

Abstract
5-Aminolevulinate synthase (ALAS), a pyridoxal-5′phosphate (PLP)-dependent enzyme, catalyzes the first step of heme biosynthesis in mammals. Circular dichroism (CD) and fluorescence spectroscopies were used to examine the effects of pH (1.0–3.0 and 7.5–10.5) and temperature (20 and 37 °C) on the structural integrity of ALAS. The secondary structure, as deduced from far-UV CD, is mostly resilient to pH and temperature changes. Partial unfolding was observed at pH 2.0, but further decreasing pH resulted in acid-induced refolding of the secondary structure to nearly native levels. The tertiary structure rigidity, monitored by near-UV CD, is lost under acidic and specific alkaline conditions (pH 10.5 and pH 9.5/37 °C), where ALAS populates a molten globule state. As the enzyme becomes less structured with increased alkalinity, the chiral environment of the internal aldimine is also modified, with a shift from a 420 nm to 330 nm dichroic band. Under acidic conditions, the PLP cofactor dissociates from ALAS. Reaction with 8-anilino-1-naphtalenesulfonic acid corroborates increased exposure of hydrophobic clusters in the alkaline and acidic molten globules, although the reaction is more pronounced with the latter. Furthermore, quenching the intrinsic fluorescence of ALAS with acrylamide at pH 1.0 and 9.5 yielded subtly different dynamic quenching constants. The alkaline molten globule state of ALAS is catalytically active (pH 9.5/37 °C), although the kcat value is significantly decreased. Finally, the binding of 5-aminolevulinate restricts conformational fluctuations in the alkaline molten globule. Overall, our findings prove how the structural plasticity of ALAS contributes to reaching a functional enzyme.
Keywords: aminolevulinate synthase, pyridoxal-5′phosphate, heme, molten globule, protein folding, intrinsically disordered proteins
1. Introduction
5-Aminolevulinate synthase (ALAS) catalyzes the condensation between glycine and succinyl-CoA to generate CoA, CO2, and ALA (1,2). This reaction represents the first, and regulated, step of heme biosynthesis in mammals, and subtle disturbances in the rate of ALA formation resulting from either loss-of-function or gain-of-function mutations in the human erythroid specific gene, ALAS2, can lead to X-linked sideroblastic anemia and X-linked dominant protoporphyria, respectively (3,4).
Murine erythroid ALAS (henceforth abbreviated as mALAS2) is a homodimer with a molecular weight of 112 KDa (5). Each of its two active sites is located at the dimeric interface, and is composed of amino acids from the individual monomers (6). Pyridoxal-5′phosphate (PLP), which serves as a cofactor during catalysis, is covalently attached to an active site lysine (K313 in mALAS2), forming an internal aldimine (7). The absorbance spectrum of mALAS2 in the absence of substrates displays maxima at 330 and 420 nm, representing different tautomeric species of the internal aldimine (8). Previously, we assigned the 420 nm-absorbance maximum to a ketoenamine species, and we utilized fluorescence spectroscopy to assign the 330 nm maximum to a substituted aldamine (8). Moreover, positive dichroic bands with maxima at approximately 330 and 420 nm are detected in the circular dichroism (CD) spectrum of mALAS2 (9,10), and since PLP by itself is not a chiral molecule (10), the two maxima indicate an equilibrium between two populations of internal aldimine species with different chiral active site environments.
The catalytic activity of mALAS2, monitored under steady and pre-steady state conditions, exhibits dependence on pH (8,11). Specifically, an increase in alkalinity is accompanied by a decrease in the kcat value (8) and a decrease in the pre-steady state rate formation of the second quinonoid intermediate (11). Temperature also affects the catalytic turnover of mALAS2. In fact, when the temperature changes from 20 to 37 °C, the kcat value is increased from 0.02 s−1 (12) to 0.6 s−1, as determined using a continuous, coupled, enzymatic assay (13). The temperature at which 50% of the enzymatic activity of mALAS2 is lost was determined to be 55.3 °C (14). Thus far, however, it is not known to what extent pH and temperature affect the structural integrity of ALAS.
The crystal structure of Rhodobacter capsulatus ALAS (Fig. 1) reveals significant content of α-helices and β-sheets that fold into three separate and well-defined domains, which constitute the tertiary structure of each monomer (15). Two different conformations have been observed: open and closed, where the active site of the former conformation is exposed to the solvent, while in the latter, the active site is largely protected as a result of segmental, structural rearrangements (15). Importantly, both conformations were observed in the native state of the holoenzyme (15), suggesting that even in the absence of substrates, interconversion between the conformations occurs; however, based on kinetic data, it was inferred that the binding of succinyl-CoA accelerates the transition of ALAS to the catalytically competent closed conformation (16,17). Conformational interconversion also dominates the kcat, and the rate limiting step of the reaction had been assigned to a transition from the closed to the open conformation during dissociation of ALA from the active site (16,17).
Figure 1.
Crystal structure of R. capsulatus ALAS. The distribution of color indicates: in pink, amino acids with ionizable side groups whose theoretical pKa is lower than 5 (i.e., Asp and Glu); in cyan, amino acids with ionizable side groups whose theoretical pKa is greater than 5 (i.e., His, Cys, Tyr, Lys, and Arg); in yellow, amino acids with hydrophobic side chains (i.e., Ala, Val, Leu, Ile, Trp, Phe, and Met) and in white, all other amino acids (i.e., Ser, Thr, Asn, Gln, Pro, and Gly) [PDB 2BWO].
Therefore, it is evident that a certain level of structural plasticity is important for effective catalysis. Yet the question remains to what extent can the structural integrity of ALAS deviate from its native state without compromising the catalytic activity. Several enzymes have been shown to retain catalytic activity (18–21), even though they assume a partially folded, molten globule state: a highly dynamic conformational ensemble, where due to absence of close packing in the protein core, the rigidity of the tertiary structure is lost, without significant disruption in secondary structure content (22). To our knowledge the list of enzymes includes: circularly permuted dihydrofolate reductase (18), double point mutant of nuclease (19), UreG (20), and monomeric chorismate mutase (21). Among the aforementioned enzymes, monomeric chorismate mutase (21) is of particular interest, because this “molten globule” enzyme, whose structure becomes ordered upon ligand binding, retains identical catalytic activity as that of natively folded, dimeric chorismate mutase (21). This indicates that catalytic turnover in some enzymes may remain unperturbed even when the rigidity of the tertiary structure in the absence of ligands is disrupted.
In this study we have examined how pH and temperature affect the structural integrity of mALAS2. While the integrity of the secondary structure is resilient to fluctuations in pH and temperature within the examined range, the rigidity of the tertiary structure is disrupted under specific alkaline and acidic conditions and the enzyme exists in a molten globule state. These structural changes proceed with concomitant modification in the chiral active site environment of the internal aldimine. Finally, we show that the enzyme retains catalytic activity, albeit reduced, in the alkaline molten globule state, as previously and experimentally defined in the absence of ligands. To the best of our knowledge, this is the first reported case of a catalytically active alkaline molten globular enzyme.
2. Materials and Methods
2.1 Materials
Acrylamide, potassium phosphate monobasic, potassium phosphate dibasic, Tris, glycine, acetylacetone, and sodium acetate trihydrate were purchased from Fisher. PLP, 8-anilino-1-naphtalenesulfonic acid (ANS), p-dimethylaminobenzaldehyde, and CAPS were from Sigma-Aldrich. AMPSO was from MP Biomedicals, and trichloroacetic acid (TCA) and 5-aminolevulinate hydrochloride were from Acros Organics.
2.2 Methods
2.2.1 Overproduction and Purification of Wild-type mALAS2
Wild-type mALAS2 was overproduced and purified as described previously (5). All purification steps were performed in one day and the purity of the protein was assessed by SDS-PAGE. The purified enzyme was concentrated to 12 mg/mL using an Amicon cell with a 30000 MW cut-off membrane and then dialyzed against 2 L of 50 mM phosphate, pH 7.5, containing 40 μM PLP. The protein was stored under liquid nitrogen for further use. The monomeric protein concentration was determined spectroscopically using an extinction coefficient of 42860 M−1 cm−1 calculated from its amino acid sequence.
2.2.2 Circular Dichroism Measurements
CD measurements were collected using a JASCO J-815 spectropolarimeter and a scan speed of 20 nm/min at either 20 or 37 °C. The temperature was regulated using a thermostatically controlled cell holder. For the measurements collected in the far-UV region (260-190 nm), the concentration of protein was 0.15 mg/ml and a cell with a 0.1 cm path length was used. The measurements were conducted at different pH values using the following buffers: 20 mM KH2PO4, pH 1.0, 20 mM KH2PO4, pH 2.0, 20 mM citrate, pH 3.0 (16.7 mM citrate dihydrate and 3.3 mM sodium citrate dihydrate), 20 mM phosphate, pH 7.5 (16 mM K2HPO4 and 4 mM KH2PO4), 20 mM TRIS, pH 8.5, 20 mM AMPSO, pH 9.5 and 20 mM CAPS, pH 10.5. The pH of the buffers was adjusted either with hydrochloric acid or sodium hydroxide. Each reported measurement in the far-UV region represents the average of four accumulated spectra.
Protein concentration of 1 mg/mL and a cell with a 1 cm path length was used for the measurements in the near-UV/visible region (500-260 nm), which were conducted in identical buffers as those used for the measurements in the far-UV region. The total volume in the cell was 2.5 mL, and once the measurements were completed, the pH was immediately verified using an Accumet AR15 pH meter to ensure that the pH of the buffer did not change as a result of the addition of protein. Each of the reported measurements in the near-UV/visible range represents the average of three accumulated spectra.
CD spectra in the near-UV/visible range (500-260 nm) were collected in the presence or absence of ALA at pH 9.5 and 37 °C. The experiments were performed in 50 mM AMPSO buffer, pH 9.5, with ALAS and ALA concentrations of 1 mg/mL and 4 mM, respectively. In order to prevent changes in the buffer pH as a result of the high acidity of ALA, 4 mM ALA (from 0.5 M stock) was neutralized with equal volume of 1 M NaOH prior to initiating the reaction. The pH of the buffer, measured after adding 4 mM ALA to the enzyme, was 9.5.
2.2.3 Reaction with ANS
Fluorescence spectra for the reaction between ANS and wild-type mALAS2 were collected using a Shimadzu RF 5301PC spectrofluorophotometer. All experiments were performed at either 20 or 37 °C and the following pH values: 1, 2, 3, 7.5, 8.5, 9.5, and 10.5. Identical buffers were used as those described in the circular dichroism measurement section. Due to the low solubility of ANS in water, stock solution of 50 mM ANS was initially prepared in dimethyl sulfoxide (DMSO), which was further diluted in deionized water to prepare a stock of 4 mM ANS. Reactions at the pH value of interest were performed by incubating 1 μM wild-type mALAS2 (0.06 mg/mL), 20 μM ANS, and 20 mM buffer for 1 hour at either 20 or 37 °C. As negative controls, reactions at the pH value of interest were performed in the absence of protein by incubating 20 μM ANS and 20 mM buffer for 1 hour at either 20 or 37 °C. Fluorescence spectra were then collected at either 20 or 37 °C by monitoring the emission in the 400–600 nm range, following excitation at 375 nm. A cell with a 0.3 cm path length was used and the temperature in the cell holder was regulated using a circulating water bath. All reactions were performed in triplicates.
2.2.4 Acrylamide Quenching of Intrinsic Protein Fluorescence
The accessibility of tryptophans in wild-type mALAS2 was examined by monitoring the fluorescence spectra resulting from the quenching of the intrinsic fluorescence of the enzyme upon reaction with acrylamide. All reactions were conducted at either 20 or 37 °C and the following pH values: 1, 7.5, and 9.5. The buffers used were: 20 mM KH2PO4, pH 1; 20 mM phosphate, pH 7.5 (16 mM K2HPO4 and 4 mM KH2PO4); 20 mM AMPSO, pH 9.5. Reactions at the pH value of interest were performed by incubating 1 μM wild-type mALAS2 (0.06 mg/mL), acrylamide, and 20 mM buffer for 5 minutes at either 20 or 37 °C. To ensure that the pH of the reaction was not modified by the addition of acrylamide, 2 M acrylamide stock solution was prepared in the following buffers: 20 mM KH2PO4, pH 1.0; 20 mM phosphate, pH 7.5 (16 mM K2HPO4 and 4 mM KH2PO4); 20 mM AMPSO, pH 9.5. Fluorescence spectra were collected on a Shimadzu RF 5301PC spectrofluorophotometer by monitoring the emission at 340 nm upon excitation at 280 nm. During the collection of the spectra, the temperature in the cell holder was maintained at either 20 or 37°C, and a cell with a path length of 0.3 cm was used. All reactions were performed in triplicates. The data resulting from the quenching of the intrinsic fluorescence were analyzed using the Stern-Volmer equation (Eq. 1),
| Eq. 1 |
where F0 is the fluorescence emission in the absence of acrylamide, F is the emission at a specific concentration of acrylamide, Ksv is the dynamic quenching constant, and Q is the concentration of acrylamide.
2.2.5 Colorimetric Assay for Determination of ALAS Activity
The catalytic activity of wild-type mALAS2 was examined using a colorimetric assay, which, briefly, involves the quantitation of the ALA-derived pyrrole upon reaction with Ehrlich’s reagent by monitoring the absorbance at 552 nm (23). All reactions were performed at 37 °C in the acidic and alkaline pH ranges. Identical buffers were used as described in the circular dichroism measurement section, with the exception that the buffer concentration was 100 mM. Preliminary studies demonstrated that the addition of substrates did not modify the pH value of the reaction, with the exception at pH 10.5, where the addition of 50 mM glycine lowered the pH to 10.3 (data not shown). The reaction assay was performed in the following way: 4 μM of wild-type mALAS2 (from a 220 μM mALAS2 stock) was added to a reaction tube containing 100 mM buffer at the pH of interest, and the sample was equilibrated at 37 °C for 10 minutes prior to addition of substrates. Then to each reaction tube, 100 mM glycine (from a 2 M stock of glycine) and 100 μM succinyl-CoA (from an 8.3 mM stock of succinyl-CoA) were added, and the sample was immediately returned to the water bath. Incubation for 30 minutes at 37 °C followed. The reactions were quenched by adding ½ reaction volume of 50 % TCA to each reaction tube. Following centrifugation at 12000 × g for 4 minutes at room temperature, aliquots were removed and placed in separate reaction tubes. Then, final concentration of 3.3 % acetylacetone/0.3 M sodium acetate was added and incubation at 80 °C for 10 minutes followed. The samples were cooled down to room temperature and equal volume of modified Ehrlich reagent (23) was added. After incubation for 15 minutes at room temperature, absorbance spectra in the 650-500 nm region were collected, and from the absorbance at 552 nm, the concentration of ALA was determined using extinction coefficient of 45 (mmol/L)−1·cm−1. For each reaction performed at a given pH a negative reaction control was included, where the reaction was quenched prior to the addition of substrate by adding ½ reaction volume of 50 % TCA. The negative controls were subjected to the same colorimetric assay procedure.
To determine the steady-state kinetic parameters of mALAS2, the steady-state activity of mALAS2 was measured at 37 °C, at either pH 7.5 or 9.5, using the above described colorimetric assay with slight modifications. The assays were performed by varying the concentration of succinyl-CoA (0–400 μM) while keeping the concentration of glycine fixed at 200 mM. In preliminary studies, we established that the pH of the buffer did not change upon addition of substrates within the range of substrate concentrations used. At physiological pH, the initial velocities were determined by incubating the wild-type enzyme (4 μM) with both substrates for 1 minute, and then quenching the reaction with ½ reaction volume of 50 % TCA and quantifying the formation of ALA. For reactions at pH 9.5, which were performed in 250 mM AMPSO buffer, the initial velocities were determined by incubating the enzyme (4 μM) with the substrates for a period of 20 minutes followed by quenching of the reaction with acid. The initial velocities, expressed as the concentration of ALA produced per minute, were plotted against the concentration of succinyl-CoA, and the data were fitted to the Michaelis-Menten equation (Eq. 2) using non-linear regression analysis. Km and kcat values were determined from the fitted data. All data were analyzed using the program SigmaPlot.
| Eq. 2 |
2.2.6 Absorbance spectra of mALAS2 and free PLP at various pH values
The absorbance spectra of wild-type mALAS2 and free PLP in several different buffers were recorded at 20 °C using a Shimadzu UV-2401 PC spectrophotometer. The buffers were: 50 mM K2HPO4, pH 1.0, 50 mM K2HPO4, pH 2.0, 100 mM citrate, pH 3.0 (83 mM citrate dihydrate, 17 mM sodium citrate dihydrate), and 50 mM phosphate, pH 7.5 (40 mM K2HPO4, 10 mM KH2PO4). Spectra were collected immediately upon diluting ALAS in the respective buffer at intervals of 5 minutes for a time period of 20 minutes. The protein concentration was 40 μM, while that of free PLP was 50 and 200 μM at physiological and acidic pH, respectively.
2.2.7 Dynamic light scattering
Dynamic light scattering measurements were conducted on a Wyatt DynaPro plate reader at 25 °C. Wild-type mALAS2 was diluted to a final concentration of 1 mg/ml in the following buffers: 50 mM K2HPO4, pH 1.0, 50 mM K2HPO4, pH 2.0, 100 mM citrate, pH 3.0, 50 mM K2HPO4, pH 7.5 and 50 mM AMPSO, pH 9.5. The protein samples and all buffers were filtered through 0.22 μm filters prior to measurements.
3. Results
3.1 Effects of pH and temperature on the secondary structure of mALAS2
CD spectra in the far-UV region (195–260 nm) were collected to examine how variations in temperature and pH affect the secondary structure of wild-type mALAS2. At physiological pH, the far-UV CD spectrum of mALAS2 resembles that of proteins with ordered secondary structure, and changing the temperature from 20 to 37 °C has no effect on the secondary structure (Fig. 2). As the pH is lowered to a value of 2.0, the protein becomes progressively disordered; under these conditions, and in comparison to physiological pH, the 208 nm elliptical minimum is blue-shifted to 204 nm and the intensity of the 222 nm minimum is decreased (Fig. 2A and B). This trend, however, does not continue with further decreases in pH. In fact, the far-UV CD spectrum of mALAS2 at pH 1.0 is similar to the spectrum collected at pH 7.5, indicating that the secondary structure is restored to near-native levels (Fig. 2A and B). Since hydrochloric acid was used to adjust the pH of the phosphate buffer, and it is present at higher concentrations at pH 1.0 than at pH 2.0, the acid-induced refolding observed in the buffer at pH 1.0 is attributed to the ability of negative chloride ions to minimize repulsion among positively charged groups of mALAS2 (see discussion). The integrity of the secondary structure is not significantly modified by changes in pH in the alkaline range, with only modest effects detected at pH 10.5 (Fig. 2C and D). All far-UV CD spectra were similar at 20 and 37 °C, indicating that moderate increase in temperature does not disrupt the secondary structure.
Figure 2.

Effects of pH and temperature on the secondary structure of mALAS2. Far-UV CD spectra collected at (A) acidic pH and 20 °C, (B) acidic pH and 37 °C, (C) alkaline pH and 20 °C, (D) alkaline pH and 37 °C. The colors of the spectra are: pH 1 (pink), pH 2 (purple), pH 3 (gray), pH 7.5 (black), pH 8.5 (red), pH 9.5 (blue), and pH 10.5 (cyan).
3.2 Effects of pH and temperature on the tertiary structure of mALAS2
The effects of pH and temperature on the tertiary structure of mALAS2 were characterized from the spectra in the near-UV CD region (310-260 nm). As the pH varies from physiological to alkaline conditions, the rigidity of the tertiary structure is disrupted, as discerned from the decreased ellipticity in the near-UV signal (Fig. 3A and B). The effect is amplified when the temperature increases from 20 to 37 °C, implying that temperature and alkalinity enhance conformational mobility and modification in ionic interactions, which significantly disrupt the tertiary structure of mALAS2 (Fig. 3A–D). This can be clearly seen at pH 9.5 upon increasing the temperature to 37 °C (Fig. 3D). Moreover, under alkaline conditions, the actual decrease in the intensity of the near-UV CD signal is far more pronounced than what it seems in Fig. 3. This is because in comparison to physiological pH, the 330 nm maximum that results from the internal aldimine of ALAS is profoundly modified in alkaline solution (Fig. S1) and thus making the ellipticity at 305 nm much more intense than that at pH 7.5 (Fig. 3 and S1). To account for this disparity and present the actual reduction in elliptical signal more clearly, we have normalized the data by subtracting the 280 nm ellipticity from that at 305 nm; the normalized data are presented in Fig. 4. In the acidic pH range, the CD signal in the near-UV region is at baseline close to zero, indicating that the enzyme has no rigid tertiary structure (Fig. 3). Importantly, the near and far-UV CD data indicate that under specific acidic (pH 1 and pH 3) and alkaline (pH 9.5/37 °C and pH 10.5) conditions, mALAS2 exists in a molten globule state, where it retains native levels of secondary structure, without having rigid tertiary structure. At pH 2.0, the enzyme is noticeably unfolded as it follows from the specific changes in the far-UV CD spectrum.
Figure 3.

Effects of pH and temperature on the tertiary structure of mALAS2. Near-UV CD spectra collected at different acidic and alkaline pH values at (A) 20 °C, and (B) 37 °C. (C) Spectra at 20 °C (solid) and 37 °C (dashed) at pH 7.5. (D) Spectra at 20 °C (solid) and 37 °C (dashed) at pH 9.5. The colors of the spectra are: pH 1 (pink), pH 2 (purple), pH 3 (gray), pH 7.5 (black), pH 8.5 (red), pH 9.5 (blue), and pH 10.5 (cyan).
Figure 4.

Dependence of CD ellipticity difference (Δθ280-305) with pH. The CD ellipticity difference (Δθ280-305) was calculated by subtracting the degree of ellipticity at 280 nm from that at 305 nm. The symbols correspond to the spectra collected at different temperatures: 20 °C (squares) and 37 °C (triangles).
3.3 Effects of pH and temperature on the PLP-binding site of mALAS2
PLP-dependent enzymes usually display a positive dichroic signal in the 500-310 nm region of the CD spectrum as a result of the positioning of the internal aldimine in a chiral, active site environment [(10) and references therein]. At physiological pH value of 7.5, the CD spectrum of mALAS2 in the 500-310 nm region displays two positive dichroic bands with maxima at 430 and 325 nm, where the intensity of the former is more pronounced (Fig. 5). However, as the alkalinity of the solution increases, the chiral environment of the internal aldimine changes (Fig. 5A and B). Starting at pH 8.5, the 325 nm dichroic band becomes more pronounced than the 430 nm band. This trend continues with further increases in pH and is accompanied by a shift of the 325 nm maximum toward shorter wavelengths, such that at pH 9.5 the maximum is shifted to 317 nm and to 312 nm at pH 10.5 (Fig. 5A and B). In the acidic pH range (pH 1–3), the CD signal that results from the internal aldimine of mALAS2 is lost, suggesting severe disruption of the active site environment or dissociation of PLP from the enzyme (Fig. 5C and D). With the exception of the spectra collected at pH 10.5, the spectra collected at all other pH values were nearly identical at either 20 or 37 °C (Fig. 5).
Figure 5.

Effects of pH and temperature on the chiral active site environment of the mALAS2 internal aldimine. CD spectra in the 500-310 nm region collected at (A) alkaline pH and 20 °C, (B) alkaline pH and 37 °C, (C) acidic pH and 20 °C, (D) acidic pH and 37 °C. The colors of the spectra correspond to: pH 1 (pink), pH 2 (purple), pH 3 (gray), pH 7.5 (black), pH 8.5 (red), pH 9.5 (blue), and pH 10.5 (cyan).
In order to examine whether PLP remains covalently bound to the active site of ALAS at acidic pH, the absorbance spectra of mALAS2 were recorded and compared to that of free PLP (Fig. S2). At physiological pH, the absorbance spectrum of free PLP displays a maximum at 390 nm, which differs from the spectrum of ALAS, where, as a result of the Schiff base linkage between PLP and K313, the maximum is red shifted to 415 nm (Fig. S2A). In contrast, PLP does not bind covalently to the active site lysine of ALAS under acidic conditions. In fact, at acidic pH, a time-dependent dissociation of PLP from the active site of ALAS was observed. The spectra of ALAS collected at time point zero and at pH values 1.0–3.0 resembled that of ALAS at physiological pH; however, within the time period of 20 minutes, the spectra of holo-ALAS under acidic conditions became similar to the spectra of free PLP in acidic solution, implying that the cofactor dissociated from the active site (Fig. S2B–D). No significant changes in the spectra of holo-ALAS were observed at physiological pH within the time period used in this study, indicating that PLP remained covalently bound to the enzyme (Fig. S2A). Further, the spectra of free PLP did not change within the experimental time period and at all examined pH values (data not shown).
3.4 Reaction with ANS
ANS is a fluorescence probe that is commonly used to characterize the exposure of hydrophobic surfaces in proteins in order to monitor the formation of partially folded molten globule-like intermediates (24). Upon reaction with hydrophobic clusters, the emission intensity of ANS intensifies and is shifted toward shorter wavelengths (24). The fluorescence spectra for the reaction between mALAS2 and ANS are shown in Fig. S3, while graphical representation of the wavelength of emission maxima and emission intensity are shown in Fig. 6A and B, respectively. In the absence of protein, ANS in the pH range between 7.5–10.5 displays emission maxima at 534 nm of equal intensity, which are not affected when the temperature changes from 20 to 37 °C. However, upon reacting with mALAS2, the fluorescence emission of ANS intensifies and the maximum is shifted toward shorter wavelengths (Fig. 6). The reactivity between ANS and mALAS2 increases as the alkalinity and temperature are increased, implying that the hydrophobic clusters of mALAS2 become more exposed to the solvent as a result of disruption in the rigidity of the tertiary structure (Figs. 6 and S3). In the acidic pH range (pH 1–3), when ANS reacts with mALAS2, the shift in the emission maximum toward shorter wavelengths is comparable to that observed under alkaline conditions; however, the emission intensity of ANS is significantly more pronounced (Figs. 6 and S3). This finding indicates potential differences in the exposure of hydrophobic clusters in acidic and alkaline solution or presence of some internal quenchers at alkaline pH.
Figure 6.
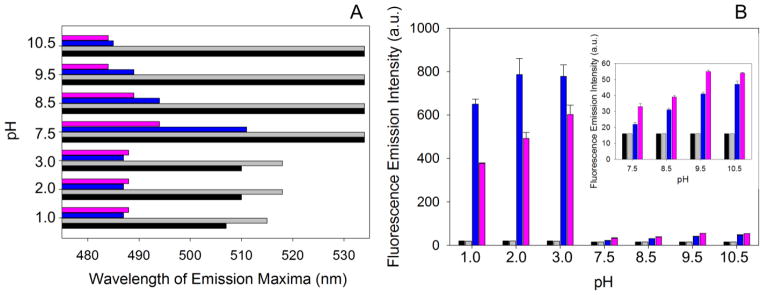
Reaction between ANS and mALAS2 at different pH values at 20 or 37 °C. (A) Wavelength of emission maxima. (B) Fluorescence emission intensity. The colors correspond to: free ANS at 20 °C (black), free ANS at 37 °C (gray), ANS in the presence of mALAS2 at 20 °C (blue), and ANS in the presence of mALAS2 at 37 °C (pink).
3.5 Acrylamide quenching of intrinsic protein fluorescence
Acrylamide quenching studies were performed to characterize how changes in pH and temperature affect the orientation of tryptophans in mALAS2. The data fitted to the Stern-Volmer equation are presented in Fig. 7, whereas the calculated dynamic quenching constants are in Table 1. At physiological pH, as the temperature changes from 20 to 37 °C, the dynamic quenching constant, Ksv, decreases from 8 to 5.1 M−1. This could imply that as the temperature increases, the protein becomes more dynamic, causing the repositioning of some tryptophans in an environment that is less accessible to the quencher. In contrast, at pH 1.0 and pH 9.5, as the temperature increases to 37 °C, the Ksv values remain identical to those observed at 20 °C. However, the Ksv values obtained at pH 1.0 and pH 9.5 are different, indicating subtle differences in the accessibility of tryptophans under acidic and alkaline conditions.
Figure 7.

Acrylamide quenching of intrinsic fluorescence of mALAS2. (A) pH 1.0 and 20 °C, (B) pH 7.5 and 20 °C, (C) pH 9.5 and 20 °C, (D) pH 1 and 37 °C, (E) pH 7.5 and 37 °C, (F) pH 9.5 and 37 °C.
Table 1.
Dynamic quenching constants at different pH and temperature values
| pH | Ksv at 20 °C M−1 | Ksv at 37 °C M−1 |
|---|---|---|
| 1.0 | 7.2 ± 0.1 | 6.7 ± 0.3 |
| 7.5 | 7.9 ± 0.2 | 5.1 ± 0.2 |
| 9.5 | 5.6 ± 0.1 | 5.7 ± 0.3 |
3.6 Dynamic light scattering
Dynamic light scattering was used to examine the propensity of ALAS to self-associate into higher oligomeric species at different pH values. The greatest propensity for self-association into higher oligomers was observed at pH 3.0, although a significant heterogeneous oligomeric population was observed at pH 2.0, and to a lesser extent at pH 1.0, where the hydrodynamic radius of the most populous species is over 2-fold greater than at physiological pH (Fig. 8). Since in the pH range 6.5-3.2, mALAS2 precipitates out of solution, forming visibly insoluble aggregates (data not shown), the extent of protein association into higher oligomers depends on the proximity of the buffer pH to the lower limit of solubility (~pH 3.1). There was no significant protein association at either pH 7.5 or 9.5 (Fig. 8), indicating that at these pH values, mALAS2 is predominantly a dimer. Interestingly, even at 25 °C, the hydrodynamic radius at pH 9.5 is increased by 12 % when compared to that at pH 7.5. This observation is in agreement with the CD and ANS data, which showed that structural perturbations are associated with increased alkalinity.
Figure 8.

Size distribution of mALAS2 at pH values of (A) pH 9.5, (B) pH 7.5, (C) pH 3.0, (D) pH 2.0, and (E) pH 1.0. All experiments were performed using 1 mg/ml protein and at 25 °C.
3.7 Is catalysis possible under conditions in which ALAS, devoid of ligands, lacks a rigid tertiary structure?
To assess if the enzyme retains catalytic activity under conditions in which its structural integrity is compromised, a colorimetric assay was used to determine the mALAS2-catalyzed formation of ALA at different acidic and alkaline pH values. No activity was detected in the acidic range, while the enzyme remains active in the alkaline pH range up to pH 9.5 (Table 2). This indicates that, although the reaction velocity is about 10-fold lower than at physiological pH, mALAS2 is active at 9.5 and 37 °C, conditions under which the enzyme adopts, in the absence of substrates, the molten globule state.
Table 2.
Velocities for the mALAS2-catalyzed formation of ALA at different pH values
N.A., no measurable activity
Comparison of the steady-state kinetic parameters of the alkaline molten globule, as described for the ALAS state at pH 9.5/37 °C, with those of natively folded ALAS (pH 7.5/37 °C) indicated 40- and 6-fold decreases in the kcat and values, respectively (Table 3). However, the most drastic reduction was in the specificity constant for succinyl-CoA, , which was 250-fold lower at pH 9.5 than at pH 7.5 (Table 3). The and values could not be determined accurately at alkaline conditions because the pH changed at concentrations of glycine greater than 200 mM.
Table 3.
Steady-state kinetic parameters of mALAS2 determined at 37 °C and pH 7.5 or pH 9.5
| pH | kcat (min−1) | (μM) | (min−1·μM−1) |
|---|---|---|---|
| 7.5 | 2.80 ± 0.10 | 16 ± 4 | 175 × 10−3 ± 50 × 10−3 |
| 9.5 | 0.07 ± 0.01 | 103 ± 32 | 0.68 × 10−3 ± 0.2 × 10−3 |
3.8 Is ligand binding to ALAS coupled to its folding in the alkaline molten globule?
To assess whether ligand binding stabilizes the tertiary structure of ALAS, CD spectra in the near-UV range were collected, at pH 9.5 and 37 °C, in the presence and absence of ALA (Fig. 9). The binding of ALA to the alkaline molten globule state of ALAS increases the elliptical signal in the near-UV range, implying that ALA binding to ALAS induces folding by reducing structural fluctuations. A similar substrate-induced stabilization of the tertiary structure is probably necessary for catalysis. Of relevance, the observed increase in ellipticity results from stabilization of the tertiary structure, rather than from unbound ALA, since the elliptical signal of ALA in the absence of enzyme in the near-UV region is near baseline (data not shown). Further, it was not possible to perform the experiments in the presence of substrates, because succinyl-CoA has an absorbance maximum at ~260 nm, and at concentrations greater than 100 μM (i.e., concentration greater than the Km value at pH 9.5) interferes with the acquisition of the CD data.
Figure 9.

Binding of ALA to the alkaline molten globule state of ALAS restricts fluctuations in the tertiary structure. Near-UV CD spectra were collected at 37 °C with the reactants in 50 mM AMPSO, pH 9.5. Dashed and solid lines correspond to mALAS2 in the absence and presence of ALA, respectively.
4. Discussion
The integrity of the secondary structure of mALAS2 is rather resilient to changes in pH in the alkaline range. In contrast, the secondary structure becomes disrupted at pH 2.0 where the enzyme exists as a stable, partially folded intermediate. Since at pH 2.0, mALAS2 is positively charged due to protonation of its aspartates and glutamates side groups, we hypothesize that the partial unfolding of the secondary structure results from increased repulsion amongst the positively charged groups. One would therefore expect greater disruption as the pH is further decreased; however, rather than becoming less structured, the secondary structure of mALAS2 at pH 1.0 is restored to nearly native levels (Fig. 2).
ALAS is by no means the only protein whose secondary structure content becomes restored under extreme acidic conditions. In fact, Goto et al. were the first to describe the phenomenon of acid-induced refolding for three different proteins: cytochrome c, apomyoglobin, and β-lactamase (25). The authors showed that the aforementioned proteins, which exist in fully unfolded forms at pH 2.0, restore their secondary structure content to nearly native levels as the pH is further decreased by increasing the concentration of hydrochloric acid (25). The refolding resulted from the ability of negatively charged chloride ions to minimize repulsion among the positively charged groups of the protein (25,26). We also propose that the refolding of the secondary structure of mALAS2 at extremely low pH results from the ability of the increased concentration of chloride ions (which are present in the buffer since the pH was adjusted with hydrochloric acid) to minimize repulsion among positively charged groups. This phenomenon of acid-induced (25,26) or anion-induced (27) refolding of acid-unfolded state was also described earlier for several proteins, such as β-lactamase (28), apomyoglobin (29), cytochrome c (30,31), various heme proteins (32), Staphylococcal nuclease (27,33), ribulosebisphosphate carboxylase (Rubisco) from Rhodospirillum rubrum (34), human serum albumin (35), α-chymotrypsinogen (36), rabbit muscle pyruvate kinase (37), jack bean urease (38), and many other proteins (39,40).
Under specific acidic (pH 1.0 and pH 3.0) and alkaline (pH 9.5/37 °C and pH 10.5) conditions, mALAS2 exists in a molten globule state, where its secondary structure is mainly preserved, while the rigidity of the tertiary structure is lost. This finding implies an important role for ionic interactions in stabilizing the tertiary structure, because upon increasing the same type of electric charges (either positive or negative), tertiary contacts are disrupted. Ultimately, this disruption leads to loosening of the side chain packing in the protein core and conversion of mALAS2 into a molten globule state. Interestingly, the acidic and alkaline molten globule states of mALAS2 are assumed independently of temperature, with the exception that at pH 9.5 the molten globule state is observed only at 37 °C. Greater structural mobility, which ensues from increases in temperature, is probably necessary for the conversion of mALAS2 into the molten globule state at pH 9.5.
ANS-mALAS2 binding experiments further corroborate that the enzyme populates the molten globule state under the aforementioned conditions, as inferred from the observation that the reactivity of the enzyme with ANS is more pronounced in the alkaline molten globule state than in the native state (pH 7.5). These findings strongly imply greater exposure of the hydrophobic clusters of mALAS2 in the alkaline solvent due to disruption of the tertiary structure.
Interestingly, in the acidic pH range, the fluorescence emission intensity of ANS upon reacting with mALAS2 is significantly more prominent than that observed in the alkaline pH range (Fig. 6). Since at acidic pH there is a greater propensity for self-association, due to the presence of partially folded intermediates, and thus heterogeneity in ALAS oligomeric species, we propose that the increase in fluorescence emission intensity observed at acidic pH results from ANS interaction with the hydrophobic surfaces of partially folded intermediates and with those of higher oligomeric species. The fluorescence emission intensity of ANS has been reported to increase upon interaction with self-associating proteins forming higher oligomers (41–43).
Quenching of the protein intrinsic fluorescence with acrylamide indicates that the tryptophans of mALAS2 at pH 1.0 and pH 9.5 have different accessibility to the quencher, as inferred from their respective Ksv values. This finding implies that the acidic and alkaline molten globule states of mALAS2 differ subtly, as also suggested by the ANS experiments. At physiological pH, changing the temperature from 20 to 37 °C proceeds with a decrease in the dynamic quenching constant. We hypothesize that an increase in temperature results in greater mobility of the conformational ensemble of mALAS2, which in turn reduces the accessibility of tryptophans to the quencher.
In the acidic pH range, the CD signal resulting from the positioning of the internal aldimine within the chiral active site environment of mALAS2 is lost (Fig. 5). The disappearance of the CD signal results from the dissociation of PLP from the active site, and this process, as validated by absorbance spectroscopy, occurs in a time-dependent manner (Fig. S2). Our results are in good agreement with those of NMR studies of the reaction between PLP and free lysine, which led Chan-Huot et al. to conclude that, at pH values below 4, PLP no longer forms a Schiff base with lysine, but rather exists as a free hydrate (44). Moreover, time-dependent dissociation of PLP has been reported for the guanidine deuterochloride denaturation of aspartate aminotransferase (45).
Increasing alkalinity causes the 330 nm dichroic band to intensify and the 420 nm band to weaken. These findings indicate that at pH values of 8.5 or higher, the orientation of the internal aldimine is different from that observed under physiological conditions, at which the corresponding 420 nm band predominates. Importantly, the chiral environment of the internal aldimine is modified in favor of the species with the 330 nm dichroic band at approximately the same pH value at which mALAS2 becomes less structured. If the alteration in the chirality of the internal aldimine stems from ionization of group(s) important for the positioning of the cofactor or through water molecules that modify previously established interactions between PLP and active site groups, then a shift to a less structured conformational subset probably assists the tautomeric interconversion by making the active site more accessible to the solvent. However, we cannot exclude the possibility that the alteration in the chiral environment of the internal aldimine results solely from conformational rearrangements. It is interesting to note that upon stabilization of a particular conformational subset, shift in the tautomeric equilibrium of the internal aldimine toward the 340 nm absorbance species was reported for tyrosine phenol lyase, when this fold type I PLP-dependent enzyme was encapsulated in wet nanoporous silica gel (46).
While the enzyme does not retain any measurable activity in the acidic pH range under the assay conditions, it remains active in the alkaline pH range up to pH 9.5. The most surprising finding is that mALAS2 is catalytically active even in the molten globule state (pH 9.5 and 37 °C), with the holoenzyme as a highly dynamic ensemble of interconverting conformations. However, the catalytic activity of mALAS2 at pH 9.5 and 37 °C is severely compromised, with 40- and 250-fold reductions in the kcat and values, respectively, in relation to those determined at physiological pH (Table 3), implying that the ability of the enzyme to effectively convert its substrates into products is impaired. Clearly, ligand binding to ALAS partly restricts the conformational fluctuations observed in the alkaline molten globule state of the holoenzyme (Fig. 9). The ligand-induced conformational stabilization is likely to determine the structural reorganization and transition to a conformation amenable for catalysis. However, even with the ligand-dependent conformational stabilization, the catalytic activity of ALAS remains hindered, suggesting an energetic penalty associated with the structural reorganization to the catalytically competent conformation that contributes, at least partly, in lowering the catalytic efficiency of the enzyme. Since no activity was detected in the acidic pH range and at pH 10.5, where ALAS also assumes a molten globule state, the abolished activity may not entirely stem from structural disorder, but rather at acidic pH, it results from dissociation of PLP from the active site, and at pH 10.5, from deprotonation of the catalytically important Lys313, which acts as a base and acid catalyst during the reaction cycle (47).
In this study we have shown that ALAS exists as an intrinsically disordered molten globule under specific acidic and alkaline conditions. Yet the question remains whether under physiologically relevant conditions, apart during the folding process, ALAS ever exists in an intrinsically disordered state. One way to address this question is to examine if any of the XLSA mutations affect the folding pathway of ALAS, where the variant fails to fold to the native state, and populates a less structured state. Of particular interest are XLSA mutations in patients responsive to pyridoxine treatment (3). About two-thirds of XLSA patients respond to treatment with pyridoxine (which, like PLP, is a B6 vitamer). While the mechanism through which pyridoxine restores the catalytic activity of ALAS is not understood (3), the presumption has been that the mutated enzymes bind PLP less tightly, and given that pyridoxine is enzymatically converted to PLP, the increased PLP concentration would lead to increased ALAS activity. Since the binding of ligands has been shown to promote structural disorder-to-order transition in certain proteins (48), it would be useful to examine if there are any XLSA mutations where the binding of PLP to the ALAS variants promotes conversion of the enzyme to a more structured and catalytically competent conformation.
Supplementary Material
Highlights.
ALAS populates a molten globule state under specific acidic and alkaline conditions.
Acid-induced refolding of the secondary structure was detected.
Changes in pH affect the chiral environment of the internal aldimine.
The holoenzyme is catalytically active in the alkaline molten globule state.
Ligand binding restricts conformational fluctuations in the tertiary structure
Acknowledgments
This work was supported by the National Institutes of Health, grant #GM080270, and the American Heart Association, Greater Southeast Affiliate, grant #10GRNT4300073, to G.C.F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fratz EJ, Stojanovski BM, Ferreira GC. Toward Heme: 5-Aminolevulinate Synthase and Initiation of Porphyrin Synthesis. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. The Handbook of Porphyrin Science. World Scientific Publishing Co; New Jersey, USA: 2013. pp. 1–78. [Google Scholar]
- 2.Hunter GA, Ferreira GC. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim Biophys Acta. 2011;1814:1467–1473. doi: 10.1016/j.bbapap.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottomley SS. Sideroblastic Anemias. In: Greer JP, Arber DA, Glader B, List AF, Means RT, Paraskevas F, Rodgers GM, editors. Wintrobe’s Clinical Hematology. 13. Lippincott WIlliams & Wilkins, a Wolters Kluwer business; Philadelphia: 2014. pp. 643–661. [Google Scholar]
- 4.Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408–414. doi: 10.1016/j.ajhg.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreira GC, Dailey HA. Expression of mammalian 5-aminolevulinate synthase in Escherichia coli Overproduction, purification, and characterization. J Biol Chem. 1993;268:584–590. [PubMed] [Google Scholar]
- 6.Tan D, Ferreira GC. Active site of 5-aminolevulinate synthase resides at the subunit interface. Evidence from in vivo heterodimer formation. Biochemistry. 1996;35:8934–8941. doi: 10.1021/bi952918m. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira GC, Neame PJ, Dailey HA. Heme biosynthesis in mammalian systems: evidence of a Schiff base linkage between the pyridoxal 5′-phosphate cofactor and a lysine residue in 5-aminolevulinate synthase. Protein Sci. 1993;2:1959–1965. doi: 10.1002/pro.5560021117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Cheltsov AV, Ferreira GC. Conversion of 5-aminolevulinate synthase into a more active enzyme by linking the two subunits: Spectroscopic and kinetic properties. Protein Sci. 2005;14:1190–1200. doi: 10.1110/ps.041258305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira GC, Vajapey U, Hafez O, Hunter GA, Barber MJ. Aminolevulinate synthase: lysine 313 is not essential for binding the pyridoxal phosphate cofactor but is essential for catalysis. Protein Sci. 1995;4:1001–1006. doi: 10.1002/pro.5560040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong J, Kay CJ, Barber MJ, Ferreira GC. Mutations at a glycine loop in aminolevulinate synthase affect pyridoxal phosphate cofactor binding and catalysis. Biochemistry. 1996;35:14109–14117. doi: 10.1021/bi961296h. [DOI] [PubMed] [Google Scholar]
- 11.Hunter GA, Zhang J, Ferreira GC. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J Biol Chem. 2007;282:23025–23035. doi: 10.1074/jbc.M609330200. [DOI] [PubMed] [Google Scholar]
- 12.Lendrihas T, Hunter GA, Ferreira GC. Targeting the active site gate to yield hyperactive variants of 5-aminolevulinate synthase. J Biol Chem. 2010;285:13704–13711. doi: 10.1074/jbc.M109.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan D, Harrison T, Hunter GA, Ferreira GC. Role of arginine 439 in substrate binding of 5-aminolevulinate synthase. Biochemistry. 1998;37:1478–1484. doi: 10.1021/bi971928f. [DOI] [PubMed] [Google Scholar]
- 14.Tan D, Barber MJ, Ferreira GC. The role of tyrosine 121 in cofactor binding of 5-aminolevulinate synthase. Protein Sci. 1998;7:1208–1213. doi: 10.1002/pro.5560070516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24:3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter GA, Ferreira GC. Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J Biol Chem. 1999;274:12222–12228. doi: 10.1074/jbc.274.18.12222. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J. Transient State Kinetic Investigation of 5-Aminolevulinate Synthase Reaction Mechanism. J Biol Chem. 2002;277:44660–44669. doi: 10.1074/jbc.M203584200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uversky VN, Kutyshenko VP, Protasova N, Rogov VV, Vassilenko KS, Gudkov AT. Circularly permuted dihydrofolate reductase possesses all the properties of the molten globule state, but can resume functional tertiary structure by interaction with its ligands. Protein Sci. 1996;5:1844–1851. doi: 10.1002/pro.5560050910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Jing G. Double point mutant F34W/W140F of staphylococcal nuclease is in a molten globule state but highly competent to fold into a functional conformation. J Biochem. 2000;128:739–744. doi: 10.1093/oxfordjournals.jbchem.a022810. [DOI] [PubMed] [Google Scholar]
- 20.Zambelli B, Stola M, Musiani F, De Vriendt K, Samyn B, Devreese B, Van Beeumen J, Turano P, Dikiy A, Bryant DA, Ciurli S. UreG, a chaperone in the urease assembly process, is an intrinsically unstructured GTPase that specifically binds Zn2+ J Biol Chem. 2005;280:4684–4695. doi: 10.1074/jbc.M408483200. [DOI] [PubMed] [Google Scholar]
- 21.Vamvaca K, Vogeli B, Kast P, Pervushin K, Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc Natl Acad Sci U S A. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 23.Lien LF, Beattie DS. Comparisons and modifications of the colorimetric assay for delta-aminolevulinic acid synthase. Enzyme. 1982;28:120–132. doi: 10.1159/000459096. [DOI] [PubMed] [Google Scholar]
- 24.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 25.Goto Y, Calciano LJ, Fink AL. Acid-induced folding of proteins. Proc Natl Acad Sci U S A. 1990;87:573–577. doi: 10.1073/pnas.87.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goto Y, Takahashi N, Fink AL. Mechanism of acid-induced folding of proteins. Biochemistry. 1990;29:3480–3488. doi: 10.1021/bi00466a009. [DOI] [PubMed] [Google Scholar]
- 27.Uversky VN, Karnoup AS, Segel DJ, Seshadri S, Doniach S, Fink AL. Anion-induced folding of Staphylococcal nuclease: characterization of multiple equilibrium partially folded intermediates. J Mol Biol. 1998;278:879–894. doi: 10.1006/jmbi.1998.1741. [DOI] [PubMed] [Google Scholar]
- 28.Goto Y, Fink AL. Conformational states of β-lactamase: molten-globule states at acidic and alkaline pH with high salt. Biochemistry. 1989;28:945–952. doi: 10.1021/bi00429a004. [DOI] [PubMed] [Google Scholar]
- 29.Goto Y, Fink AL. Phase diagram for acidic conformational states of apomyoglobin. J Mol Biol. 1990;214:803–805. doi: 10.1016/0022-2836(90)90334-I. [DOI] [PubMed] [Google Scholar]
- 30.Hamada D, Hoshino M, Kataoka M, Fink AL, Goto Y. Intermediate conformational states of apocytochrome c. Biochemistry. 1993;32:10351–10358. doi: 10.1021/bi00090a010. [DOI] [PubMed] [Google Scholar]
- 31.Sinibaldi F, Piro MC, Coletta M, Santucci R. Salt-induced formation of the A-state of ferricytochrome c – effect of the anion charge on protein structure. FEBS J. 2006;273:5347–5357. doi: 10.1111/j.1742-4658.2006.05527.x. [DOI] [PubMed] [Google Scholar]
- 32.Goto Y, Fink AL. [1] Acid-induced folding of heme proteins. In: Johannes Everse KDVRMW, editor. Meth Enzymol. Academic Press; 1994. pp. 3–15. [DOI] [PubMed] [Google Scholar]
- 33.Fink AL, Calciano LJ, Goto Y, Nishimura M, Swedberg SA. Characterization of the stable, acid-induced, molten globule-like state of staphylococcal nuclease. Protein Sci. 1993;2:1155–1160. doi: 10.1002/pro.5560020710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Vies SM, Viitanen PV, Gatenby AA, Lorimer GH, Jaenicke R. Conformational states of ribulosebisphosphate carboxylase and their interaction with chaperonin 60. Biochemistry. 1992;31:3635–3644. doi: 10.1021/bi00129a012. [DOI] [PubMed] [Google Scholar]
- 35.Muzammil S, Kumar Y, Tayyab S. Anion-induced refolding of human serum albumin under low pH conditions. Biochim Biophys Acta. 2000;1476:139–148. doi: 10.1016/s0167-4838(99)00226-5. [DOI] [PubMed] [Google Scholar]
- 36.Khan F, Khan RH, Muzammil S. Alcohol-induced versus anion-induced states of α-chymotrypsinogen A at low pH. Biochim Biophys Acta. 2000;1481:229–236. doi: 10.1016/s0167-4838(00)00129-1. [DOI] [PubMed] [Google Scholar]
- 37.Edwin F, Jagannadham MV. Anion-Induced Folding of Rabbit Muscle Pyruvate Kinase: Existence of Multiple Intermediate Conformations at Low pH. Archi Biochem Biophys. 2000;381:99–110. doi: 10.1006/abbi.2000.1968. [DOI] [PubMed] [Google Scholar]
- 38.Bhowmick R, Jagannadham MV. Multiple Intermediate Conformations of Jack Bean Urease at Low pH: Anion-induced Refolding. Protein J. 2006;25:399–410. doi: 10.1007/s10930-006-9026-3. [DOI] [PubMed] [Google Scholar]
- 39.Fink AL, Calciano LJ, Goto Y, Kurotsu T, Palleros DR. Classification of acid denaturation of proteins: intermediates and unfolded states. Biochemistry. 1994;33:12504–12511. doi: 10.1021/bi00207a018. [DOI] [PubMed] [Google Scholar]
- 40.Uversky VN, Goto Y. Acid denaturation and anion-induced folding of globular proteins: multitude of equilibium partially folded intermediates. Curr Protein Pept Sci. 2009;10:447–455. doi: 10.2174/138920309789352029. [DOI] [PubMed] [Google Scholar]
- 41.Lindgren M, Sörgjerd K, Hammarström P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J. 2005;88:4200–4212. doi: 10.1529/biophysj.104.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Povarova OI, Kuznetsova IM, Turoverov KK. Differences in the pathways of proteins unfolding induced by urea and guanidine hydrochloride: molten globule state and aggregates. PLoS One. 2010;5:e15035. doi: 10.1371/journal.pone.0015035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshimura Y, Lin Y, Yagi H, Lee YH, Kitayama H, Sakurai K, So M, Ogi H, Naiki H, Goto Y. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc Natl Acad Sci (USA) 2012;109:14446–14451. doi: 10.1073/pnas.1208228109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan-Huot M, Sharif S, Tolstoy PM, Toney MD, Limbach HH. NMR studies of the stability, protonation states, and tautomerism of 13C- and 15N-labeled aldimines of the coenzyme pyridoxal 5′-phosphate in water. Biochemistry. 2010;49:10818–10830. doi: 10.1021/bi101061m. [DOI] [PubMed] [Google Scholar]
- 45.Osés-Prieto JA, Bengoechea-Alonso MT, Artigues A, Iriarte A, Martinez-Carrión M. The nature of the rate-limiting steps in the refolding of the cofactor-dependent protein aspartate aminotransferase. J Biol Chem. 2003;278:49988–49999. doi: 10.1074/jbc.M309922200. [DOI] [PubMed] [Google Scholar]
- 46.Pioselli B, Bettati S, Demidkina TV, Zakomirdina LN, Phillips RS, Mozzarelli A. Tyrosine phenol-lyase and tryptophan indole-lyase encapsulated in wet nanoporous silica gels: selective stabilization of tertiary conformations. Protein Sci. 2004;13:913–924. doi: 10.1110/ps.03492904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunter GA, Ferreira GC. Lysine-313 of 5-aminolevulinate synthase acts as a general base during formation of the quinonoid reaction intermediates. Biochemistry. 1999;38:3711–3718. doi: 10.1021/bi982390w. [DOI] [PubMed] [Google Scholar]
- 48.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.