

Abstract
Reprogramming to pluripotency is a low-efficiency process at the population level. Despite notable advances to molecularly characterize key steps, several fundamental aspects remain poorly understood, including when the potential to reprogram is first established. Here, we apply live-cell imaging combined with a novel statistical approach to infer when somatic cells become fated to generate downstream pluripotent progeny. By tracing cell lineages from several divisions before factor induction through to pluripotent colony formation, we find that pre-induction sister cells acquire similar outcomes. Namely, if one daughter cell contributes to a lineage that generates induced pluripotent stem cells (iPSCs), its paired sibling will as well. This result suggests that the potential to reprogram is predetermined within a select subpopulation of cells and heritable, at least over the short term. We also find that expanding cells over several divisions prior to factor induction does not increase the per-lineage likelihood of successful reprogramming, nor is reprogramming fate correlated to neighboring cell identity or cell-specific reprogramming factor levels. By perturbing the epigenetic state of somatic populations with Ezh2 inhibitors prior to factor induction, we successfully modulate the fraction of iPSC-forming lineages. Our results therefore suggest that reprogramming potential may in part reflect preexisting epigenetic heterogeneity that can be tuned to alter the cellular response to factor induction.
Keywords: cell fate decisions, live-cell imaging, reprogramming
Introduction
Somatic cells can be reprogrammed to a pluripotent state by overexpression of defined transcription factors, Oct4, Sox2, Klf4, and c-Myc (OSKM) 1, 2, 3, 4. The reprogramming process is characterized by widespread epigenetic changes that generate induced pluripotent stem cells (iPSCs) with the functional and molecular characteristics of embryonic stem cells (ESCs) derived from the early embryo 3, 5, 6, 7, 8, 9. Generation of iPS cells is a robust and highly reproducible procedure, yet it is exceedingly inefficient at the per-cell level and requires an extended latency before autonomous pluripotency is acquired 10. Different models have been suggested to explain these two notable attributes 11. On one extreme, a fully stochastic model suggests that every cell division essentially constitutes a coin toss in which the cell ‘decides’ whether or not to reprogram. In this model, all cells are equally likely to reprogram at any time after factors have been induced, independent of their history prior to the time of induction. On the other extreme, a deterministic ‘elite’ model posits that the initial cell population contains a subpopulation that is predisposed or fated to successfully reprogram. Experimental work designed to test these models have offered different perspectives that vary between these two extremes. For instance, low-frequency stochastic reprogramming is inherent to any continuously proliferating lineage given enough time and cellular divisions 12. We have previously described an early decision point after which the trajectory to successful reprogramming becomes defined, suggesting that the initial response to factor induction may determine the downstream trajectory 13. By sampling single-cell transcription over the reprogramming timeline, high cell-to-cell variability in gene expression was found to describe early reprogramming, after which a deterministic hierarchical phase is acquired 14. Finally, a recent study suggested that only select subpopulations of granulocyte–monocyte progenitors can reprogram, and do so with higher efficiency, while the majority of cells remain intransigent 15.
To investigate this in more detail, we used a live imaging approach to characterize key decision points and contributing factors during the reprogramming process. We find evidence suggesting that the potential to reprogram is largely pre-established within somatic cells before reprogramming factors are induced. We show that responding cells differ in their pre-induction properties from non-responding ones and that perturbing the epigenetic state of the somatic population prior to reprogramming can alter the potential of single cells to generate iPSC-forming lineages. Our findings emphasize the relevance of preexisting cell-to-cell variability in reprogramming, expanding prior studies that pointed to the early stages following factor induction as critical to the final outcome. These observations will eventually lead to a better molecular definition of cellular state that includes a given cell's potential to respond to transcriptional perturbation and has implications to other processes beyond reprogramming to pluripotency.
Results
We employed the NGFP2 MEF secondary reprogramming system, where all somatic cells contain identical integrations of OSKM factors under doxycycline (dox)-inducible promoters 13, 16, and therefore, phenotypic variability must be non-genetic in origin. In all experiments, we focused on colonies generated within a 2-week reprogramming timeline to concentrate on the initial wave of iPSC colony formation, which is generally more defined than colonies that emerge later 12, 13. To assist in cell lineage tracking, we transduced the MEFs with lentiviral vectors that constitutively express fluorescent proteins prior to reprogramming to create populations of uniquely labeled cells. To minimize efficiency calculation errors resulting from previously described satellite colonies that emanate from primary reprogramming lineages 13 and do not represent de novo acquisition of pluripotency, we used a colony-counting method which is estimated exclusively from colonies that can be traced back to the original fibroblast (see Materials and Methods).
Cells are predisposed to major cell fate decisions before factor induction
To determine when the potential to successfully generate iPSC colonies is established, we devised a strategy inspired by the Luria–Delbrück experiment. The original experiment demonstrated that acquisition of resistance through mutation precedes selection by employing a pre-growth period prior to screening for mutants 17. In our version, we begin with a known number of MEFs and allow them to divide several times prior to factor induction, increasing the number of cells per well while holding the number of lineages constant (Fig1A). If the potential to reprogram is largely predetermined, the fraction of iPSC containing wells will depend on the initial population size, assuming that the potential is inherited in daughter cells over the short term. In a post-determined model, reprogramming will depend only on the number of cells at the time of induction, increasing the fraction of iPSC containing wells as a function of population number.
Figure 1.
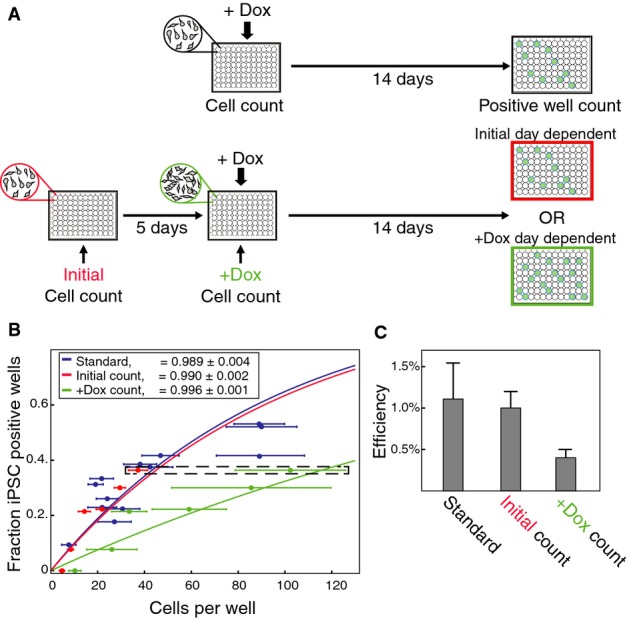
- A Schematic of the Luria–Delbrück inspired experiment. Doxycycline (dox) was administered after either no delay or 5 days following plating. Cells in each well were counted both after plating and at dox induction. The number of GFP+ wells at the end of 14 days was used to distinguish between different potential acquisition models (see text).
- B Reprogramming efficiencies measured as fraction of wells with GFP+ colonies as a function of cells per well. Dark blue mark denotes mean and standard deviation of one 96-well plate experiment where dox was administered immediately after plating. Red and green marks denote wells that were induced to reprogram 5 days after plating binned according to their cell number as demonstrated in Supplementary Table S1. For each group: red mark, initial cell count; green mark, cell count at day of dox induction. A pair of marks with the same y-value corresponds to the same group of wells. The dashed box highlights the specific wells exemplified in Supplementary Table S1. The solid curve of each color represents the theoretical efficiency for the corresponding reprogramming probability parameter, calculated as the maximum-likelihood estimator according to each data set separately (see Supplementary Text S2). The data are based on two independent experiments.
- C Reprogramming efficiency parameters corresponding to the three models shown in (B). Error bars represent standard deviation computed using sample bootstrapping (see Supplementary Text S2). Efficiency computed from initial count is more similar to the standard than that computed from count at day of dox induction.
We seeded cells at different low densities in 96-well plates (n + 17) and initiated reprogramming after 0 days (13 plates) or 5 days (4 plates), counting the exact number of cells in each well both at the day of plating and at the time of OSKM induction. After 2 weeks, we assessed the fraction of wells containing iPSC-marker-positive colonies and inferred the per-cell efficiency (Materials and Methods). By counting wells rather than colonies, we avoid inaccurate scoring of satellite colonies as unique reprogramming events when estimating efficiency 13. Given an expected reprogramming efficiency of 1% for our system, we seeded only a small number of cells per well (10–100) to ensure that the fraction containing iPSCs will be within the dynamic range (i.e., lower than 96 wells) to precisely measure per-cell efficiency. As it has been reported that reprogramming potential in fibroblasts is diminished by progressive passaging 18, we separately tested reprogramming efficiency in cells that were expanded for 5 days before being replated and induced by OSKM (Supplementary Text S1, Supplementary Fig S1). We found no effect for the 5-day expansion period, ruling out a possible confounding effect of reduced efficiency due to a later generation.
We first calculated the reprogramming efficiency using plates in which dox was applied on day 0 (with no delay, Fig1A, top). In these plates, the starting cell number and the number on the day of induction are trivially the same, so the estimated efficiency would be applicable to both models. By computing the fraction of positive wells for a certain starting number, the standard efficiency can be easily calculated (Fig1B, blue). After verifying that reprogramming efficiency does not depend on the location of the well on the plate (Supplementary Fig S2), we determined the ‘standard failure rate’ parameter, which represents the probability that a cell does not produce any reprogrammed progeny, to be 0.989 ± 0.004 (maximum-likelihood estimator, see Supplementary Text S2), consistent with previous lineage normalized estimates using this system 13. A similar efficiency was obtained using labeled subpopulations within a standard cell density (Supplementary Fig S3), verifying the low density in this experiment does not affect efficiency.
We next estimated the efficiency in wells where the addition of dox followed a 5-day expansion period of our restricted starting cell populations (Fig1A, bottom). To increase the distinction between the two tested models, we only used wells that at least doubled their cell number between the two counts (n + 71 wells). We estimated the failure rate parameter separately according to starting cell count (0.990 ± 0.002) or count at time of dox (0.996 ± 0.001) using bootstrap sampling over a maximum-likelihood estimator (see Supplementary Text S2). The failure rate parameter according to starting cell count is closer to the standard failure rate parameter computed above, where the number of reprogramming lineages matches the number of cells at the time of induction. In fact, the efficiency as estimated from the number of cells at the time of induction (‘+ dox count’) is 2.5-fold lower than expected if the likelihood of reprogramming were stochastically distributed to all cells equally at that time (Fig1C). To visualize the reprogramming efficiency according to each model, we divided the 71 wells to 6 groups of 10–13 wells, where each group has a similar initial cell count (Supplementary Table S1), and plotted the average efficiency for each group according to either initial count or dox-day count (Fig1B). Efficiency as a function of starting cell number (Fig1B, red) is closer to the day 0 ‘scaling points’ (Fig1B, blue) than the efficiency as a function of cell number at time of induction (Fig1B, green). This suggests that starting cell count (day 0) is a better predictor of reprogramming efficiency than the number of cells at the time of induction, favoring a predetermined per-lineage model over any post-determined model, including a fully stochastic one. To rule out the possibility that a higher local density after 5 days could reduce the per-cell exposure to dox, or deleteriously bias reprogramming in any way, we specifically disrupted cellular position within each well by brief trypsinization prior to factor induction, resulting in no effect on the apparent lineage dependence of reprogramming outcome (Supplementary Fig S4).
Predetermined potential is symmetrically maintained over the short term
The results above suggest the potential to reprogram is determined before OSKM induction, with limited acquired potential generated during ensuing divisions. However, we do not know how the potential is inherited within the lineage or how stable it is. For example, it could be restricted in sequential steps along the lineage, similar to fate restriction during early development. Alternatively, it could be equally inherited during each cell division. A third option is that cells within the potentiated lineage interconvert between ‘amenable’ and ‘recalcitrant’ states. To better understand how reprogramming potential is restricted and to validate the point of its appearance, we analyzed the fate statistics of lineage pairs of fluorescently labeled secondary MEFs. Cells were tracked from 2 days before induction of reprogramming. After the first division, prior to dox induction, two sister cells were tagged as paired lineages. We designated the fate of each cellular lineage into one of three categories: iPSC (Nanog-GFP+ forming), fast dividers (FD, Nanog-GFP-, indicative of transformation without reprogramming), and non-responder (NR, which do not acquire rapid proliferation or exhibit overt changes in fibroblast morphology, see Materials and Methods for full definitions). We then independently assigned these fates to both lineages within a pair to examine the possible combinations between them (Fig2A). Notably, in each of the pairs examined, the two lineages adopted the same fate (Fig2B, Supplementary Movie S1). We prospectively counted 58 pairs of FD-FD lineages, 6 pairs of iPS-iPS lineages, and 79 pairs of NR-NR lineages. We did not observe any pair of lineages acquiring mixed fates (such as iPS-FD, where only one of the lineages contributes to iPSC and the other is transformed). Repeating the same experiment at a higher cell density resulted in similar within-pair correlations, verifying the effect is not density dependent (Supplementary Fig S5).
Figure 2.
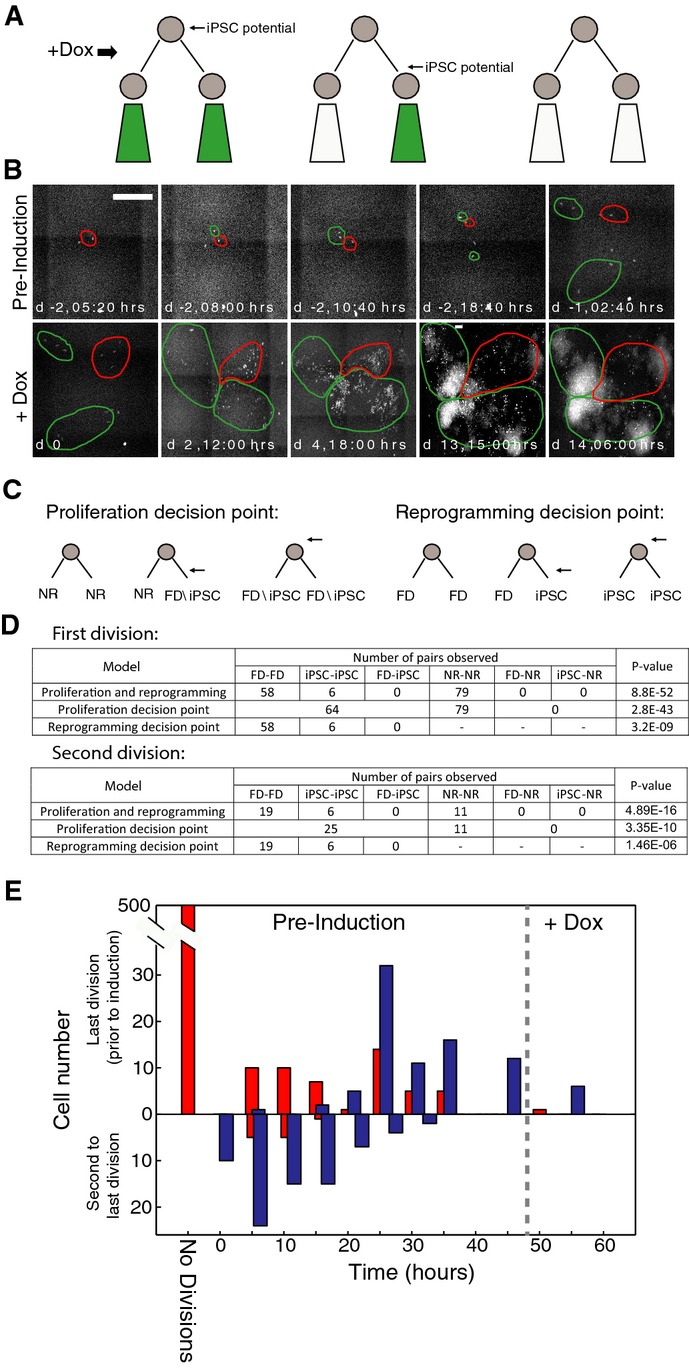
- A Schematic of the ‘paired lineage’ concept, depicting the last cell division prior to induction of OSKM (black arrow). The paired lineages can be classified into three categories with respect to when the potential to become an iPSC is obtained: potential may be acquired before the first division and both sub-lineages will include iPSC colony forming events (left); potential may be acquired in one sub-lineage (or lost in its sibling lineage) after the first division, resulting in two different fates (center); or no potential is acquired over the timeline (right). Green color denotes a sub-lineage that will form iPSCs.
- B Snapshots from imaging a cell lineage originating from a single MEF where the first cell division occurred 8 h after imaging. From that division on, the paired lineages (marked red and green) were traced. After 18:40 h, the green lineage divided again and its sub-lineages were tracked separately. Dox was added at day 0. The final GFP+ colonies are composed of cells from different sub-lineages (see also Supplementary Movie S1). Scale bar, 500 μm.
- C Possible paired lineage outcomes for two different decision points under a model assuming sequential acquisition of proliferation and reprogramming potentials. Left: possible pairs given the point of obtaining the potential for fast proliferation (marked by an arrow). Right: possible pairs given the point of obtaining the potential for reprogramming, assuming the cell already has fast proliferation potential.
- D Counts of tracked lineage pairs from the 1st or 2nd division with each corresponding fate combination. No mixed-fate pairs (e.g., iPS-FD) were observed. P-values are shown for each post-division fate decision model as described in text, rejecting all three post-division decision models.
- E Division times during the 48 h that cells were tracked before initiating reprogramming, grouped according to their response as proliferating (either iPSC or FD, blue) and non-responding (NR, red) lineages. The ‘no division’ bar represents cells that did not divide prior to dox induction.
We used these lineage pair counts to rule out different models in which potential is acquired or lost after the first division. For example, by taking the observed counts of all 6 possible paired lineage combinations for the three fates (FD, NR, or iPSC), we can reject a model in which cells ‘decide’ their fate after the initial division (here, the term decision refers to gain or loss of a fate potential). It is possible, however, that multiple fate decisions may occur within discrete steps. For example, cells may or may not decide to proliferate in response to OSKM, and only as a second decision may proliferating cells acquire full reprogramming potential (Fig2C). The time of acquiring each of these potentials would be reflected statistically within our lineage pair counts. A model in which cells acquire the potential to proliferate (shared between iPSC and FD fates) only after the first division can be ruled out by computing a P-value of iPSC and FD versus NR lineage pairs, which represents the probability of getting the observed count or higher of same-fate lineage pairs from our data compared to an alternate random pairing model. Given random pairing, if potential is acquired independently after the initial division, the observed combinations of pairs (in this case NR-NR, NR-FD/iPS, or FD/iPS-FD/iPS) will follow a random distribution (see Supplementary Text S3). A model in which proliferative cells acquire or lose reprogramming potential after the initial division can also be ruled out by computing the P-value for which our FD versus iPSC lineage data reflect the random acquisition of FD-FD, FD-iPS, and iPS-iPS pairs. Using our empirical paired lineage counts, we can reject all three of these models at high significance (Fig2D, top). Repeating the same analysis for pairs resulting from the second observed division resulted in similar statistics (Fig2D, bottom), suggesting fate potential is maintained (not gained or lost) over at least two divisions. Pairs from later divisions are harder to track, but some loss (or partial fulfillment) of fate potential in parts of the sub-lineages is observed (Fig2B, Supplementary Movie S1).
The apparent predisposition toward different reprogramming fates suggests there are different internal states in the somatic cell population, which may be reflected by other cellular properties. We first tested whether the proliferation rate of the cells prior to induction correlates with their response to reprogramming by marking the number of divisions in each lineage during the 48 h prior to OSKM induction (Fig2E). While proliferating cells of either the iPSC or FD fates could not be distinguished in this manner, both are likely to divide more times before induction than non-responding cells. Most responding cells divide at least twice during this period, while the majority of NR cells do not divide at all during the same period of time, and about 10% of them divide once. Thus, while division rate may distinguish between senescence-prone cells and proliferative cells, it cannot sufficiently predict whether responding cells will successfully navigate to pluripotency or simply acquire features associated with transformation. While we cannot rule out that the source of proliferation rate heterogeneity present in MEFs prior to factor induction could be effected by a mosaically represented genetic component, previous evidence converting non-responding to reprogramming cells by Mbd3 inhibition suggest these differences can be altered epigenetically 3.
Reprogramming potential is independent of local neighborhood and of early OSKM levels
What are the mechanisms that predetermine a cell's response during the reprogramming process and what enables a predisposed lineage to realize this potential? One option is that environmental cues—such as reinforcing signals coming from neighboring cells—affect the future fate of the colonies. As cells within a lineage reside in close proximity to one another, they could respond similarly as a consequence of a shared local environment. Support from neighboring cells should be reflected by some preferential relative locations of future iPSC lineages to a specific type of lineage. To test this possibility, we examine the distribution of distances between starting cells in a reprogramming experiment (Fig3A) and how it statistically depends on the final fates of their progeny. We observed no significant difference between the distance distributions of iPSC to FD, iPSC to NR, or FD to FD progenitors (Fig3B), suggesting the relative location of starting cells does not affect their future fate. However, the reprogramming lineage itself could also supply a self-supportive local niche, which supports identical fates within each pair of sub-lineages. To temporarily remove the possible effect of lineage niche, we replated a CFP-labeled population of reprogramming MEFs at specific time points onto YFP-labeled cells reprogrammed in parallel (Fig3C). In this system, reprogramming CFP cells are isolated from their original spatial niche, which includes both their lineage mates and neighboring lineages, and are randomly distributed among YFP lineages of different fates. After replating of the CFP cells, we followed the lineages for an additional 10–14 days and annotated the terminal fate of both CFP and YFP lineages. We then computed the distribution of distances between different lineage types (Fig3D and E). At both early (days 2–6) or late (days 8–12) replating time points, we could not find any spatial effect—the distance distributions between all lineage types are similar. These results suggest that though signals from other colonies in the well may provide supportive and essential signals for successful reprogramming, the local proximity to specific neighboring lineages does not distinguish between different lineage fates.
Figure 3.
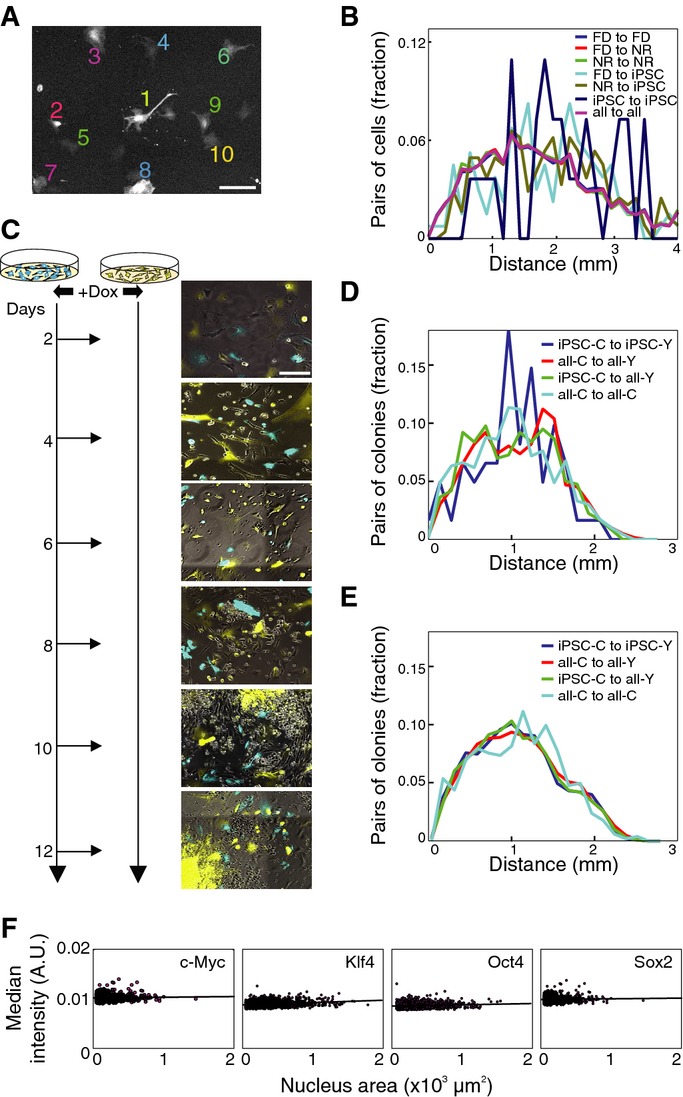
- A, B All cells in a standard reprogramming experiment were annotated by final lineage fate, and distances between them at the time of induction were computed. Sample frame (A) at day 0 with 10 annotated cells. Scale bar, 100 μm. Histograms of cell-to-cell distances (B) between cells with different terminal fates show no significant relationship between proximity and outcome.
- C Schematic of replating experiments. CFP- and YFP-labeled MEF cells were reprogrammed separately and in parallel. At specific time points within the 2nd to 12th day of reprogramming, CFP-labeled cells were replated onto stage-matched YFP cells. Scale bar, 200 μm.
- D, E Histograms of distances between colonies of various fates replated after days 2–6 (D) or 8–12 (E). iPSC-C, iPSC-Y: CFP- or YFP-labeled iPSC colony, respectively; all-C, all-Y: CFP- or YFP-labeled colony of any type, respectively. The similarity between all distance distributions shows that there is no preference for the relative location of iPSC colonies.
- F Correlation between the level of each factor on day 2 of reprogramming and morphological response. Plots show size of nuclear immunostaining signal for a given factor within induced cells against their median fluorescence intensity.
It is possible that cell replating during the course of reprogramming can disrupt the process. For example, it has been shown that a mesenchymal to epithelial transition (MET) occurs early during reprogramming 19, 20. Replating during this phase may disturb MET mechanically and consequently perturb downstream events. To study the effect of replating in isolation, we repeated the experiment differently by plating over empty or feeder-covered wells. We estimated final colony counts for different replating days (2–12) as well as time of colony appearance after replating (Supplementary Fig S6). Replating early in the process (days 2–4) resulted in both marked delay in appearance of colonies and lower number of final iPSC colonies compared to the non-replated case. Alternatively, replating during the later stages of the process (days 8–12) resulted in increasingly higher number of iPSC colonies that form with minimal delay after replating, suggesting that by this stage, iPSC-forming cells are more likely to maintain their route. For these later time points, replating may increase colony number as a trivial reflection of multiple iPSC-fated cells from each iPSC lineage being spatially distributed to different positions 14.
The per-cell expression level of the OSKM factors represents an ectopically induced cue that could also affect reprogramming potential. Despite the clonal origin of our secondary system, differential activation within single cells is possible. Different epigenetic states at sequence features of the lentiviral vectors could affect factor induction from their Tet-responsive promoters and lead to different fates in a simple way. For example, OSKM level could positively correlate to reprogramming outcome, and previous reports have shown that refractory reprogramming lineages with low factor expression can be rescued by elevating OKSM levels 21. We found that OKSM levels are much higher in the NGFP2 inducible system than in the polycistronic OKSM cells used in Polo et al and that efficiency is not increased by additional supplementation (Supplementary Fig S7). To test whether the different behaviors are caused by different nuclear concentrations of the factors early in the reprogramming process, we examined the correlation between OSKM protein levels and the behavior of cells after induction. After 2 days of reprogramming, cells undergo consistent changes in morphology, usually resulting in a decrease in cell size 13 as well as nucleus size (Supplementary Fig S8). Using this behavior, we can distinguish cells that respond positively to factor induction (FD/iPSC) from those that do not. We stained reprogramming cells on days 0, 2, 4, and 6 days after induction using antibodies against OSKM. We indeed observe a variable level for each of the factors from day 2 onward, but found no negative correlation between nucleus size and the level of fluorescence (Fig3F, Supplementary Fig S9). Together, these results suggest that the variable response to reprogramming is not due to obvious differences in OSKM factor levels at early stages.
Perturbing H3K27 or H3K4 methylation pre-induction alters future lineage fates
With exogenous explanations for these fated responses discounted, we hypothesized that differences in reprogramming potential may be epigenetic in origin and reflect innate differences in nuclear state. Discrete MEF responses may be a consequence of different chromatin states, either global or at the level of specific genes, that could permit constructive factor engagement at target sites upon their induction. Perturbation of chromatin modifiers has been extensively screened over the reprogramming process itself, some targets of which contribute to population level effects in reprogramming efficiency 3, 19, 22, 23. Our findings suggest that such treatments could also be effective when limited to a period preceding OSKM induction, if they alter the cell's epigenome in a manner that changes its predisposition to reprogram. To test this hypothesis, we subjected our secondary system to a panel of drugs that affect epigenetic modifications for either 24 or 48 h during the 2 days that precede factor induction (Fig4A). We compared the NGFP+ colony count at day 14 to an untreated control. Of the drugs tested, both Lsd1 and Ezh2 inhibitors showed the most significant increase in efficiency, increasing the number of Nanog-positive colonies ∽3-fold compared to DMSO-treated and untreated controls (Fig4B). Ezh2 is a histone methyltransferase that catalyzes repressive H3K27 methylation 24 (Supplementary Fig S10), while Lsd1 is a histone demethylase removing H3K4 mono- and di-methylation 25. Subsequently, inhibition of either may result in a permissive chromatin state that could enable otherwise recalcitrant cells to switch to a reprogramming amenable state.
Figure 4.
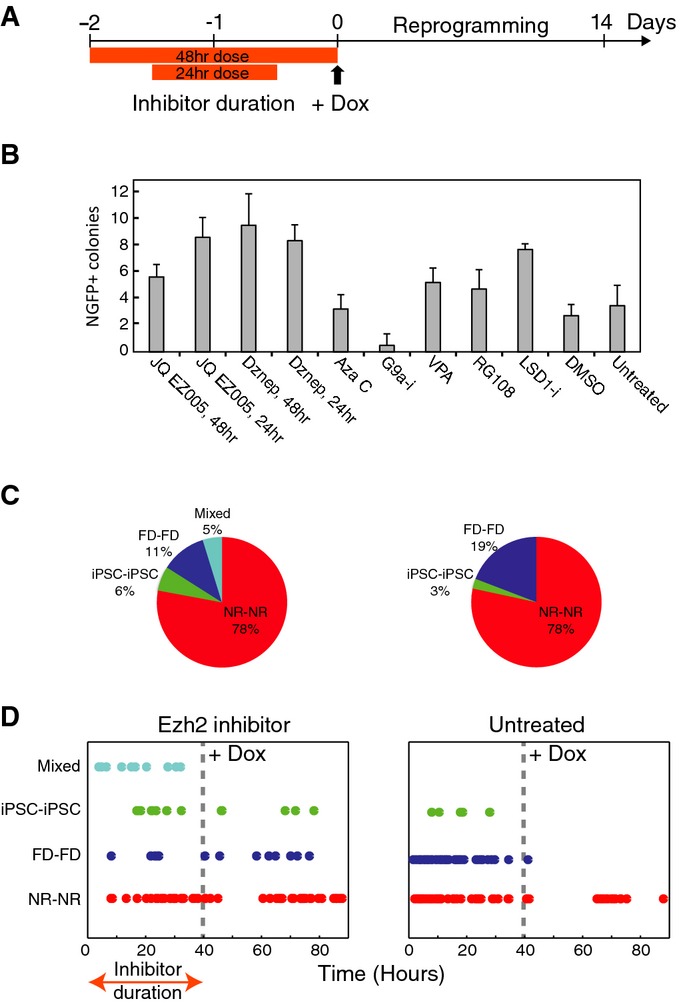
- A Schematic of inhibitors experiment. Inhibitors were added during the 48 h preceding dox induction, for either 24 or 48 h.
- B Effect of pre-treatment with different inhibitors on reprogramming efficiency. Shown are mean and standard deviation for imaged colony counts over 6 replicate wells (see counting procedure in Materials and Methods).
- C Fraction of lineage pairs with each corresponding fate combination after transient Ezh2 inhibition in the somatic population (left) or after no treatment (right, taken from Fig2).
- D Time of first division for the different lineage pairs under Ezh2 pre-dox inhibition (left) or no treatment (right).
We further studied the effect of Ezh2 inhibitor pre-treatment on reprogramming. We hypothesized that Ezh2 inhibition could improve reprogramming by several different mechanisms: it could alter the number of reprogramming amenable MEFs, enable downstream stochastic fate switching, or simply amplify predisposed lineages, such that the final colony count is higher but the per-cell efficiency would be unchanged. To distinguish between these options, we repeated our ‘paired lineage’ experiment on cells treated with the Ezh2 inhibitor during the 2-day window where cell lineages are traced prior to dox induction. We reasoned that a delayed stochastic switch would enable the appearance of mixed pairs (e.g., FD-iPS), which would result in only one of the branches acquiring reprogramming potential. Alternatively, if all lineage pairs remain symmetrical in terms of their fates, as was observed in the untreated case, but with a higher fraction of pairs becoming iPSCs, then Ezh2 inhibition acts to increase the number of amenable cells within the population. Under the third scenario, the same number of lineages would reprogram, but would divide faster during the treatment period, creating more colonies of a secondary nature that increase the efficiency estimate artifactually. Supplementary Movie S2 shows a representative 16-day time lapse with lineage tracking for one such well. 27% of the wells (19/71) generated iPSC colonies, compared to 11% (11/96) in the no-treatment experiment (Fig2), consistent with the global efficiency calculated during our screen (Fig4B). Our data indicate the effect of Ezh2 inhibition is not on proliferation. Instead, the distribution of pair types (Fig4C) suggests the majority of additional iPSC pairs may come from converted FD pairs. Though the majority of pairs we followed were still symmetrical, about 10% (10/98) were asymmetrical (4 NR-iPS pairs and 6 NR-FD pairs). These results suggest the Ezh2 inhibitor treatment increases the number of cells that generate iPSC-forming lineages, with some asymmetric lineage pairs between proliferative and non-proliferative fates possibly reflecting some cytotoxic effect. We compared the time of first division (that generates each pair) between Ezh2 inhibition and no pre-treatment conditions (Fig4D). The treatment appears to delay division time, causing a significant fraction of cells to divide for the first time only after OSKM induction. Interestingly, all mixed pairs divide early during the treatment period, while their following divisions occur mostly after dox induction and drug withdrawal (not shown). These pairs were exposed to Ezh2 inhibition for a longer period as individual cells than symmetrically fated pairs, allowing more time for the treatment to act differentially on isolated daughter cells, possibly allowing modulation between different responses.
Discussion
Our main results show that reprogramming potential is inherent to somatic cells prior to factor induction and that this potential is shared between a pair of lineages originating from the same pre-induced progenitor cell. These data suggest that reprogramming potential is set at least several divisions before induction and is heritable in the short term. Consistently, we do not find any effect of local signaling from neighboring lineages on the fate adopted by cells. Recently, an early stochastic phase was proposed to exist during reprogramming, based on high cell-to-cell variability in expression of specific genes in the same inducible system as the current study 14. Our results suggest that this variability may stem from preexisting differences in cell states, rather than stochastic switching between different states after reprogramming is initiated 26. Stochastic steps leading to potential loss or realization within each iPSC lineage may still occur at later divisions, but the likelihood of realization has to be high enough such that each pair member contributes to iPSC colonies within the allotted time window. The results imply that some predictive early marker could be identified and potentially used to isolate cells that will respond positively to factor induction. Our efforts to see whether Thy1 21, a fibroblast specific marker heterogeneously present in the MEF population, can be used as such a marker indicate that Thy1 expression at day 0 is not predictive of lineage fate (Supplementary Fig S11). The stability of the transcriptional state may only be one marker for a cell's response, while other relevant characteristics could include the presence of supportive (or the absence of deleterious) cofactors or the epigenetic configuration of target enhancer sequences. Genetic variants in the starting MEF population could also contribute to a cell's potential to reprogram. However, efforts to characterize specific genetic variation within iPSC colonies generated from fibroblast pools have found only very rare instances of overrepresented polymorphisms 27, suggesting that genetic contributions to this process would fall below the overall frequency of ∽1% that we observe for fibroblasts that successfully reprogram over our time course.
Our spatial dependence analysis shows no substantial contribution from the local niche, in terms of signaling from neighboring cells, to the final lineage fate. This suggests paired lineages do not adopt similar fates because of local ‘nurturing’ external effects but rather because of internal cell state. Additionally, the differential effect in colony formation between early and late replating during reprogramming suggests that from around day 6, cells are less prone to disturbance in their route to an iPSC fate, even though molecular markers associated with complete reprogramming have not yet been activated.
Effects of epigenetic perturbations on reprogramming efficiency have been demonstrated previously 3, 22. Here, we show that such a perturbation of H3K27 methylation through Ezh2 inhibition can change reprogramming potential by altering cellular state prior to factor induction. Furthermore, the shift in fates adopted by inhibitor treated cells suggests that the difference in potential may be chromatin-related, consistent with recent results where a major barrier to reprogramming is how transcription factors modify target chromatin once engaged 3. Ezh2 inhibition increased the total number of iPSC-forming lineages at the expense of ‘fast dividers’, the other continuously proliferating response to factor induction, and not of senescence-prone ‘non-responding’ fates, contrary to other methods of improving reprogramming efficiency that act by altering the population of dividing cells only 12, 18. Paired lineages with mixed fates also arose solely under inhibitor treatment, but usually for cells that divided early during dosage, and as such spent more time under treatment as separate cells. This suggests an asymmetry either in the cumulative effect of Ezh2 inhibition on sister cells, or in the sisters' internal states after division. All mixed-lineage pairs are between either iPSC- or FD-forming lineages and senescent non-responders, and never between alternate proliferating fates. As such, they may represent compounding effects between drug toxicity and the oncogenic stress of OSKM induction. Finally, the effect of Ezh2 inhibition is likely not mediated through a change in OSKM levels, as these levels are unaltered by the inhibitor (Supplementary Fig S12).
Characterizing the molecular events that prescribe successful reprogramming is challenged by the low efficiency and extended latency of the process. As such, most studies have generally relied on inferences from static population sampling or via lineage tracing with a limited number of reporters. The methodology we present here seeks to address fundamental aspects of reprogramming lineages with minimal preconceived assumptions about the exact molecular mechanisms in play. This strategy may be used to track decision time points along other complex cellular lineages where little is known, such as in vitro differentiation or cancer progression. Using live imaging and statistical analysis, we show that reprogramming potential in MEFs is preset and can be manipulated epigenetically. With a greater understanding of the key determinants through which reprogramming lineages are first established, future experiments may be designed to identify the underlying mechanisms that enable somatic cells to change fates in a directed fashion.
Materials and Methods
Cell culture
Secondary Nanog-GFP (NGFP2) MEFs derived from isolated E13.5 doxycycline-inducible murine fibroblasts as previously described 1 and cultured in ES cell medium, DMEM (Invitrogen) supplemented with 15% FBS, L-glutamine, penicillin–streptomycin, nonessential amino acids (Biological Industries), β-mercaptoethanol (Sigma), and 1,000 U/ml leukemia inhibitory factor (LIF, Millipore). All experiments were conducted after three passages from isolation. Collagen-OKSM-Oct4-EGFP MEFs 28 were grown in the same conditions as the NGFP2 MEFs.
Reprogramming and image acquisition
Nanog-GFP (YFP, or H2B-Cerulean labeled)-inducible MEFs were plated on gelatin-coated 24-well, 12-well, and 6-well plates (TC-treated polystyrene plate) at a density of 5,000, 10,000, and 20,000 cells per well, respectively. In 96-well plates, cells were seeded on feeder cells, either at low densities of 30–200 cells per well, or at high density, mixing 20–40 CFP-labeled cells with 1,000 YFP-labeled cells per well, as denoted in the text. Imaging started about 16 h after plating. Cells were cultured under serum starvation conditions (0.5% FBS) for ∽16 h before switching into standard mouse ES medium supplemented with 2 μg/ml doxycycline (Sigma) for all experiments to ensure all traced lineages began reprogramming from G1. Cells were kept on doxycycline for the duration of all imaging experiments. Growth medium (supplemented with dox) was replaced every 24–48 h. On day 12, the medium was switched to N2B27 + LIF + 2i + dox medium, containing neurobasal medium, DMEM/F12, B27, BSA (Invitrogen), Ndiff (Millipore), 3 μM CHIR 99021 (Biovision), 1 μM PD0325901 (Santa Cruz). After 14 days, cells were fixed and immunostained against pluripotency markers. Inducible MEFs were imaged using a Nikon TiE epi-fluorescence microscope equipped with a motorized XY stage (Prior) and taken within a connected 6 × 6 or 7 × 7 spatial range at 10× magnification in up to three fluorescent wavelengths and phase contrast using NIS Elements software. Acquisitions were taken every 2–4 h for 14–18 days.
Image analysis
Tracking cell divisions during the first few generations, as well as tracking lineage dynamics at later generations, was done manually using ImageJ. Cell segmentation was done using CellProfiler 29. Cell counting as well as final colonies counting was done automatically using CellProfiler and verified manually. In replating experiments, CFP and YFP colony identification was done using CellProfiler. iPSC-positive colonies were determined by coordinate comparison with the red channel, containing the Nanog staining data.
Cell fate classification
Cells were traced from 2 days before dox induction. All cells, at first division, if occurred during that 48 h period, were segmented as paired lineages. All lineages as well as cells that did not divide during that time were traced to their final fates. Final fate of a cell or a lineage was assigned to one of three categories: (1) non-responder (NR)—cells that did not divide at all after dox induction or divide slowly prior to death or senescence within 4 days of induction; (2) fast dividers (FD—can also be referred to as partly reprogrammed cells)—cells that divide quickly after dox induction, creating a spread out lineage with morphological features of fibroblasts, and do not survive after switching into N2B27 medium (day 12) or do not stain for E-cadherin and Nanog. A lineage is classified FD only if it yields no iPSC colonies; and (3) IPSC—cells that divide to form a condensed colony that resolves to a clear Nanog-GFP positive colony after switching into N2B27 + LIF + 2i conditions at the end of the experimental time course. These colonies are confirmed by positive staining for E-cadherin and Nanog (for some experiments, alkaline phosphatase activity was also measured). A lineage is classified as iPSC if it yields any iPSC colony, even if some cells within the subsequent lineage are FD.
Pre-treatment with chromatin modifiers inhibitors
Ezh2 methyltransferase was inhibited with two different inhibitors: 3-deazaneplanocin A (DZNep, Sigma, 5 μM) which inhibits the expression of Ezh2, and JQ EZ005, an Ezh2 inhibitor that was kindly provided by J. Bradner. Each inhibitor was added to ES medium at concentration of 5 μM. Other chromatin modifiers tested included the LSD1 inhibitor RN-1 (Millipore, 0.01–1 μM), the DNA methyltransferase inhibitors RG108 (Cayman chemical, 10 μM), and 5-azacytidine (Sigma, 2 μM), the histone deacetylase inhibitor valproic acid (VPA, Sigma, 1 mM) and the G9a inhibitor BIX01294 (Stemgent, 1 μM). Effects of these inhibitors on reprogramming efficiency were measured against untreated and vehicle treated (0.1%DMSO, Sigma) controls. Cells were grown and plated for experiment as described above. During the 2 days before dox induction, cells were treated with either inhibitors for 2 different time periods (see Fig4A). The concentration and duration of treatment was calibrated to identify conditions that maintain viability, enhance reprogramming, and aberrate their target epigenetic modification as determined by immunofluorescence or taken from the literature.
Additional experimental methods as well as description of colony counting are provided in the Supplementary Materials and Methods. Full description of the statistical methods employed is given in the Supplementary Information.
Acknowledgments
We thank J. Bradner for contributing an Ezh2 inhibitor and C. Sindhu for help with cell sorting. This study was supported in part by the Human Frontiers Scientific Program (HFSP RGY0080/2011), the Binational Science Foundation (BSF 2009403), and the Edmond J. Safra Center for Bioinformatics at Tel Aviv University.
Author contributions
MP, IP, ZDS, AM, and IN conceived or designed the experiments. MP and IP performed the experiments. MP and RR analyzed the data. MP, IP, ZDS, AM, and IN wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Movie S1
Supplementary Movie S2
Review Process File
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Jaenisch R. Nuclear reprogramming and pluripotency. Nature. 2006;441:1061–1067. doi: 10.1038/nature04955. [DOI] [PubMed] [Google Scholar]
- Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. doi: 10.1038/nature12587. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M, Creyghton MP, Steine EJ, Cassady JP, Foreman R, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cells. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K. Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. , . [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Yamanaka S. A fresh look at iPS cells. Cell. 2009;137:13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- Yamanaka S. Elite and stochastic models for induced pluripotent stem cell generation. Nature. 2009;460:49–52. doi: 10.1038/nature08180. [DOI] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, Jaenisch R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Nachman I, Regev A, Meissner A. Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat Biotechnol. 2010;28:521–526. doi: 10.1038/nbt.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zi X, Schulz VP, Cheng J, Zhong M, Koochaki SH, Megyola CM, Pan X, Heydari K, Weissman SM, et al. Nonstochastic reprogramming from a privileged somatic cell state. Cell. 2014;156:649–662. doi: 10.1016/j.cell.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Lengner CJ, Hanna J, Lodato MA, Steine E, Foreman R, Staerk J, Markoulaki S, Jaenisch R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat Biotechnol. 2008;26:916–924. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–1148. doi: 10.1038/nature08285. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, Woltjen K, Nagy A, Wrana JL. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- Snijder B, Pelkmans L. Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol. 2011;12:119–125. doi: 10.1038/nrm3044. [DOI] [PubMed] [Google Scholar]
- Young MA, Larson DE, Sun CW, George DR, Ding L, Miller CA, Lin L, Pawlik KM, Chen K, Fan X, et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell. 2012;10:570–582. doi: 10.1016/j.stem.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Borkent M, Hochedlinger K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat Methods. 2010;7:53–55. doi: 10.1038/nmeth.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Movie S1
Supplementary Movie S2
Review Process File