

Abstract
The mitogen-activated protein (MAP) kinases ERK1 and ERK2 are critical intracellular signaling intermediates; however, little is known about their isoform-specific functions in vivo. We have examined the role of ERK2 in neural development by conditional inactivation of the murine mapk1/ERK2 gene in neural progenitor cells of the developing cortex. ERK MAP kinase (MAPK) activity in neural progenitor cells is required for neuronal cell fate determination. Loss of ERK2 resulted in a reduction in cortical thickness attributable to impaired proliferation of neural progenitors during the neurogenic period and the generation of fewer neurons. Mutant neural progenitor cells remained in an undifferentiated state until gliogenic stimuli induced their differentiation, resulting in the generation of more astrocytes. The mutant mice displayed profound deficits in associative learning. Importantly, we have identified patients with a 1 Mb microdeletion on chromosome 22q11.2 encompassing the MAPK1/ERK2 gene. These children, who have reduced ERK2 levels, exhibit microcephaly, impaired cognition, and developmental delay. These findings demonstrate an important role for ERK2 in cellular proliferation and differentiation during neural development as well as in cognition and memory formation.
Keywords: MAP kinase, neurogenesis, cortical development, associative learning, intermediate progenitor cell, neural progenitor cell
Introduction
The extracellular signal-regulated kinases ERK1/2 are among the most prominent signal transduction molecules through which extracellular stimuli are propagated from the cell surface to cytoplasmic and nuclear effectors. ERK1 (mapk3) and ERK2 (mapk1) exhibit 84% sequence identity and are uniquely and coordinately activated through the sequential phosphorylation of the classical three-tier mitogen-activated protein kinase (MAPK) cascade. Significantly, although both ERK1 and ERK2 are expressed throughout the brain (Selcher et al., 2001; Mazzucchelli et al., 2002), genetic inactivation of ERK1 has only subtle phenotypic effects, whereas ERK2 inactivation results in early embryonic lethality (Aouadi et al., 2006).
The orchestration of cortical development occurs primarily through the actions of growth factors that signal through receptor tyrosine kinases (RTKs) to control cell cycle initiation, progression, and cell fate decisions in neural progenitor cells (NPCs). The mammalian neocortex arises from self-renewing multipotent NPCs that initially generate neurons and subsequently produce glia. During neurogenesis, NPCs within the ventricular zone (VZ) divide symmetrically to produce two neurons or asymmetrically to generate a self-renewing progenitor and a daughter cell, which becomes either a neuron or an intermediate progenitor cell (IPC). The nascent IPC continues to undergo asymmetric renewal or, more frequently, divides symmetrically, yielding two neurons (Noctor et al., 2004; Pontious et al., 2008). Gliogenesis commences late in embryogenesis, with undifferentiated NPCs responding to stimuli secreted by newly born neurons (Miller and Gauthier, 2007). Disruption of the dynamics of NPC or IPC proliferation and differentiation result in alterations in cell number, cellular identity, and organization of the cortex (Dehay and Kennedy, 2007).
Signal transduction through the sequential activation of elements of the ERK MAPK cascade directs neurogenesis while concurrently suppressing gliogenesis (Miller and Gauthier, 2007). It has been demonstrated that alteration of MAPK signaling abrogates the generation of a fully populated, normal size cortex (Vaccarino et al., 1999; Menard et al., 2002; Barnabe-Heider and Miller, 2003; Dono, 2003; Ohkubo et al., 2004; Zheng et al., 2004; Paquin et al., 2005; Thomson et al., 2007). Inhibition of the upstream activator of the MAPKs, MEK1, causes NPCs to remain in the subventricular zone (SVZ)/VZ in an undifferentiated state, blocking neurogenesis (Menard et al., 2002; Barnabe-Heider and Miller, 2003; Paquin et al., 2005). Similarly, mutation or inactivation of scaffolding proteins such as FRS2 and SHP-2 that link RTKs to MAPK activation have analogous effects on cortical progenitors (Hadari et al., 1998; Yamamoto et al., 2005; Gauthier et al., 2007; Ke et al., 2007).
It has been appreciated recently that a group of genetic disorders termed neuro-cardio-facial-cutaneous syndromes (including cardio-facio-cutaneous, Costello, Leopard, and Noonan syndromes) are caused by mutations in upstream elements of the ERK/MAPK signaling cascade (Bentires-Alj et al., 2006; Roberts et al., 2006) and are collectively characterized by distinctive cardiac and craniofacial defects, developmental delay, and mental retardation. The mutation of downstream elements in the ERK cascade has similarly been associated with mental retardation syndromes (Weeber and Sweatt, 2002). These observations suggest that perturbations in ERK MAPK signaling underlie a diverse range of neurodevelopmental syndromes.
To determine the role of ERK2 in cortical development and function, we generated a murine model in which mapk1/ERK2 undergoes conditional inactivation at the peak of neurogenesis. The loss of ERK2 resulted in the generation of fewer neurons and many more astrocytes in the cortex. This change in cell fate is attributable to alterations in the dynamics of NPC proliferation and specifically with a reduction in IPC division. Behaviorally, the mutant mice display profound impairments in cued and contextual fear conditioning. The identification of individuals with haploinsufficiency for the MAPK1/ERK2 gene as a result of distal microdeletions at 22q11.2 (Shaikh et al., 2007) enabled the first analysis of humans with reduced expression levels of ERK2. These patients exhibit microcephaly and neurodevelopmental deficits, consistent with the phenotype observed in the murine models presented here. Together, these studies provide direct evidence for the unique actions of the ERK2 isoform in neural development and suggest its involvement in cognitive function.
Materials and Methods
Generation of a conditional ERK2 allele
A targeting construct was designed using the loxP system to excise exon 2 in the mapk1 gene, resulting in a null allele. A neo cassette flanked by loxP sites was inserted into an ApaI site upstream of exon 2, and a loxP site was cloned into an XbaI site between exons 2 and 3. The βADT diphtheria toxin cassette was used for negative selection. The construct was linearized and electroporated into 129 embryonic stem cells (ES) cells, and clones were selected after treatment with G418. Clones positive for homologous recombination were further transfected with a Cre-expressing vector, and resultant clones were analyzed for deletion of the Neo cassette. One clone had deletion of only the Neo cassette and was expanded and injected in C57BL/6 blastocysts. Resultant chimeras were mated to establish germ-line transmission, and heterozygote (Mapk1flox/+) animals were intercrossed to establish an Mapk1flox/flox line of mice. hGFAP–cre mice on an FVB/N background were obtained from Dr. Albee Messing (University of Wisconsin-Madison, Madison, WI) and interbred with the ERK2 floxed line to generate Mapk1flox/flox;hGFAPcre/+ animals in which the ERK2 gene has been conditionally knocked out (ERK2 CKO). All experiments were performed in accordance with the Case Western Reserve University Institutional Animal Care and Use Committee.
Southern blotting
Genomic DNA derived from cortex was digested with Xba, resolved on a 0.8% agarose gel, and transferred to a nylon membrane. The DNA was cross-linked and hybridized to a 32P radiolabeled, 500 bp 3′ probe (located distal to the targeted region of the mapk1 locus, between exons 3 and 4) using ExpressHyb (Clontech).
Cell culture
Cortical progenitor cell culture.
Cortical progenitor cells were cultured from embryonic day 14.5 (E14.5) mouse embryos using protocols described previously by Miller and colleagues (Menard et al., 2002; Barnabe-Heider and Miller, 2003). Briefly, cortices were dissected from E14.5 mouse embryos in ice-cold HBSS and transferred to Neurobasal medium containing 500 μm l-glutamine, 2% B27 supplement, 1% penicillin–streptomycin (Invitrogen), and 40 ng/ml FGF2 (BD Biosciences Discovery Labware). The tissue was mechanically triturated with a plastic pipette and plated into eight-well chamber slides (Nalge Nunc International) precoated with 2% laminin (BD Biosciences Discovery Labware) and 1% poly-d-lysine (Sigma) at a cell density of 50,000 cells per well. Cells were fed every 2 d. For experiments with CNTF, cells were plated in FGF2 for 12 h, and then CNTF (3 ng/ml; PeproTech) was added to the media for an additional 2 or 4 d.
Astrocyte culture.
Astrocytes were derived from the brains of neonatal mice as described previously but with some modifications (Giulian and Baker, 1986). Cerebral cortices were isolated from postnatal day 1 (P1) to P3 mice. Cortices were minced, and cells were dissociated in PBS containing 0.25% trypsin and 1 mm EDTA. Digestion was terminated by adding an equal volume of DMEM/F-12 medium (Invitrogen) containing 20% FCS, and cells were triturated to obtain a single-cell suspension. Cells were plated, and media was replaced the next day with DMEM/F-12 containing 20% FCS.
Cell counts.
Quantification of immunocytochemical staining was performed on dissociated cells or cortical progenitor cell cultures. Counts were made from at least three embryos of each genotype obtained from a minimum of three litters. For each condition, >300 cells from nine fields of a randomly selected reference space per well were counted and analyzed from two wells per embryo. Digital image acquisition was performed on a Leitz DM R B microscope with Leica DC 500 camera and the software of the manufacturer.
Viability assay.
Cellular viability was assessed by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (Sigma) assay. In brief, primary CKO and wild-type (WT) astrocytes (5 × 103/well) were seeded in a 96-well plate. At 2 and 4 d, 10 μl of MTT (5 mg/ml PBS) was added to each well, and plates were incubated at 37°C for 2 h. Medium was then removed, and cells were resuspended in 100 μl of DMSO. Cell viability was assessed by colorimetric change using the SpectraMax 340 PC plate reader (Molecular Devices) at λ = 550 nm.
Immunocytochemistry.
Immunocytochemistry of cultured cells was performed by washing the cultures with HEPES-buffered saline (HBS), followed by fixation with 4% paraformaldehyde for 15 min. The cells were permeabilized with 0.2% NP-40 in HBS, followed by incubation in blocking with buffer containing 6% normal goat serum (NGS) and 0.5% bovine serum albumin (BSA) for 1–2 h at room temperature. Cells were then incubated with primary antibodies in HBS containing 3% NGS and 0.25% BSA at 4°C overnight. After washing with HBS, cells were incubated with secondary antibodies prepared in HBS containing 3% NGS and 0.25% BSA at room temperature for 1 h. Cells were then washed with HBS and counterstained with 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI) for 2 min. Primary antibodies used were anti-ERK2 (1:200; BD Biosciences Discovery Labware), anti-Nestin (1:200; Millipore Bioscience Research Reagents), and anti-βIII tubulin (1:4000), anti-MAP2 (1:200), anti-S100β (1:200), anti-vimentin IgM (1:200; each from Sigma).
Immunohistochemistry
For immunohistochemistry, 10 μm cryosections were rinsed in PBS, blocked, and permeabilized with 0.1% Triton X-100, 10% goat serum in PBS. Sections were incubated with primary antibodies at 4°C overnight, washed with PBS, and incubated with secondary antibodies at room temperature for 1 h. The sections were then counterstained with DAPI for 2 min. The primary antibodies used were anti-Tbr1 (1:2000; a gift from Dr. Robert Hevner, University of Washington, Seattle, WA), anti-Otx1 (1:10; Developmental Studies Hybridoma Bank), anti-Brn1 (1:50; Santa Cruz Biotechnology), anti-GFAP (1:500; DakoCytomation), anti-neuronal-specific nuclear protein (NeuN) (1:300; Millipore Bioscience Research Reagents), anti-phospho-histone H3 (pH3) (1:200; Millipore), anti-Zebrin (1:50), anti-Reelin (1:50; Santa Cruz Biotechnology), anti-GABA (1:500; Sigma), and anti-caspase 3 (1:100; Cell Signaling Technology). The secondary antibodies used were Alexa fluorophore-conjugated IgG or IgM. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining was performed as described by the protocol of the manufacturer (DeadEnd Fluorometric TUNEL staining kit; Promega). Golgi staining was performed using the Rapid GolgiStain kit from FD Neurotechnologies.
For confocal analysis of sections, images were obtained using a Zeiss LSM 510 confocal laser microscope using argon and helium–neon lasers (excitation lines, 488 and 594, respectively) and Plan-Neofluar 10×, numerical aperture (N.A.) 0.3, Plan-Neofluar 20×, N.A. 0.5, and C-Apochromat 40×, N.A. 1.2 water immersion objectives. Photomicrographs were processed using Adobe Photoshop, and cell number was quantified with Image ProPlus software.
Cell counts.
Analysis was performed on at least eight tissue sections obtained from four mice in each group. Counts were made within the frontal ∼2.0 mm proximal to bregma) and parietal lobes (∼1.25 mm distal to bregma) because of the robust recombination in these areas. The primary motor cortex (“medial”) and primary somatosensory cortex (“lateral”) were identified in the same section of each lobe using standard stereotaxic coordinates (Paxinos and Franklin, 2001). The reference space was fixed by the reticule of the microscope objective, measuring 150 μm in width. Cell layers were identified based on morphological distinctions and distance from the pial surface. Quantification of cell numbers was performed using the Image ProPlus software by blinded observer.
Western blot analysis
Cells were washed with ice-cold HBSS and lysed by sonication in lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% NP-40, 10% glycerol, 1 mm EDTA, 1.5 mm MgCl2, 20 mm NaF, and 20 mm β-glycerophosphate) supplemented with protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mm PMSF, and 1 mm Na3VO4). Lysates were cleared by centrifugation, and protein concentration was determined using the bicinchoninic acid assay (Pierce) using BSA as a standard. Equal amounts of protein were boiled in sample buffer, separated by SDS-PAGE gels, and transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore). Membranes were blocked in 3% BSA [or 5% skim-milk powder for anti-glyceraldehyde-3-phosphate dehydrogenase (G3PDH)] in TBS and 0.1% Tween 20 (TBS-T) for 2 h at room temperature and incubated overnight at 4°C with primary antibodies. The primary antibodies used were rabbit anti-pERK (1:1000; Cell Signaling Technology), mouse anti-ERK2 (1:3000; BD Biosciences Discovery Labware) directed to the C terminus of ERK2 or mouse anti-ERK2 antibody that detects the N terminus (0.5 μg/ml; Acris Antibodies), mouse anti-ERK1 (1:1000; Zymed Laboratories), anti-CrkL (1:500; Santa Cruz Biotechnology), anti-G3PDH (1:5000; Trevigen), anti-β-tubulin (1:5000; Santa Cruz Technology), and anti-c-Src (1:500; Santa Cruz Technology). After washing with TBS-T, membranes were incubated with HRP-conjugated goat anti-mouse or anti-rabbit (1:5000; GE Healthcare) secondary antibodies in TBS-T with 5% milk for 2 h at room temperature. Blots were washed with TBS-T, and detection was performed using either Millipore or Pierce chemiluminescence reagents using BioMax MR x-ray film (Eastman Kodak). Densitometric analysis was performed using Adobe Photoshop histogram function, and statistical analysis was done with GraphPad Prism software. Patient samples and clinical data were collected after informed consent under an Institutional Review Board-approved protocol at the Children's Hospital of Philadelphia.
Bromodeoxyuridine labeling and analysis
Pregnant mice were injected intraperitoneally with bromodeoxyuridine (BrdU) at 100 mg/kg body weight. The animals were killed 30 min after the injection. BrdU-positive (BrdU+) cells were detected by immunostaining as described by the instructions of the manufacturers with heat-mediated antigen retrieval in citrate buffer followed by 2 N HCl treatment. Sections were incubated with a rat anti-BrdU antibody (1:1000; Abcam) overnight at 4°C. Quantification of BrdU+ cells during embryogenesis was performed by dissociation of individual cortices and fixation in 4% paraformaldehyde immediately after the animals were killed. Cells were plated at 50,000 cells per well and immunostained and counted as described above. For BrdU labeling of astrocytes, cells were plated at 50,000 cells per well in eight-well chamber slides overnight. Astrocytes were then treated with 1 μg/ml BrdU for 24 h and processed as described above.
Statistical analysis
Statistical analysis was performed using either two-way ANOVA with the Bonferroni's post hoc test or the Student's t test, as indicated in the figures. In all graphs, the error bars indicate SD.
Behavioral analysis
Animals used for behavioral analysis were bred at the University of Alabama at Birmingham. They were housed with same sex siblings and with ad libitum access to food and water, and they were maintained on a 12 h light/dark cycle. Male wild-type and ERK2 CKO littermates were compared in all behavioral experiments. All experiments were performed in accordance with the University of Alabama at Birmingham Institutional Animal Care and Use Committee. All experiments were performed with the experimenter blind to genotype.
Baseline sensory tests.
The following assessments were performed similarly to those described previously (Selcher et al., 2001). Adult mice (14–40 weeks of age) were tested for baseline locomotor behavior and anxiety-like behavior using an open field test (Med Associates), which consisted of a 17 × 17 inch square polycarbonate box with three 16-beam infrared arrays (measuring x-, y-, and z-axis movement). Mice were placed into the center of the open field to start the test, and activity was recorded for 15 min under fluorescent lighting. Total distance moved was used as a measure of locomotor activity, and thigmotaxis (quantified by calculating the percentage of total ambulatory distance that was traveled in the center zone only of the open field) was used as a measure of baseline anxiety-like behavior. Nociception and visual ability were performed as described previously (Crawley, 1999). Nociception was measured with a shock threshold test. One-second-duration shocks were administered in increasing increments (0.08, 0.10, 0.15, 0.25, 0.35, 0.45, and 0.50 mA), each separated by 15-s, and mice were observed for flinching, jumping, running, and vocalization at each increment. The test was terminated when mice performed all four behaviors or when the highest shock increment was reached, whichever came first. Visual ability was measured using a visual cliff test. All mice were observed during fear conditioning experiments for their startle response to the onset of the tone used as a conditioned stimulus.
Fear conditioning.
Mice were tested for cued and contextual fear conditioning as described previously (Selcher et al., 2001) with some modifications. For training, animals were placed in a conditioning chamber with a metal grid floor (Video Fear Conditioning; Med Associates) for 8 min. Training consisted of a 3 min exploration period followed by three tone–shock pairings consisting of a 30-s-long 90 dB tone, with a 1 s, 0.5 mA shock occurring during the last second of the tone. Each tone–shock pairing was followed by a 60 s rest, except after the third pairing, which was followed by a 90 s rest period leading up to the conclusion of the training session. Twenty-four hours after training, each mouse was placed back into the training chamber for a 5 min contextual learning test, during which time freezing behavior was assessed in the absence of a tone or foot shock. One hour after the contextual test, cued learning was assessed by placing the mouse back into the chamber after the context of the chamber had been masked by altering its size, shape, color, and odor. After a 3 min exploration period of the novel context, the 90 dB conditioned stimulus tone was presented for 3 min, during which time freezing behavior was assessed. For the repeated-training paradigm, animals were treated as noted, except that the training procedure was performed three times in the same day. Contextual and cued learning was then assessed 24 h after the second training trial, using the procedure described above.
Results
ERK2 is required for normal development of the murine cerebral cortex
To directly assess the role of ERK2 in neural development, we generated a conditional mapk1/ERK2 null mutation. The mapk1flox allele was produced by introducing loxP sites into positions flanking exon 2 of the mapk1/ERK2 gene (Fig. 1A). Exon 2 is within the ERK2 open reading frame and contains the highly conserved subdomain II, which is essential for catalytic activity of the kinase (Robinson et al., 1996). Transgenic mice in which cre recombinase expression is driven by the human glial fibrillary acidic protein (hGFAP) promoter (Zhuo et al., 2001) were interbred with the mapk1flox/flox mice, resulting in deletion of ERK2 within cortical NPCs. During activation of the hGFAP promoter in these animals, cre expression led to the deletion of a 2 kb genomic region that includes exon 2 of the mapk1/ERK2 gene. Cre-mediated recombination resulted in production of a 9.5 kb fragment on the Southern blots (Fig. 1B). Western blot analysis of adult cortical brain homogenates confirmed that ERK2 protein levels in the cortex were reduced. No truncated fragments of ERK2 were detected by immunoblot analysis using an antibody directed to the N terminus of the protein, confirming that the targeting strategy resulted in a null allele (Fig. 1D). Expression of ERK1 was analyzed as a control and was not altered (Fig. 1C).
Figure 1.
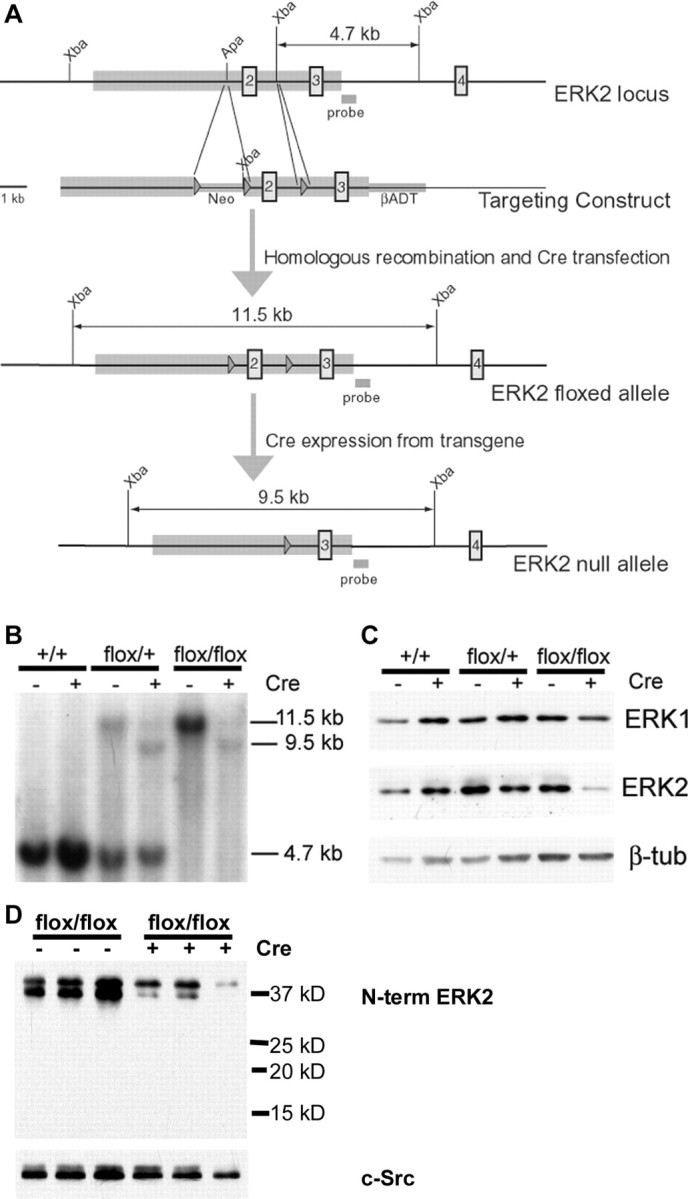
Generation of ERK2 conditional knock-out mice. A, Schematic representation of gene targeting strategy. The shaded boxes on the mapk1/ERK2 locus and the targeting construct designate homologous regions identified for recombination. Numbered boxes represent mapk1 exons 2–4. The targeting construct contains a floxed Neo cassette upstream of exon 2 and a loxP site between exons 2 and 3. A βADT cassette was inserted downstream of exon 3 for negative selection. Relevant restriction sites and the 3′ probe used for Southern blot analysis are indicated. Recombinant ES cells were transfected with a Cre plasmid to remove the Neo cassette, giving rise to the floxed mapk1 allele. Cre recombinase expression from the hGFAP–cre transgene produces a null allele. B, Southern blot of genomic brain DNA. After Xba digestion of DNA, the 3′ probe identifies the wild-type allele as a 4.7 kb fragment and the mutant allele as 11.5 kb before and 9.5 kb after recombination. C, Western blot analysis of adult cortical brain homogenates. Tissue was homogenized, separated by SDS-PAGE, and immunoblotted with anti-ERK1, anti-ERK2, and β-tubulin antibodies. D, Western blot analysis of embryonic cortical homogenates. Tissue was lysed, separated by SDS-PAGE, and immunoblotted with an antibody directed to an epitope at the N terminus of ERK2. A lack of bands below 42 kDa in both flox/flox and flox/flox cre+ samples demonstrates that no truncated protein fragments were produced by recombination. An antibody for c-Src was used as a loading control.
Mapk1flox/flox; hGFAPcre/+ mice are viable and fertile mice with normal lifespans. Macroscopic examination of these ERK2 CKO brains at P2 revealed no gross abnormalities (Fig. 2A) or differences in brain mass (CKO, 0.157 g, n = 6; WT, 0.167 g, n = 7; unpaired t test, p = 0.1466). Despite the normal overall morphology and size of the ERK2 CKO cortex, we observed a significant reduction in cortical thickness throughout the dorsal telencephalon of CKO mice compared with wild-type littermates. A detailed regional analysis revealed a reduction in thickness of primary motor (0.8968), somatosensory (0.8673), auditory (0.8872), and visual (0.9042) cortices (Fig. 2B). No deficits in the organization of specific cortical lamina were recognized; however, cellular density appeared greater throughout the ERK2 CKO cortex (Fig. 2C). ERK1 null mice were analyzed and were not different from wild-type controls (Selcher et al., 2001), establishing that specific inactivation of ERK2 causes the cortical thinning (Fig. 2D).
Figure 2.
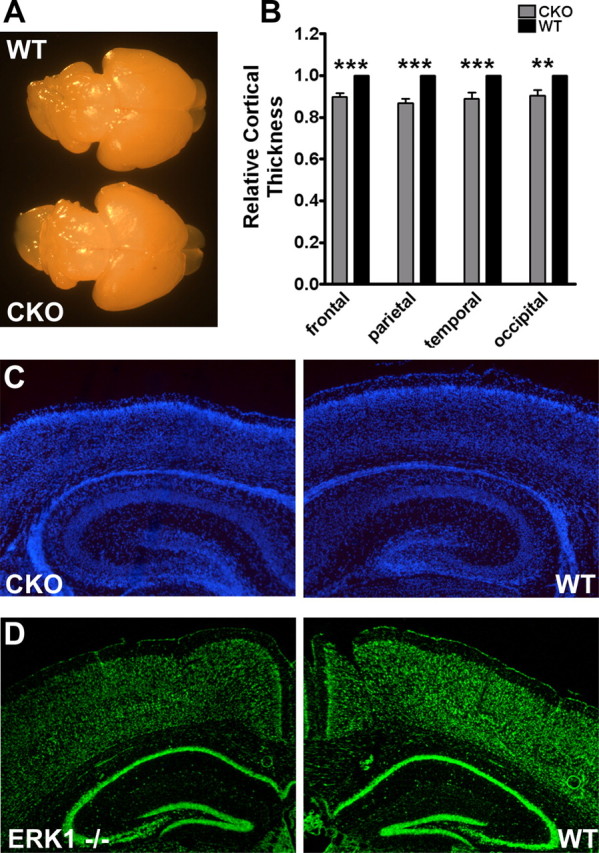
ERK2 CKO mice display reduced cortical thickness. A, Dorsal view of P2 CKO (bottom) and wild-type (top) littermates. B, Quantitative analysis of cortical thickness. Corresponding coronal sections from CKO and wild-type brains were used to measure thickness of the frontal, parietal, temporal, and occipital lobes corresponding to the motor, somatosensory, auditory, and visual cortices. Measurements were taken from the ventricular zone to the pial surface (n = 5). Two-way ANOVA, p < 0.0001 with Bonferroni's post hoc tests, **p < 0.01, ***p < 0.001. C, Representative coronal sections of CKO and wild-type brains from P2, stained with DAPI. Note the difference in cellular density apparent between genotypes. D, Coronal sections of adult ERK1−/− and wild-type brains stained with the fluorescent Nissl stain Neurotrace.
The hGFAPcre/+ transgene is expressed in NPCs beginning at embryonic day 13.5 (Zhuo et al., 2001; Malatesta et al., 2003). This population of NPCs gives rise to >90% of projection neurons and astrocytes populating the neocortex and hippocampus (Malatesta et al., 2003), which we verified by interbreeding of Rosa26 reporter (R26R) mice (Soriano, 1999) with mice carrying the hGFAPcre/+ transgene and subsequent LacZ staining in the adult brain of cre+ progeny (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). ERK2 is deleted throughout the dorsal telencephalon with only a minority of cells retaining expression. The ERK2-expressing cells include Tbr1+ cells populating layer VIb and Reelin+ Cajal-Retzius cells, both of which are generated before induction of cre expression. The majority of ERK2-expressing cells in the mature cortex of the CKO mice are GABAergic interneurons, which arise and migrate from the ganglionic eminence that does not express the hGFAPcre/+ transgene (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). ERK2 expression was absent in the hippocampus of the mutant mice; however, expression in the amygdala was similar to that of wild-type animals, because the hGFAPcre/+ transgene is not expressed in this structure (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
We quantified cortical ERK2 protein levels at E14.5 and found a 50% reduction in ERK2 levels and a corresponding decrease in the levels of activated, phospho-ERK2 in the mutant mice. We did not detect any change in ERK1 expression (Fig. 3A,B) or activation status at this time (Fig. 3C,D). Evaluation of these animals at E16.5 revealed that the expression levels of ERK2 were reduced to 25% of wild-type levels, reflecting the ongoing recombination of the mapk1/ERK2 gene and the half-life of the protein. The level of active ERK2 was found to be 75% lower in the ERK2 CKO cells than those from the wild-type animals (Fig. 3A,C) at E16.5. Although no compensatory increase in ERK1 expression was found, phosphorylated ERK1 levels were dramatically (fourfold) higher in the ERK2 CKO compared with the wild-type cortex at this time (Fig. 3B,D). These data provide evidence that, although ERK1 expression was not elevated to compensate for the loss of ERK2, there was a significant increase in ERK1 activity at E16.5.
Figure 3.

Loss of ERK2 expression and activity in ERK2 CKO cortices. A–D, E14.5 and E16.5 cortices were microdissected and prepared for Western blot analysis immediately during dissection. The levels of ERK2, ERK1, and phospho-ERK1/2 were detected by immunoblotting, and representative blots are shown. Densitometric analysis of the bands, normalized to β-tubulin, was performed for quantification and expressed as fold difference relative to wild-type. For A, quantification of ERK2 levels at E14.5 (CKO, n = 15; WT, n = 21; p < 0.0001) and E16.5 (CKO, n = 7; WT, n = 8; p < 0.0001). B, Quantification of ERK1 expression levels at E14.5 (CKO, n = 13; WT, n = 18; p = 0.4019) and E16.5 (CKO, n = 7; WT, n = 8; p = 0.7772). C, Relative phospho-ERK2 activation at E14.5 (CKO, n = 12; WT, n = 18; p = 0.0093) and E16.5 (CKO, n = 7; WT, n = 8; p < 0.0001). D, Relative phospho-ERK1 activation at E14.5 (CKO, n = 11; WT, n = 17; p = 0.3594) and E16.5 (CKO, n = 4; WT, n = 4; p < 0.0001). **p < 0.001; ***p < 0.0001.
ERK2 inactivation alters the cellular composition of the cortex
To determine the basis for the cortical thinning and assess the cellular density within the cortex (Fig. 2B,C), we quantified both total number of DAPI+ cells per cortical region as well as the number of neurons (NeuN+ cells) in ERK2 CKO and wild-type littermates at postnatal day 10. The area from the VZ/SVZ boundary to the pial surface of the cortex was analyzed in 150 μm sectors from sections of the frontal and parietal lobes. We found no difference in DAPI+ cells per cortical region between genotypes, a finding consistent with the observation of increased cellular density and reduced cortical thickness. Strikingly, we found 10% fewer NeuN+ neurons per region in the ERK2 CKO compared with wild-type littermates (Fig. 4A). However, there was a 40% increase in non-neuronal [DAPI+; NeuN-negative (NeuN−)], presumptive glial cells. These data indicate that ERK2 inactivation resulted in the production of fewer neurons and more non-neuronal cells, which was associated with an overall reduction in cortical thickness.
Figure 4.
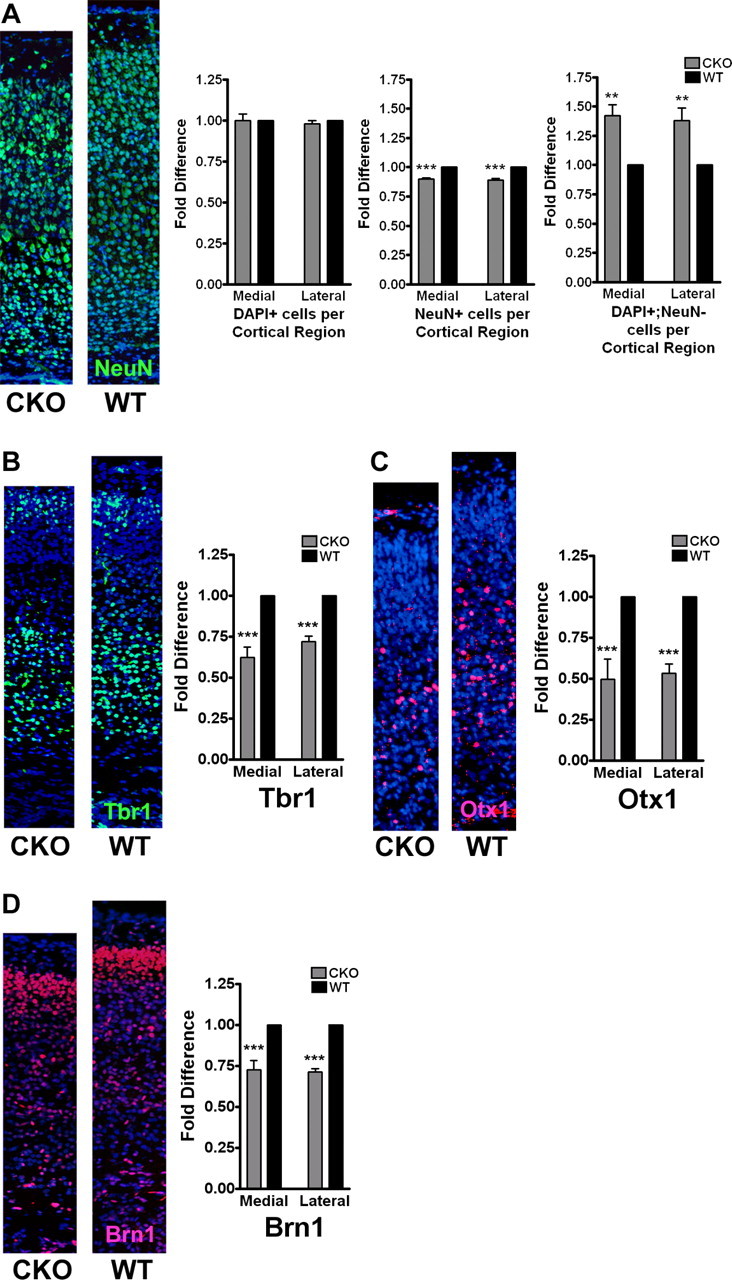
Inactivation of ERK2 in neural progenitor cells results in the generation of fewer cortical neurons. A, Analysis of cell density and cell fate in CKO and wild-type littermates at P10. Corresponding coronal sections of P10 cortices were immunostained with NeuN (green) and counterstained with DAPI (blue). Total cell number per cortical region from the VZ to the pial surface was calculated by counting DAPI+ cells in both medial (primary motor cortex, CKO, 1386.167 average cells per region; WT, 1398.167 average cells per region; for each n = 3) and lateral (primary somatosensory cortex, CKO, 1534.833 average cells per region; WT, 1571.167 average cells per region; for each n = 3) sections. Two-way ANOVA, p = 0.6179, and Bonferroni's post hoc test, p > 0.05. The fold difference in NeuN+ cells was calculated by two-way ANOVA, p < 0.0001, Bonferroni's post hoc test, p < 0.0001. Fold difference in DAPI; NeuN− cells was calculated. Two-way ANOVA, p = 0.0004, Bonferroni's post hoc test, p < 0.01. B–D, Corresponding coronal sections of P2 CKO (left) and WT (right) littermates were stained with anti-Tbr1 (B, green), anti-Otx1 (C, red), and anti-Brn1 (D, red) antibodies and counterstained with DAPI. Cortical lamina were identified based on morphology and distance from the pial surface. Immunoreactive cells were counted from two independent reference spaces of the cortex from at least eight sections per genotype. For each, two-way ANOVA, p < 0.0001, and Bonferroni's post hoc tests, p < 0.0001, were performed (n = 4). **p < 0.001; ***p < 0.0001.
To determine whether neuronal identity was affected by the loss of ERK2, a detailed examination of the number of neurons within each cortical lamina was performed. Cortices from P2 wild-type and ERK2 CKO littermates were stained with antibodies to transcription factors whose expression is limited to specific cortical layers. We found a 35% reduction in number of Tbr1+ neurons in layer VI and the cortical preplate (Fig. 4B) of ERK2 CKO animals. Similarly, we detected approximately half as many Otx1+ neurons populating layer V in the ERK2 CKO compared with wild-type animals (Fig. 4C). Evaluation of layers II–IV using Brn1 revealed an analogous, genotype-dependent 30% decrease in neurons populating this layer (Fig. 4D). No significant differences were found between medial and lateral regions of the cortex. Thus, inactivation of ERK2 was associated with an overall reduction in the numbers of neurons populating the cortex.
To confirm the identity of the non-neuronal (DAPI+; NeuN−) cells, we performed immunohistochemistry with astrocyte-specific markers. We found a dramatic increase in Zebrin II+ (Fig. 5A) and GFAP+ (Fig. 5B) astrocytes throughout the cortex and subventricular zone. To assess the proliferative capacity of ERK2 CKO astrocytes compared with wild-type astrocytes, we cultured these cells from each genotype and evaluated their proliferation by measuring BrdU incorporation, Ki67 immunoreactivity, and change in cell number (Fig. 5C–E, respectively). We also assessed astrocyte metabolism and viability using an MTT assay (Fig. 5F). No significant differences were found in any of these parameters. We interpret these data to suggest that loss of ERK2 does not significantly affect astrocyte viability or their intrinsic ability to proliferate. We did not observe increasing numbers of astrocytes in older animals.
Figure 5.
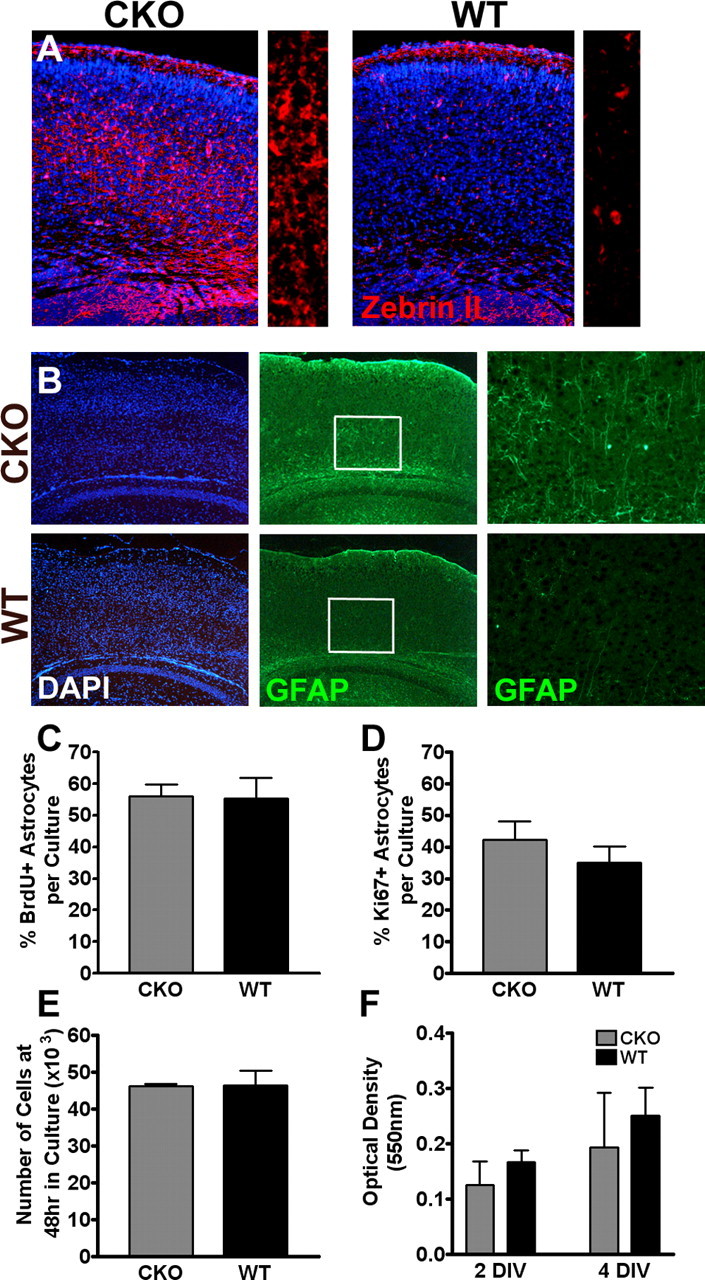
Inactivation of ERK2 in neural progenitor cells results in the presence of more astrocytes within the cerebral cortex. A, B, Representative images of corresponding coronal sections from neonatal (A) and mature (B) ERK2 CKO and wild-type littermates immunostained with anti-Zebrin II (red) anti-GFAP (green) antibodies and counterstained with DAPI. A higher-magnification image is shown on the right. C, Primary astrocyte cultures from CKO and wild-type P2 pups were prepared and maintained for 5 DIV. Cultures were incubated with BrdU overnight and fixed. Quantification of percentage BrdU immunoreactivity (CKO, n = 4; WT, n = 3; p = 0.9084). D, Quantification of Ki67-immunoreactive cells (CKO, n = 3; WT, n = 3; p = 0.4166). E, Characterization of astrocyte proliferative index (CKO, n = 2; WT, n = 2; p = 0.9827). F, MTT viability assay (CKO, n = 4; WT, n = 5; p = 0.4556).
Thus, these data demonstrate that the loss of ERK2 from NPCs results in the reduction of neurons populating those cortical layers that are generated subsequent to its inactivation. We further document that the increase in cellular density is caused by reductions in cortical thickness as the total number of DAPI+ cells is equivalent between genotypes. We also demonstrate that many more astrocytes are present in the ERK2 CKO cortex. We next investigated whether the reduction in neuronal cell number resulted from enhanced apoptosis. Analysis of E17 embryos by immunostaining for cleaved caspase 3 and by TUNEL assay revealed no differences between genotypes (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These data suggest that loss of ERK2 does not provoke neuronal death. Additionally, we assessed whether incomplete neuronal differentiation might contribute to the reduction in cortical thickness. Brains from 4-month-old CKO and wild-type littermates were Golgi stained, and individual projection neurons within layers 3 and 5 of the primary motor cortex were traced and reconstructed with Neurolucida. We did not observe any differences in dendritic length, branching, volume, or surface area (supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). We also did not identify any structural changes in axonal projections.
ERK2 inactivation alters the dynamics of neurogenesis
The ERKs play critical roles in regulating cellular proliferation (Chambard et al., 2006) and neuronal differentiation of cortical NPCs (Menard et al., 2002; Barnabe-Heider and Miller, 2003; Paquin et al., 2005). To determine when the loss of ERK2 affects cell fate determination, we investigated the proliferation of NPCs by evaluation of BrdU incorporation. Pregnant dams were injected with BrdU, embryos were harvested 0.5 h after injection, and coronal sections of embryonic brains were immunostained with anti-BrdU and anti-phospho-histone H3 antibodies. During S phase, BrdU is incorporated into NPCs that are actively replicating DNA. pH3 marks mitotic cells during G2 and M phase of the cell cycle. At E12.5 (Fig. 6A), before the induction of the hGFAPcre/+ transgene, the incorporation of BrdU and the number of ventricular cells undergoing mitosis was indistinguishable between genotypes. However, when pregnant dams were injected on E14.5, the peak of the neurogenic period, and 1 d after initiation of hGFAP–cre expression, we observed significantly less BrdU incorporation and pH3 immunoreactivity in the ERK2 CKO cortex (Fig. 6B). To quantify the difference, individual embryonic cortices were dissected 30 min after injection, dissociated, plated, and immediately processed for immunostaining. We found a 44% reduction in the number of BrdU+ cells in the E14.5 ERK2 CKO cortex (CKO, 6.7%; WT, 11.87%, n = 3 for each; unpaired t test, p < 0.0001). Importantly, we observed a dramatic reduction in the number of abventricular pH3+ cells after ERK2 inactivation. These cells were identified as intermediate progenitor cells by colabeling with Tbr2, a marker of this cellular population (Hevner et al., 2006) (Fig. 6D). There was no significant genotype-related difference in the total number of mitotic pH3+ cells (CKO, 101; WT, 104.3, for each n = 3; unpaired t test, p = 0.6005) as counted in tissue sections. Similarly, we did not observe any significant difference in the number of Tbr2+ IPCs or in Pax6+ progenitors (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). However, the number of dividing IPCs (pH3+; Tbr2+) was significantly reduced (by 37%) in the ERK2 CKO cortex (CKO, 13%; WT, 20.67% per section, n = 3 animals per genotype; unpaired t test, p = 0.0192).
Figure 6.

ERK2 CKO mice display changes in the dynamics of NPC proliferation. A–C, Pregnant mice were given intraperitoneal injections of BrdU at E12.5, E14.5, and E16.5, respectively. Embryos were fixed 30 min after BrdU injection and corresponding coronal sections of ERK2 CKO and wild-type littermates were double labeled with anti-pH3+ (red) and anti-BrdU (green) antibodies followed by DAPI counterstaining (blue). Confocal images were taken at 10× and 20× (A, B; scale bar, 50 μm) and 10× and 40× (C; scale bar, 20 μm). D, E, Corresponding coronal sections of E14.5 (D) and E16.5 (E) embryos were immunostained with Tbr2 and phosphorylated histone H3 followed by counterstaining with DAPI. Scale bar, 20 μm.
In contrast to the results at E14.5, analysis of NPC proliferation at E16.5 revealed dramatically more BrdU incorporation (Fig. 6C). Quantification of dissociated cortices revealed 2.5-fold more BrdU+ cells in the CKO at E16.5 when compared with wild-type littermates (CKO, 9.41%; WT, 4.18%, n = 4 animals per genotype; unpaired t test, p = 0.005). A moderate increase in the numbers of pH3+, mitotic cells lining the ventricle (Fig. 6C,E) was also observed in the ERK2 CKO; however, no increase in abventricular pH3+ IPCs was identified (Fig. 6E). Wild-type animals have passed through the neurogenic period and exhibit few proliferating or dividing cells at this time. Together, these findings suggest that ERK2 inactivation delays NPC proliferation until E16.5 and suppresses the division of IPCs at the peak of the neurogenic period, resulting in fewer neurons populating the ERK2 CKO cortex. The data also suggest a reinitiation of progenitor proliferation during the period of normal gliogenesis in the mutant cortex, resulting in more astrocytes.
ERK2 CKO neural progenitors generate fewer neurons in vitro
To evaluate whether ERK2 inactivation affected intrinsic cell fate determination by NPCs, we performed in vitro analysis of cortical progenitor cell cultures (Menard et al., 2002; Paquin et al., 2005). Cortical progenitor cells from ERK2 CKO and wild-type littermate embryos were cultured at E14.5 and grown under proliferating conditions in Neurobasal media supplemented with FGF2 for 2 d. Importantly, we observed a significant increase (1.3-fold) in the number of uncommitted, Nestin+ NPCs (Fig. 7A1,A2,E) in the ERK2 CKO cultures compared with those of wild-type littermates. Furthermore, there were fewer immature βIII-tubulin+ (0.8-fold) and mature MAP2+ (0.7-fold) neurons in the ERK2 CKO cultures compared with those from wild-type animals (Fig. 7B,C,E). To assess whether ERK2 CKO progenitors became committed to glial fates in vitro, we counted S100β+ cells. ERK2 CKO cultures have 20% fewer S100β+ cells than wild-type cultures (Fig. 7D,E), indicating that NPCs lacking ERK2 persist in an undifferentiated state. Our finding are consistent with a previous report demonstrating that NPCs transfected with a dominant-negative MEK1-construct remain in an undifferentiated state until gliogenic signals induce astrocyte differentiation (Paquin et al., 2005).
Figure 7.

ERK2 CKO cortical progenitor cells exhibit reduced neuronal generation. A, Cortical progenitor cell cultures from ERK2 CKO and wild-type E14.5 embryos were grown in Neurobasal with FGF2 for 2 d in vitro. Cells were then fixed and immunostained. Progenitors were identified by Nestin immunoreactivity (A; n = 3). Cultures were assayed for immature neurons with βIII-tubulin (B; n = 3), mature neurons with MAP2 (C; n = 4), and astrocyte precursors with S100β (D; n = 4). E, Analysis of cellular identity in culture with FGF2. Fold difference is the comparison of average of positive cells/total cell number relative to wild-type. Student's t test, p < 0.001 for Nestin; p = 0.014 for βIII-tubulin; p = 0.0003 for MAP2; and p = 0.0018 for S100β. *p < 0.05; **p < 0.001; ***p < 0.0001.
It has been suggested that ERK1/2 activation actively suppresses gliogenesis elicited by CNTF/leukemia inhibitory factor (Bonni et al., 1997; Paquin et al., 2005). To assess whether the loss of ERK2 affects cellular fate in a gliogenic environment, we added CNTF to cultures 12 h after plating. A significant (1.4-fold) increase in vimentin+ astrocytes in the ERK2 CKO cultures was observed after treatment for either 2 or 4 d with CNTF (Fig. 8A1–A4,D). Similar results were found at both 2 and 4 d in vitro (DIV) when cultures were assessed with the mature astrocyte marker GFAP (1.7-fold at 2 DIV, 1.6-fold at 4DIV) and the immature astrocyte marker S100β (1.6-fold at 2 DIV, 1.3-fold at 4 DIV) (Fig. 8B–F). These data indicate that ERK2 normally acts to suppress gliogenesis and that this isoform alone may be sufficient for directing cell fate decisions. In summary, our in vitro analysis confirms and validates the conclusion that ERK2 participates in neurogenic cell divisions. Furthermore, loss of ERK2 does not change the differentiative potential of the NPCs, because they remain in an undifferentiated Nestin+ state until gliogenic stimuli induce astrocytic differentiation.
Figure 8.

ERK2 CKO cortical progenitors generate more astrocytes in the presence of gliogenic stimuli. Cortical progenitor cell cultures from ERK2 CKO and wild-type E14.5 embryos were grown in Neurobasal media. Twelve hours after plating, cells were treated with CNTF to induce astrocyte differentiation. Cells were fixed and immunostained with anti-vimentin (A), anti-GFAP (B), and anti-S100β (C) antibodies after 2 d in vitro (1, 2) and 4 d in vitro (3, 4). D–F, Analysis of cellular identity with CNTF treatment. Fold difference in astrocyte generation was measured by the average of marker immunopositive cells/total cell number relative to wild-type at 2 DIV and 4 DIV (n = 4 for each). Two-way ANOVA, p < 0.0001 with Bonferroni's post hoc tests, p < 0.0001 for each. **p < 0.001; ***p < 0.0001.
Mature ERK2 CKO mice display impaired associative learning
Because of the changes in cellular composition observed in the ERK2 CKO cortex and the critical roles the ERKs play in learning and memory (Selcher et al., 2001; Mazzucchelli et al., 2002), we tested whether ERK2 CKO mice showed impairments in long-term memory using two associative learning tests. Cued and contextual fear conditioning are sensitive to disruption of the MAPK signaling pathway as MAP kinase kinase (MEK) or MAPK inhibitors have been shown to attenuate both forms of memory (Walz et al., 1999, 2000; Sweatt, 2001; Shalin et al., 2004). ERK2 CKO mice exhibited significant deficits in long-term memory for both contextual and cued conditioning compared with littermate wild types (Fig. 9A,B). Additional testing was conducted to determine whether overtraining could rescue the deficits in contextual and cued conditioning; however, despite repeated training, ERK2 CKO mice continued to display significantly less freezing behavior during testing compared with littermate wild types (t(15) = 3.65 and 3.35, respectively; p < 0.05) (Fig. 9B). These data indicate a striking deficit in associative fear memory in ERK2-deficient animals. Differences in freezing behavior could not be explained by deficits in baseline sensory responses and baseline activities, because wild-type and CKO mice showed no differences in locomotor behavior (CKO, 3300 cm; WT = 2450 cm; t(15) = −1.536) or general anxiety-like behavior measured by thigmotaxis (CKO, 73.386%; WT, 77.116%; t(15) = 0.027) in the open field, in threshold to respond to a foot shock (flinch: CKO, 0.083 mA; WT, 0.087 mA; t(15) = 0.98; jump: CKO, 0.369 mA; WT, 0.389 mA; t(15) = 0.42; run: CKO, 0.141 mA; WT, 0.183 mA; t(15) = 1.27; vocalize: CKO, 0.275 mA; WT, 0.2560 mA; t(15) = −0.45) or in visual ability (CKO, 71.429%; WT, 66.6670%; t(15) = −0.19), which was measured with a visual cliff test (n = 8 for CKO, n = 10 for WT; p > 0.05 for all baseline experiments). We conclude that the selective loss of the mapk1/ERK2 gene results in a pronounced associative memory deficit, in the absence of generalized alterations in sensory responses.
Figure 9.

Male ERK2 CKO mice have deficits in associative learning. Male ERK2 CKO mice displayed deficits in cued and contextual fear conditioning. A, At 24 h after training, CKOs showed significantly less freezing behavior when tested for contextual and cued recall (t(16) = 2.27 and 2.95, respectively; p < 0.05). B, Repeated training, or “overtraining,” did not overcome the deficit seen in cued and contextual fear conditioning. CKOs showed significantly less freezing behavior in both recall tests when tested the next day (t(16) = 3.65 and 3.35, respectively; p < 0.05). *p < 0.05.
MAPK1/ERK2 deficiency in humans is associated with learning deficits
The human MAPK1 gene encoding ERK2 is positioned on the long (q) arm of chromosome 22. Chromosomal microdeletions frequently occur at 22q11.2 because of the presence of highly homologous intrachromosomal low-copy repeats (LCRs) that mediate aberrant recombination events (Emanuel et al., 2001). The most common recurrent deletion syndrome in humans involves the proximal LCRs (A–D) and leads to 22q11 Deletion/DiGeorge syndrome (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Recently, a number of unrelated patients have been identified with a 1 Mb deletion mediated by the distal LCRs D–E (Rauch et al., 1999; Saitta et al., 1999; Shaikh et al., 2007; Ben-Shachar et al., 2008). These deletions include MAPK1/ERK2, and, although they do not overlap the proximal 3 Mb deletion causing DiGeorge syndrome, they independently exhibit a similar spectrum of craniofacial abnormalities, cardiac defects, and neurodevelopmental defects. Several other genes including HIC2, YPEL1, PPIL2, UBE2L3, SDF2L1, and PPM1F occupy the D–E interval. A cluster of genes encoding the immunoglobulin λ light chain are also located in the distal part of the deletion as well; however, none of these genes have been associated with human disorders (S. C. Saitta, unpublished data). Thus far, the characteristics of eight individuals that have DiGeorge-like symptoms but which carry atypical distal deletions have been identified (Rauch et al., 1999; Saitta et al., 1999; Shaikh et al., 2007; Ben-Shachar et al., 2008). We have analyzed two patients with identical distal 1 Mb deletions. Patient 1 is a male with microcephaly (head circumference consistently <3% for age and sex-matched norms measured over 4 years) and neurocognitive deficits (Saitta et al., 1999) that include delays in fine motor function, expressive speech development, and in complex language comprehension. Patient 2 is a recently diagnosed female with microcephaly (head circumference <3% for age and sex-matched norms), a history of delays in expressive speech development and decreased academic performance overall. Analysis of cellular levels of ERK2 in lymphoblasts obtained from these patients with the distal deletion exhibit decreased levels of ERK2 expression relative to either normal individuals (n = 4) or those with a typical 3 Mb DiGeorge deletion (n = 1). Densitometric analysis of Western blots revealed that ERK2 expression in the distal microdeletion patients was 0.60876 of normal levels (p = 0.0026). In contrast, levels of CRKL, encoded by a gene located within the 3 Mb classical deletion interval of DiGeorge syndrome, were normal in patients with the distal 22q11 deletions. These data demonstrate that patients with chromosomal microdeletions of distal chromosome 22q have a phenotype that includes microcephaly and neurodevelopmental and neurocognitive deficits but is not attributable to genes in the DiGeorge interval. These children, instead, have a small microdeletion that includes the MAPK1/ERK2 gene, show reduced ERK2 levels, and display cognitive deficits. These findings are in line with our data from the murine model generated in this study in which cortical hypoplasia and cognitive deficits in associative learning are found with genetic deletion of mapk1/ERK2.
Discussion
We have identified a specific role for ERK2 in the development of the cerebral cortex and shown that its deletion in the developing telencephalon is associated with the loss of higher-order functionality in the mature brain. These findings provide concrete evidence for isoform-specific actions of the ERKs and follow from our previous studies demonstrating that mice in which the mapk3/ERK1 gene was knocked out did not have an overt phenotype and exhibited only modest behavioral changes (Pages et al., 1999; Selcher et al., 2001; Mazzucchelli et al., 2002).
The developing cortex is exquisitely sensitive to perturbations of the tightly regulated patterns of cellular proliferation and differentiation. The conditional inactivation of ERK2 within neural progenitors at the beginning of the neurogenic period resulted in fewer neurons populating the cortex. This effect resulted from the dramatic reduction in both the number of progenitors proliferating within the ventricular zone and the number of mitotic abventricular intermediate progenitor cells at E14.5. Importantly, the loss of ERK2 appears only to delay initiation or stall the progression of neural progenitors through the cell cycle during the neurogenic period, because these cells neither die nor migrate prematurely. It is of importance in this regard that isolated NPCs maintain their ability to undergo properly timed mitotic divisions, generating successive and appropriately specified lineages of neurons (Shen et al., 2006). Our findings demonstrate that loss of ERK2 does not disrupt the lineage specification of neurons populating any of the cortical lamina. These findings are consistent with in vitro studies that have shown that ERK2 regulates initiation and progression through the G1 phase of the cell cycle (Liu et al., 2004; Chambard et al., 2006; Meloche and Pouyssegur, 2007). The suppression of NPC proliferation as a consequence of ERK2 inactivation is transient, because we observed a robust increase in S phase cells 2 d later (E16.5). The effects observed in the ERK2 CKO may reflect specific actions of this isoform or alternatively might result from a net reduction in overall ERK activity. Of particular relevance, Brambilla and colleagues have argued that ERK1 acts to functionally antagonize the actions of ERK2 in stimulating cellular proliferation, thus slowing progression of the cell cycle (Vantaggiato et al., 2006). However, Lenormand and colleagues have shown that each isoform is indiscriminately activated and contend that the overall abundance of ERK1 and ERK2 dictates biological outcomes including cellular proliferation. Interestingly, the same report demonstrates that ERK2 is 13 times more abundant than ERK1 in the superficial cortex of nude mice (Lefloch et al., 2008). Our findings are consistent with the hypothesis that net ERK1/2 activity governs cellular behaviors. The striking increase in the levels of activated forms of ERK1 in the cortex of our ERK2 CKO mice at E16.5, after the near complete loss of ERK2 protein, suggests that the increase in ERK1 activity might reflect a compensatory response to the loss of ERK2, and this is correlated with a resumption of NPC proliferation. An analogous compensatory effect on ERK2 activation was reported in a number of tissues in the ERK1 knock-out mice, including the brain (Pages et al., 1999; Selcher et al., 2001; Mazzucchelli et al., 2002). These findings support the possibility that the biological actions of the ERKs are dictated by their spatial and temporal expression rather than the specific activity of one isoform or the other. However, there is compelling evidence that isoform-specific actions of the ERKs may arise from their selective association with molecular scaffolds that direct their interaction with protein substrates and with phosphatases (Pouyssegur et al., 2002). These issues are under active investigation.
The change in cellular generation by NPCs during mapk1/ERK2 inactivation confirms and expands the findings from experimental manipulation of other components of the MAPK signaling cascade. Inhibition of the upstream activator of the ERKs, MEK1, causes NPCs to remain in the SVZ/VZ in an undifferentiated state, blocking neurogenesis (Menard et al., 2002; Barnabe-Heider and Miller, 2003; Paquin et al., 2005). Similarly, mutation or inactivation of scaffolding proteins that link receptor tyrosine kinases to ERK activation have analogous effects on cortical progenitors. Mutations in the ERK cascade scaffolding protein FRS2 result in animals that exhibit nearly 30% reductions in cortical thickness and fewer cortical neurons (Hadari et al., 1998; Yamamoto et al., 2005). This study confirmed that loss of cortical neurons was attributable to reduced numbers of proliferating IPCs and was a consequence of dramatically reduced levels of ERK activation. Two independent studies have recently found that deletion or mutation of SHP-2, an upstream element of the ERK cascade, in cortical progenitors leads to the inhibition of neuronal differentiation through ERK-dependent mechanisms (Gauthier et al., 2007; Ke et al., 2007). It has been proposed recently that IPCs are the only progenitors to undergo symmetric neurogenic divisions, contributing neurons to all cortical lamina throughout neurogenesis (Pontious et al., 2008). Our findings, which document a decrease in neurons occupying each cortical lamina and a specific reduction in mitotic IPCs, indicate that ERK2 may play a critical role in regulation of IPC division. These data warrant additional investigation into the precise roles that ERK2 plays in IPC cell cycle progression and terminal differentiation.
The conditional knock-out of ERK2 also resulted in more astrocytes populating the neonatal and mature cortex. Bonni and colleagues originally demonstrated that ERK activation blocked the differentiation of NPCs into astrocytes (Bonni et al., 1997), and this conclusion has been validated both in vitro (Menard et al., 2002) and in animal models (Paquin et al., 2005). Our findings are therefore consistent with the postulated action of the ERK cascade in suppressing glial fate and promoting neurogenesis in the developing cortex (Menard et al., 2002; Barnabe-Heider and Miller, 2003; Paquin et al., 2005; Gauthier et al., 2007).
The ERKs are known to play critical roles in learning and memory, and the absence of a robust behavioral phenotype in ERK1 null mice has suggested that ERK2 is the principal isoform responsible for learning behaviors that are dependent on the neocortex and associated structures (Selcher et al., 2001; Mazzucchelli et al., 2002). Fear conditioning requires ERK-dependent interactions within and between the hippocampus, cortex, and amygdala. We demonstrate that the loss of ERK2 within the telencephalon resulted in significant impairment on cued and contextual fear conditioning tests. Our findings are similar to effects of pharmacological inhibition of the ERKs (Sweatt, 2004) or expression of dominant-negative MEK on fear conditioning (Shalin et al., 2004). Thus, the present findings provide direct evidence that ERK2 is the principal isoform required for learning in this behavioral task; however, it is unclear whether the deficit is attributable to developmentally related structural changes of the cortex or the loss of ERK2 in mature neurons. It is important to note that glutamatergic or electrical stimulation of neurons selectively stimulates ERK2 activation (English and Sweatt, 1996; Mazzucchelli et al., 2002). Our results compliment a recent report by Satoh et al. (2007) describing the behavior of an ERK2 hypomorphic mouse that exhibits deficits in long-term memory in contextual and cued fear conditioning paradigms.
We have identified individuals with deletion of a single MAPK1/ERK2 allele and exhibit decreased ERK2 protein levels. These patients' clinical presentation include congenital anomalies and neurocognitive deficits reflected by developmental delays, microcephaly, and learning disabilities (Shaikh et al., 2007). Our findings in mutant mice suggest that these abnormalities may arise from perturbation of neural developmental as well as impaired ERK2 signaling required for memory and cognition. We postulate that the cognitive deficits observed in the patients and in the mice result from a perturbation in signal transduction through the ERK signaling cascade. We further suggest that these findings may have broader significance given the recent recognition that genetic mutations within either upstream elements (Bentires-Alj et al., 2006) or downstream targets (Weeber and Sweatt, 2002) of the ERK cascade are associated with developmental delay or mental retardation.
Footnotes
This work was supported by National Science Foundation Grant IBN97-23147, National Institutes of Health Grants NS032779, MH57014, NS13546, NS057098, and NS020591, and National Alliance for Research on Schizophrenia and Depression. I.S.S. was supported by Kirschstein National Research Service Award F31-MH074241. S.C.S. was supported by National Institutes of Health Grant HL04487. We thank Natalie Cherosky, Forrest Gulden, Kevin Moran, Caroline Garber, Joanna Pucilowska, Rockell Pankey, and Maryanne Pendergast for technical assistance and Dr. Sara Shalin for her support on this project. We thank Dr. Freda Miller, Andree Gauthier, and Annie Paquin for their assistance in generating the cortical cultures. We thank Dr. Albee Messing for providing the hGFAP-cre mice, Dr. Robert Hevner for the Tbr1 antibody, and Dr. Beverly Emanuel for samples from patients with DiGeorge syndrome. We are grateful to Drs. Urs Rutishauser and Terry Magnuson for their critical roles in the initiation of this work and to Dr. Carolyn Schanen for her assistance and advice.
References
- Aouadi M, Binetruy B, Caron L, Le Marchand-Brustel Y, Bost F. Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie. 2006;88:1091–1098. doi: 10.1016/j.biochi.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Barnabe-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, Patel MS, Hummel M, Amato S, Tartaglia N, Berg J, Sutton VR, Lalani SR, Chinault AC, Cheung SW, Lupski JR, Patel A. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet. 2008;82:214–221. doi: 10.1016/j.ajhg.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–285. doi: 10.1038/nm0306-283. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2006;1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci. 2007;8:438–450. doi: 10.1038/nrn2097. [DOI] [PubMed] [Google Scholar]
- Dono R. Fibroblast growth factors as regulators of central nervous system development and function. Am J Physiol Regul Integr Comp Physiol. 2003;284:R867–R881. doi: 10.1152/ajpregu.00533.2002. [DOI] [PubMed] [Google Scholar]
- Emanuel BS, McDonald-McGinn D, Saitta SC, Zackai EH. The 22q11.2 deletion syndrome. Adv Pediatr. 2001;48:39–73. [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron. 2007;54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadari YR, Kouhara H, Lax I, Schlessinger J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol. 1998;18:3966–3973. doi: 10.1128/mcb.18.7.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, Klein R, Curran T, Ranscht B, Feng GS. Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol. 2007;27:6706–6717. doi: 10.1128/MCB.01225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefloch R, Pouyssegur J, Lenormand P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2008;28:511–527. doi: 10.1128/MCB.00800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yan S, Zhou T, Terada Y, Erikson RL. The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene. 2004;23:763–776. doi: 10.1038/sj.onc.1207188. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Gotz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci. 2004;24:6057–6069. doi: 10.1523/JNEUROSCI.1140-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Paquin A, Barnabe-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. Ed 2. San Diego: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Pontious A, Kowalczyk T, Englund C, Hevner RF. Role of intermediate progenitor cells in cerebral cortex development. Dev Neurosci. 2008;30:24–32. doi: 10.1159/000109848. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- Rauch A, Pfeiffer RA, Leipold G, Singer H, Tigges M, Hofbeck M. A novel 22q11.2 microdeletion in DiGeorge syndrome. Am J Hum Genet. 1999;64:659–666. doi: 10.1086/302235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43:833–842. doi: 10.1136/jmg.2006.042796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Harkins PC, Zhang J, Baer R, Haycock JW, Cobb MH, Goldsmith EJ. Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry. 1996;35:5641–5646. doi: 10.1021/bi952723e. [DOI] [PubMed] [Google Scholar]
- Saitta SC, McGrath JM, Mensch H, Shaikh TH, Zackai EH, Emanuel BS. A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am J Hum Genet. 1999;65:562–566. doi: 10.1086/302514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, O'Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC. Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Zirrgiebel U, Honsa KJ, Julien JP, Miller FD, Kaplan DR, Sweatt JD. Neuronal MEK is important for normal fear conditioning in mice. J Neurosci Res. 2004;75:760–770. doi: 10.1002/jnr.20052. [DOI] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, Temple S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci. 2006;9:743–751. doi: 10.1038/nn1694. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Thomson RE, Pellicano F, Iwata T. Fibroblast growth factor receptor 3 kinase domain mutation increases cortical progenitor proliferation via mitogen-activated protein kinase activation. J Neurochem. 2007;100:1565–1578. doi: 10.1111/j.1471-4159.2006.04285.x. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz R, Roesler R, Barros DM, de Souza MM, Rodrigues C, Sant'Anna MK, Quevedo J, Choi HK, Neto WP, DeDavid e Silva TL, Medina JH, Izquierdo I. Effects of post-training infusions of a mitogen-activated protein kinase kinase inhibitor into the hippocampus or entorhinal cortex on short- and long-term retention of inhibitory avoidance. Behav Pharmacol. 1999;10:723–730. doi: 10.1097/00008877-199912000-00003. [DOI] [PubMed] [Google Scholar]
- Walz R, Roesler R, Quevedo J, Sant'Anna MK, Madruga M, Rodrigues C, Gottfried C, Medina JH, Izquierdo I. Time-dependent impairment of inhibitory avoidance retention in rats by posttraining infusion of a mitogen-activated protein kinase kinase inhibitor into cortical and limbic structures. Neurobiol Learn Mem. 2000;73:11–20. doi: 10.1006/nlme.1999.3913. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Sweatt JD. Molecular neurobiology of human cognition. Neuron. 2002;33:845–848. doi: 10.1016/s0896-6273(02)00634-7. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yoshino I, Shimazaki T, Murohashi M, Hevner RF, Lax I, Okano H, Shibuya M, Schlessinger J, Gotoh N. Essential role of Shp2-binding sites on FRS2alpha for corticogenesis and for FGF2-dependent proliferation of neural progenitor cells. Proc Natl Acad Sci U S A. 2005;102:15983–15988. doi: 10.1073/pnas.0507961102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Nowakowski RS, Vaccarino FM. Fibroblast growth factor 2 is required for maintaining the neural stem cell pool in the mouse brain subventricular zone. Dev Neurosci. 2004;26:181–196. doi: 10.1159/000082136. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]