’ combinations targeting different oncogenes in order to see any benefit; this may be due either to comutation, rapid feedback upregulation, or other mechanisms. As examples, combined up-front BRAF and EGFR inhibition may blunt the rampant feedback pathway signaling present in BRAF mutant colorectal cancers 57; co-inhibition of the MAPK and PI3K pathways may be required to see clinical benefit in many cancers with co-activation of these pathways 134; and intrinsic resistance to EGFR-targeted therapies may occur through baseline upregulation of potentially targetable factors such as CRIPTO1 135 Finally, improved knowledge of oncogenic signaling in B-cell malignancies provides the rationale for BTK inhibition, which has shown dramatic clinical benefit; analysis of resulting resistance mutations further suggests vertical combinations within the same pathway that could address or hopefully prevent resistance 136.
Make data-driven decisions on optimal dosing regimens
While complete inhibition of oncogene pathways is a key goal, this does not always warrant continuous exposure to a drug, particularly if complete pathway inhibition engenders an oncogenic shock response. In these cases, intermittent dosing might effectively kill tumor cells while also reducing any on- or off-target side effects that could occur through continuous dosing. To this point, the BCR-ABL inhibitor dasatinib can induce apoptosis upon pulse dosing in BCR-ABL mutant CML cells, both in preclinical models and in CML patients 137. These findings in CML were also extended, at least preclinically, to EGFR mutant cells treated with the EGFR inhibitor erlotinib 137. Together, these results suggest high pulse doses of inhibitors could indeed unleash oncogenic shock phenotypes in cancer cells. Intermittent dosing could also prevent the induction of bypass resistance mechanisms. A recently reported primary melanoma xenograft model of vemurafenib resistance shows that in some instances, melanomas can become vemurafenib dependent. Vemurafenib withdrawal causes regression of such tumors, and an intermittent dosing strategy keeps both the drug-sensitive and drug-resistant cancer cell populations in check, leading to extended efficacy 138. This finding is not unique to BRAF or the preclinical setting—resistance mutations in AR cause antagonists such as flutamide to become partial agonists 139. Also, patients with acquired EGFR T790M resistance mutations who are taken off drug demonstrate loss of this mutation in their tumors, which may explain why these patients respond upon retreatment with EGFR inhibitors that do not target the T790M mutation 105, 140. Similar retreatment effects are also seen with crizotinib 141, 142. Such findings must be interpreted carefully, as drug holidays prior to the acquisition of resistance may actually speed the resistance process 143; however, in total, these results suggest the optimization of dosing regimens for oncogene-targeted therapeutics is never ‘one size fits all.’ Optimal dosing schedules should be actively explored in preclinical models and inform the clinical testing of distinct dosing hypotheses in patients.
Challenge our notions of what is druggable
Cancer drug discovery has been rightly focused on kinases due to their clear genetic links to cancer, as well as their “druggability”—which simply means a proven ability to be inhibited by small molecules or antibodies. However, without the perspective of history, kinase druggability is not self-evident—the kinase pocket is highly conserved in paralogous proteins and the high intracellular concentration of ATP substrate could raise concerns for at least ATP-competitive inhibitors. Similar questions can be raised for virtually any novel target, and a priori notions of druggability should be avoided. While new classes of oncogenes will certainly not be easy to target, challenging mechanisms of target inhibition such as protein–protein interactions have yielded important breakthroughs with the discovery of p53/Mdm2 interaction inhibitors 144, BCL2 inhibitors 145, and SMAC mimetics 146, among others. The discovery of allosteric or induced-fit pockets on a protein may be a path forward for challenging cancer targets such as phosphatases 147 and metabolic enzymes 148. This warrants a fresh look at well-validated but historically undruggable oncogene targets such as Ras oncogenes 149 and transcription factors 150, 151. If a target truly proves undruggable, systematic efforts to find potential synthetic lethal signaling nodes, as described below, are highly warranted.
Comprehensively identify all liabilities of oncogene-addicted cells
While a plethora of functional genomics screens have reported novel synthetic lethal targets to exploit oncogene (and non-oncogene) addiction, most studies rely on addressing single hypothesis in only one or a few cell lines. It is not uncommon that many such discoveries do not pan out in additional cell line or tumor models. In addition, many functional genomics studies rely on an RNAi approach—due to the known off-target effects of this method, it is essential that studies are sufficiently powered with multiple RNAi constructs per gene to avoid false-positive results. Most studies are underpowered in this regard. As such, the industry validation rate of many novel targets identified through ‘one-off’ functional approaches has been poor 152. A few studies have begun to address such issues by either increasing the number of cell lines used 153 or increasing the number of RNAi constructs used per gene 154. Studies that combine both, while labor-intensive, will be better powered to identify the best targets within and across cancers. In addition, novel approaches such as gene editing via the CRISPR/CAS9 system 155 could and should be explored as target identification methods, as the off-target profile of such approaches may be reduced or at least different from RNAi, and complete genetic knockout may yield novel targets that require a higher level of inhibition than partial RNAi gene knockdown can afford. Such approaches, performed at scale, will be critical for the identification of robust synthetic lethal or ‘non-oncogene’ targets for oncogene and tumor suppressor gene mutations 3, 156.
Make well-characterized disease-relevant models, and make them available
Robust validation of genetic cancer dependencies in cell line and animal models is critical to progressing therapeutics toward the clinic. To facilitate this work, it is essential to have detailed characterization of available cell line and primary patient-derived xenograft models with respect to features including mutation, copy number, gene expression, and compound vulnerability, and to make such data publicly available to the research community, as has been done for the CCLE, CGP, and other cell line collections 157, 158, 159. One limit to available cell line datasets is that some tumor lineages (e.g., prostate cancer) and genotypes (e.g., IDH1/2 mutation) are underrepresented. It will be critical for the research community to identify these gaps in currently available models, and work to develop and characterize new in vitro and in vivo models for disease. Most importantly, the models should be readily available to the research community for use. With the increased use of PDX models 160, and the advent of alternate cell culture techniques that could facilitate the outgrowth of traditionally difficult tumor types such as glioma and prostate 161, 162, 163, comprehensive screens for oncogene vulnerabilities in currently underrepresented tumor types may soon be feasible.
Conclusions
Oncogene addiction is readily apparent from the remarkable responses seen in patients treated with drugs that target key mutated oncogenic drivers of their cancers. Potent inhibition of oncogenic signaling pathways can result in oncogenic shock, a robust apoptotic response that results in sustained remissions, not just transient growth inhibition. While acquired resistance to therapy remains a key challenge, clinically observed resistance mutations largely demonstrate that most cancers retain addiction to their original oncogenic signaling pathways, if not always the mutated oncogene itself. In addition to acquired resistance mutations, the induction of paradoxical bypass pathways that reactivate growth factor-dependent signaling (commonly through relief of MEK- or PI3K-dependent feedback inhibition) upon oncogene inhibition is likely pervasive across cancers and should be anticipated. Together, these findings underscore that many resistance mechanisms fall into predictable and therapeutically tractable themes, and can be effectively targeted with rationally designed combination therapies. Recognizing that this work is not completed until patients are cured of their disease, we have outlined some key items that must be addressed to take advantage of our growing knowledge of oncogene addiction. Not discussed in this review, but also essential to our ability to optimally kill cancers, are at least two additional approaches that should be combined with therapies that exploit oncogene addiction. First is the exploitation of somatic loss-of-function tumor suppressor gene mutations through the synthetic lethal targeting of ‘non-oncogene-addicted’ signaling nodes 3. Second is the utilization of our emerging ability to reactivate immune responses to tumors 164, 165. As we learn to effectively combine these emerging therapeutic options, the road toward a cure is becoming clear.
Sidebar A: In need of answers.
What therapeutic combinations can best exploit oncogene addiction and result in oncogenic shock (versus a transient response)?
How can therapeutics be optimized and/or combined to prevent resistance?
As paradoxical bypass pathway activation is a pervasive response to many therapies, what accounts for the differences in the activation of these pathways (e.g., oncogenic shock for BCR-ABL inhibition vs. common bypass pathway activation for BRAF inhibition)? Can bypass signaling mechanisms be anticipated and therapeutically exploited?
What actionable oncogenic mutations and pathways remain to be found across all cancers?
What are the best ways to tackle inter- and intratumor heterogeneity?
Acknowledgments
We thank Frank Stegmeier, Sreenath Sharma, Alex Gaither, Nick Keen, Zineb Mounir, Darrin Stuart, Julian Levell, Young Shin Cho, Andrew Wylie and Pascal Fortin for critical review of this manuscript. We acknowledge that many studies defining and characterizing oncogene addiction are not cited herein, and apologize for omissions due to limitations of both space and scope.
Glossary
- ADC
antibody–drug conjugate
- AKT
Ak thymoma kinase, key member of the PI3K pathway
- ALK
anaplastic lymphoma kinase, activated by gene translocation in cancer
- APL
acute promyelocytic leukemia
- AR
androgen receptor, lineage driver of prostate cancer
- ATP
adenosine triphosphate
- ATRA
all-trans retinoic acid
- BCR-ABL
B-cell receptor–Abelson kinase, oncogenic fusion protein that drives CML
- BRAF
v-Raf murine sarcoma viral oncogene homolog B, activated by point mutations in cancer
- BRD4
bromodomain containing 4, chromatin modulator activated by translocation in cancer
- BTK
Bruton's tyrosine kinase, key member of oncogenic B-cell receptor signaling
- CCLE
cancer cell line encyclopedia
- CGP
cancer genome project
- CML
chronic myelogenous leukemia
- CRAF
v-Raf murine sarcoma viral oncogene homolog B
- CRISPR/CAS9
clustered regularly interspaced short palindromic repeats/Cas9 nuclease; eukaryotic gene editing technology derived from a prokaryotic viral immune editing system
- DM1
maytansine derivative, toxic payload often linkered to ADC's
- DNA
deoxyribonucleic acid
- DUSP
dual specificity phosphatase, negative regulator of MAPK pathway
- EGFR
epidermal growth factor receptor, activated by point mutation and small deletions in cancer
- ER
estrogen receptor, lineage driver for breast and other cancers
- ERK
extracellular signal-regulated kinase, member of MAPK pathway
- EZH2
enhancer of zeste homolog 2, activated by point mutation in cancer
- FGFR
fibroblast growth factor receptor, activated by gene fusion and point mutation in cancer
- FLT3
Fms-like tyrosine kinase 3, activated by ITD and point mutation in cancer
- GIST
gastrointestinal stromal tumor
- HER2
human epidermal growth factor receptor 2
- HER3
human epidermal growth factor receptor 3
- IDH1
isocitrate dehydrogenase 1, activated by point mutation in cancer
- IDH2
isocitrate dehydrogenase 2, activated by point mutation in cancer
- IGF-1R
insulin-like growth factor 1 receptor, feedback activator of oncogenic signaling
- IL-6
interleukin 6 cytokine
- ITD
internal tandem duplication, a common mutation of the FLT3 oncogene
- KIT
v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog
- KRAS
Kirsten RAS viral oncogene homolog, activated by point mutation in cancer
- MAP2K1
mitogen-activated protein kinase kinase 1
- MAP2K2
mitogen-activated protein kinase kinase 2
- MEK
MAPK/ERK kinase, alternate common name for MAPK2K1, MAPK2K2, and/or MAPK pathway
- MAPK
mitogen-activated protein kinase
- MITF
microphthalmia-associated transcription factor, lineage driver for melanoma
- MYB
v-myb avian myeloblastosis viral oncogene homolog
- NRAS
neuroblastoma RAS viral oncogene homolog, activated by point mutation in cancer
- NSCLC
non-small cell lung cancer
- NSD2
Wolf–Hirschhorn syndrome candidate 1
- PI3K
phosphoinositide 3-kinase (pathway)
- PIK3CA
phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha, activated by point mutation in cancer
- PTEN
phosphatase and tensin homolog, tumor suppressor and negative regulator of PI3K pathway
- RAS
rat sarcoma viral oncogene; generic name for KRAS, NRAS, HRAS oncogenes, and/or the signaling pathway
- RNAi
RNA interference
- ROS1
ROS proto-oncogene 1
- RTK
receptor tyrosine kinase
- SPRY
Sprouty homologs (e.g., SPRY1, SPRY2), negative regulators of RTK signaling
- TERT
telomerase reverse transcriptase, activated by promoter point mutations in cancer
Author contributions
RAP, WS, and WRS all contributed to the concept and writing of this manuscript.
Conflict of interest
RAP, WS, and WRS are employees of the Novartis Institutes for BioMedical Research, Inc.
References
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- Bartram CR, de Klein A, Hagemeijer A, van Agthoven T, Geurts van Kessel A, Bootsma D, Grosveld G, Ferguson-Smith MA, Davies T, Stone M, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277–280. doi: 10.1038/306277a0. [DOI] [PubMed] [Google Scholar]
- Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- Tomita A, Kiyoi H, Naoe T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O 3) in acute promyelocytic leukemia. Int J Hematol. 2013;97:717–725. doi: 10.1007/s12185-013-1354-4. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45:1386–1391. doi: 10.1038/ng.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E, Garcia-Manero G, Rizzieri DA, Savona M, Tibes R, Altman JK, Jongen-Lavrencic M, Döhner H, Armstrong S, Pollock RM, et al. The DOT1L inhibitor EPZ-5676: safety and activity in relapsed/refractory patients with MLL-rearranged leukemia. Blood. 2014;124:387. [Google Scholar]
- Stein E, Tallman M, Pollyea DA, Flinn IW, Fathi AT, Stone RM, Levine RL, Agresta S, Schenkein D, Yang H, et al. Clinical safety and activity in a phase I trial of AG-221, a first in class, potent inhibitor of the IDH2-mutant protein, in patients with IDH2 mutant positive advanced hematologic malignancies. 2014. 2014 Apr 5–9; San Diego, CA. Philadelphia (PA): AACR; 2014. Abstract nr CT103 In Proceedings of the 105th Annual Meeting of the American Association for Cancer Research.
- Copeland RA, Keilhack H, Italiano A, Knutson SK, Yokoi A, Kawano S, Minoshima Y, Huang KC, Warholic NM, Johnston LD, et al. EZH2 Inhibitor EPZ-6438 (E7438) in Non-Hodgkin Lymphoma: Pre-Clinical Models and Early Clinical Observations. 2014. ASH Meeting on Lymphoma Biology 2014.
- Pollyea DA, de Botton S, Fathi AT, Stein EM, Tallman MS, Agresta S, Bowden C, Fan B, Prah M, Yang H, et al. Clinical safety and activity in a phase 1 trial of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in patients with IDH1 mutant positive advanced hematological malignancies. Eur J Cancer. 2014;50(Suppl 6):195. [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, Di Cosimo S, Matias-Guiu X, Ramon y Cajal S, Arribas J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99:628–638. doi: 10.1093/jnci/djk134. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–349. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, Hugo W, Yu CC, Ng C, Chodon T, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4:69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AY, Man CH, Kwong YL. FLT3 inhibition: a moving and evolving target in acute myeloid leukaemia. Leukemia. 2013;27:260–268. doi: 10.1038/leu.2012.195. [DOI] [PubMed] [Google Scholar]
- Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5:65–77. doi: 10.1177/2040620714532123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Warmuth M, Sellers WR, Dorsch M. Targeted cancer therapies in the twenty-first century: lessons from imatinib. Clin Pharmacol Ther. 2010;87:543–552. doi: 10.1038/clpt.2009.297. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Settleman J. Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock”. Biochem Pharmacol. 2010;80:666–673. doi: 10.1016/j.bcp.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Gajowniczek P, Way IP, Lee DY, Jiang J, Yuza Y, Classon M, Haber DA, Settleman J. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–435. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol. 2004;5:441–450. doi: 10.1038/nrm1400. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26:207–221. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- Asmussen J, Lasater EA, Tajon C, Oses-Prieto J, Jun YW, Taylor BS, Burlingame A, Craik CS, Shah NP. MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction. Cancer Discov. 2014;4:200–215. doi: 10.1158/2159-8290.CD-13-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab SS, Meier FA, Zarbo RJ, Jensen DC, Geisinger KR, Booth CN, Krishnamurti U, Stone CH, Janosky JE, Grzybicki DM. The “Big Dog” effect: variability assessing the causes of error in diagnoses of patients with lung cancer. J Clin Oncol. 2006;24:2808–2814. doi: 10.1200/JCO.2005.04.3661. [DOI] [PubMed] [Google Scholar]
- Blackhall F, Ranson M, Thatcher N. Where next for gefitinib in patients with lung cancer? Lancet Oncol. 2006;7:499–507. doi: 10.1016/S1470-2045(06)70725-2. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59:1190–1199. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
- Willyard C. ‘Basket studies’ will hold intricate data for cancer drug approvals. Nat Med. 2013;19:655. doi: 10.1038/nm0613-655. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506:445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Hodis E, Jasmine M, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46:1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci USA. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes CB, Rocha RM, Buzelin MA, Balabram D, de Souza Foureaux F, Porto SS, Gobbi H. False positivity in HER2 testing of breast cancer: novel paths for approaching an old dilemma. J Clin Pathol. 2013;66:946–950. doi: 10.1136/jclinpath-2013-201647. [DOI] [PubMed] [Google Scholar]
- Little SE, Popov S, Jury A, Bax DA, Doey L, Al-Sarraj S, Jurgensmeier JM, Jones C. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012;72:1614–1620. doi: 10.1158/0008-5472.CAN-11-4069. [DOI] [PubMed] [Google Scholar]
- Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20:810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA. 2012;109:3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Glaberman U, Dayao Z, Royce M. HER2-targeted therapy for early-stage breast cancer: a comprehensive review. Oncology (Williston Park) 2014;28:281–289. [PubMed] [Google Scholar]
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Rea D, Etienne G, Nicolini F, Cony-Makhoul P, Johnson-Ansah H, Legros L, Huguet F, Tulliez M, Gardembas M, Bouabdallah K, et al. First-line imatinib mesylate in patients with newly diagnosed accelerated phase-chronic myeloid leukemia. Leukemia. 2012;26:2254–2259. doi: 10.1038/leu.2012.92. [DOI] [PubMed] [Google Scholar]
- Palandri F, Castagnetti F, Testoni N, Luatti S, Marzocchi G, Bassi S, Breccia M, Alimena G, Pungolino E, Rege-Cambrin G, et al. Chronic myeloid leukemia in blast crisis treated with imatinib 600 mg: outcome of the patients alive after a 6-year follow-up. Haematologica. 2008;93:1792–1796. doi: 10.3324/haematol.13068. [DOI] [PubMed] [Google Scholar]
- Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–1043. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl P, Tedesco D, Chenchik A. Use of RNAi screens to uncover resistance mechanisms in cancer cells and identify synthetic lethal interactions. Drug Discov Today Technol. 2014;11:11–18. doi: 10.1016/j.ddtec.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci USA. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6:1152–1161. doi: 10.1097/JTO.0b013e318216ee52. [DOI] [PubMed] [Google Scholar]
- Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014;4:60. doi: 10.3389/fonc.2014.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liegl B, Hornick JL, Antonescu CR, Corless CL, Fletcher CD. Rhabdomyosarcomatous differentiation in gastrointestinal stromal tumors after tyrosine kinase inhibitor therapy: a novel form of tumor progression. Am J Surg Pathol. 2009;33:218–226. doi: 10.1097/PAS.0b013e31817ec2e6. [DOI] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–827. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, Pasquini R, Clark RE, Hochhaus A, Hughes TP, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783–1796. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, Morcos PN, Lee RM, Garcia L, Yu L, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15:1119–1128. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- Burtness B, Anadkat M, Basti S, Hughes M, Lacouture ME, McClure JS, Myskowski PL, Paul J, Perlis CS, Saltz L, et al. NCCN Task Force Report: Management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(Suppl 1):S5–S21. doi: 10.6004/jnccn.2009.0074. quiz S22–24. [DOI] [PubMed] [Google Scholar]
- Herbst RS, LoRusso PM, Purdom M, Ward D. Dermatologic side effects associated with gefitinib therapy: clinical experience and management. Clin Lung Cancer. 2003;4:366–369. doi: 10.3816/clc.2003.n.016. [DOI] [PubMed] [Google Scholar]
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjin Tham Sjin R, Lee K, Walter AO, Dubrovskiy A, Sheets M, Martin TS, Labenski MT, Zhu Z, Tester R, Karp R, et al. In vitro and in vivo characterization of irreversible mutant-selective EGFR inhibitors that are wild-type sparing. Mol Cancer Ther. 2014;13:1468–1479. doi: 10.1158/1535-7163.MCT-13-0966. [DOI] [PubMed] [Google Scholar]
- Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- Ishii N, Harada N, Joseph EW, Ohara K, Miura T, Sakamoto H, Matsuda Y, Tomii Y, Tachibana-Kondo Y, Iikura H, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050–4060. doi: 10.1158/0008-5472.CAN-12-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gomez H, Dinh P, Fauria K, Van Dooren V, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379:633–640. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Dieras V, Guardino E, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- Garner AP, Bialucha CU, Sprague ER, Garrett JT, Sheng Q, Li S, Sineshchekova O, Saxena P, Sutton CR, Chen D, et al. An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res. 2013;73:6024–6035. doi: 10.1158/0008-5472.CAN-13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395–1409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- Park KS, Raffeld M, Moon YW, Xi L, Bianco C, Pham T, Lee LC, Mitsudomi T, Yatabe Y, Okamoto I, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest. 2014;124:3003–3015. doi: 10.1172/JCI73048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Staudt LM. Ibrutinib treatment of CLL: the cancer fights back. Cancer Cell. 2014;26:11–13. doi: 10.1016/j.ccr.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NP, Kasap C, Weier C, Balbas M, Nicoll JM, Bleickardt E, Nicaise C, Sawyers CL. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008;14:485–493. doi: 10.1016/j.ccr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, Balk SP. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997;3:1383–1388. [PubMed] [Google Scholar]
- Riely GJ, Kris MG, Zhao B, Akhurst T, Milton DT, Moore E, Tyson L, Pao W, Rizvi NA, Schwartz LH, et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res. 2007;13:5150–5155. doi: 10.1158/1078-0432.CCR-07-0560. [DOI] [PubMed] [Google Scholar]
- Schrodl K, von Schilling C, Tufman A, Huber RM, Gamarra F. Response to chemotherapy, reexposure to crizotinib and treatment with a novel ALK inhibitor in a patient with acquired crizotinib resistance. Respiration. 2014;88:262–264. doi: 10.1159/000364949. [DOI] [PubMed] [Google Scholar]
- Browning ET, Weickhardt AJ, Camidge DR. Response to crizotinib rechallenge after initial progression and intervening chemotherapy in ALK lung cancer. J Thorac Oncol. 2013;8:e21. doi: 10.1097/JTO.0b013e31827a892c. [DOI] [PubMed] [Google Scholar]
- Foo J, Chmielecki J, Pao W, Michor F. Effects of pharmacokinetic processes and varied dosing schedules on the dynamics of acquired resistance to erlotinib in EGFR-mutant lung cancer. J Thorac Oncol. 2012;7:1583–1593. doi: 10.1097/JTO.0b013e31826146ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Ray A, Barrett R, Nelson E, Christie AL, Porter D, Straub C, Zawel L, Daley JF, Lazo-Kallanian S, et al. Smac mimetics: implications for enhancement of targeted therapies in leukemia. Leukemia. 2010;24:2100–2109. doi: 10.1038/leu.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin AG, Faitg TH, Richter M, Groy A, Seefeld MA, Darcy MG, Peng X, Federowicz K, Yang J, Zhang SY, et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol. 2014;10:181–187. doi: 10.1038/nchembio.1427. [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Hurov J, Cianchetta G, Murray S, Dang L. Action at a distance: allostery and the development of drugs to target cancer cell metabolism. Chem Biol. 2014;21:1143–1161. doi: 10.1016/j.chembiol.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat Med. 2009;15:750–756. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop MS, Stransky N, Garvie CW, Theurillat JP, Hartman EC, Lewis TA, Zhong C, Culyba EK, Lin F, Daniels DS, et al. A small molecule that binds and inhibits the ETV1 transcription factor oncoprotein. Mol Cancer Ther. 2014;13:1492–1502. doi: 10.1158/1535-7163.MCT-13-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci USA. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci USA. 2014;111:3128–3133. doi: 10.1073/pnas.1316793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, Zubovych I, Seo BY, Kim J, Eskiocak B, Chung H, et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell. 2013;155:552–566. doi: 10.1016/j.cell.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11:24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, Moiraghi B, Shen Z, Mayer J, Pasquini R, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- Roumiantsev S, Shah NP, Gorre ME, Nicoll J, Brasher BB, Sawyers CL, Van Etten RA. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc Natl Acad Sci USA. 2002;99:10700–10705. doi: 10.1073/pnas.162140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour EJ, Cortes JE, Kantarjian HM. Resistance to tyrosine kinase inhibition therapy for chronic myelogenous leukemia: a clinical perspective and emerging treatment options. Clin Lymphoma Myeloma Leuk. 2013;13:515–529. doi: 10.1016/j.clml.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traer E, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, Martinez J, Tyner JW, Wong M, Druker BJ. Ponatinib overcomes FGF2-mediated resistance in CML patients without kinase domain mutations. Blood. 2014;123:1516–1524. doi: 10.1182/blood-2013-07-518381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Wang WL, Conley A, Reynoso D, Nolden L, Lazar AJ, George S, Trent JC. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2011;67(Suppl 1):S15–S24. doi: 10.1007/s00280-010-1513-8. [DOI] [PubMed] [Google Scholar]
- Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–481. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, Iafrate AJ, Takeuchi K, Taiji M, Okuno Y, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20:5686–5696. doi: 10.1158/1078-0432.CCR-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, Florin A, Ozretic L, Lim D, Wang L, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20:1027–1034. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Carbonell X, Castaneda-Soto NJ, Clemens M, Green M, Harvey V, Morales S, Barton C, Ghahramani P. Phase II study of efficacy, safety, and pharmacokinetics of trastuzumab monotherapy administered on a 3-weekly schedule. J Clin Oncol. 2005;23:2162–2171. doi: 10.1200/JCO.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Kaufman B, Trudeau M, Awada A, Blackwell K, Bachelot T, Salazar V, DeSilvio M, Westlund R, Zaks T, Spector N, et al. Lapatinib monotherapy in patients with HER2-overexpressing relapsed or refractory inflammatory breast cancer: final results and survival of the expanded HER2+ cohort in EGF103009, a phase II study. Lancet Oncol. 2009;10:581–588. doi: 10.1016/S1470-2045(09)70087-7. [DOI] [PubMed] [Google Scholar]
- Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17:1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012;18:6784–6791. doi: 10.1158/1078-0432.CCR-12-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk MD, Timofeevski S, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368:2395–2401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degos L, Dombret H, Chomienne C, Daniel MT, Miclea JM, Chastang C, Castaigne S, Fenaux P. All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia. Blood. 1995;85:2643–2653. [PubMed] [Google Scholar]
- Gallagher RE, Moser BK, Racevskis J, Poire X, Bloomfield CD, Carroll AJ, Ketterling RP, Roulston D, Schachter-Tokarz E, Zhou DC, et al. Treatment-influenced associations of PML-RARalpha mutations, FLT3 mutations, and additional chromosome abnormalities in relapsed acute promyelocytic leukemia. Blood. 2012;120:2098–2108. doi: 10.1182/blood-2012-01-407601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381:805–816. doi: 10.1016/S0140-6736(12)61963-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]