

Abstract
Loss of function mutations in granulin (GRN) are linked to two distinct neurological disorders, frontotemporal lobar degeneration (FTLD) and neuronal ceroid lipofuscinosis (NCL). It is so far unknown how a complete loss of GRN in NCL and partial loss of GRN in FTLD can result in such distinct diseases. In zebrafish, there are two GRN homologues, Granulin A (Grna) and Granulin B (Grnb). We have generated stable Grna and Grnb loss of function zebrafish mutants by zinc finger nuclease mediated genome editing. Surprisingly, the grna and grnb single and double mutants display neither spinal motor neuron axonopathies nor a reduced number of myogenic progenitor cells as previously reported for Grna and Grnb knock down embryos. Additionally, grna−/−;grnb−/− double mutants have no obvious FTLD- and NCL-related biochemical and neuropathological phenotypes. Taken together, the Grna and Grnb single and double knock out zebrafish lack any obvious morphological, pathological and biochemical phenotypes. Loss of zebrafish Grna and Grnb might therefore either be fully compensated or only become symptomatic upon additional challenge.
Introduction
Granulin (GRN) is a pleiotropic growth factor, which plays a role in wound healing, cancer, and inflammation [1]. Heterozygous loss of function mutations in GRN are linked to frontotemporal lobar degeneration (FTLD-TDP/GRN) [2, 3]. Furthermore, two patients with neuronal ceroid lipofuscinosis (NCL) have been reported to be homozygous for loss of function mutations in GRN (NCL/GRN) [4]. Neuropathologically, FTLD-TDP/GRN patients present with extensive micro- and astrogliosis as well as TAR DNA binding protein 43 (TDP-43) and ubiquitin-positive intracellular inclusions [5–7]. Biochemical studies revealed that lysosomal proteins such as Cathepsin D (CTSD) are increased in brain samples from FTLD-TDP/GRN patients [5] suggesting lysosomal dysfunction upon loss of GRN. Moreover, skin biopsies of NCL/GRN patients revealed the typical fingerprint profile of lipofuscin aggregates [4]. Grn knock out (KO) mouse models are viable and fertile [1]. Neuropathological examinations of KO mice show also a pronounced micro- and astrogliosis, accumulation of ubiquinated proteins and increased lipofuscinosis [8–13]. Biochemically, Grn −/− but not Grn +/− mice displayed elevated levels of Ctsd [5, 8, 11], recapitulating features of lysosomal dysfunction. Despite intensive research in the past years, the exact function of GRN and GRN-associated signalling pathways as well as the underlying pathomechanisms in FTLD-TDP/GRN and NCL/GRN are still elusive. We used zebrafish as a less complex vertebrate model organism with the potential for high throughput drug screening to investigate GRN function in health and disease.
In zebrafish there are two orthologues of GRN, granulin A (grna), granulin B (grnb) (Fig. 1A). Additionally, zebrafish have two shorter paralogues to grna and grnb with only one and a half granulin domains referred to as granulin 1 (grn1), and granulin 2 (grn2) [14] (Fig. 1A). Since FTLD-TDP/GRN patients have less functional GRN [15–17] and NCL/GRN patients have no GRN [4] loss of function models are suitable approaches to mimic aspects of FTLD-TDP/GRN and NCL/GRN. Grna and Grnb knock down (KD) in zebrafish was previously reported to result in spinal motor neuron (SpMN) axonopathies [18, 19] and a reduced number of myogenic progenitor cells (MPCs) [20]. To obtain stable mutants without phenotypic variability due to the KD procedure, we generated Grna and Grnb KO mutants by genome editing using the zinc finger nuclease (ZFN) technology. Here, we report a genetic, phenotypic and biochemical analysis of Grna and Grnb single and double KO zebrafish.
Fig 1. Generation of Grna and Grnb KOs using ZFNs.

A: Schematic illustration of human GRN and zebrafish Granulins. Human GRN has 7 ½ granulin domains, while 12 granulin domains are found in Grna, 9 in Grnb, and 1 ½ in Grn1 and Grn2. Grey: signal peptide. Black numbers: amino acids. Darker colour and white letters/numbers: granulin domains. B-C: Localisation of ZFN target sequences in grna and grnb and predicted protein sequence of selected alleles. The genomic structure of grna and grnb is depicted. ZFNs targeting grna and grnb are located in the first and fourth coding exon, respectively. ZFN-induced genomic lesions in grna can be detected with the restriction enzyme (RE) Eco91I and in grnb with the RE XcmI. Grey boxes: untranslated region (UTR). Coloured boxes: coding region. Light blue: ZFN binding sites in grna. Light red: ZFN binding sites in grnb. Green lines: binding sites of the RE. Dashed green line: cut site of the RE. Protein sequences of wildtype (wt) grna and 4 grna mutation alleles as well as wt grnb and 3 grnb mutation alleles are shown. *: Stop. D-E: Grna and Grnb protein is lost in all mutants. D: Grna signal is lost in all adult kidney samples from grna−/− mutants, whereas a signal is present in wt. A Calnexin blot serves as a loading control. E: The Grnb signal observed in wt is lost in all 1.5dpf samples from grnb −/− mutants. Injection of grnb mRNA leads to an increase in signal. The loading control α-tubulin is present in all samples.
Results
Generation and characterization of Grna and Grnb KO zebrafish
Human and mouse genomes have one GRN gene in contrast to the zebrafish genome, which harbours two genes with high homology to mammalian GRN (grna and grnb) and two short granulins (grn1 and grn2) (Fig. 1A). Grna and Grnb are similar to each other and most similar to mammalian GRN, but are expressed in distinct tissues. At 24 hours post fertilization (hpf) grna is most prominently expressed in the intermediate cell mass where precursors of blood and immune cells reside, consistent with the mammalian expression pattern [21], whereas grnb is predominantly expressed in various regions of the brain [14]. Grn1 and Grn2 are very short with only one and a half granulin domains (Fig. 1A) and might have a similar function as the proteolytically processed GRN peptides in mammals. We therefore choose Grna and Grnb for the GRN loss of function analysis in zebrafish. Targeted genome editing was performed using ZFNs. The grna ZFNs target the first coding exon of grna (Fig. 1B), and the grnb ZFNs the fourth coding exon of grnb (Fig. 1C). ZFN mRNAs were injected at one-cell stage. The embryos were raised to adulthood (P0 generation) and then outcrossed to wildtype zebrafish. The resulting F1 generation was screened for successful germline transmission of induced mutations. The efficiency of ZFNs to introduce mutations in the germline was 75% (27/36 P0 fish) for grna and 62% (29/47 P0 fish) for grnb. The adult F1 generation was further analysed for ZFN-induced genomic lesions by sequencing.
Offspring from 13 grna and 8 grnb P0 founder fish were analysed for induced mutations and we detected up to 5 and 3 distinct mutations originating from a single founder fish, respectively. In total 13 different mutations (deletions, insertion and indels) were isolated for grna of which 6 result in a frameshift in the coding sequence. For grnb 15 different mutations were isolated of which 11 lead to a frameshift in the coding sequence. 4 grna and 3 grnb frameshift mutations that result in a premature translation termination codon were selected for further characterization (Fig. 1B, C). mRNAs containing a premature translation termination codon are known subjects to nonsense-mediated mRNA decay (NMD), one of the cells endogenous quality control mechanisms that prevents the synthesis of truncated, non-functional proteins [22]. In all grna −/− mutants analysed this results in approx. 50% reduction of grna mRNAs and in grnb −/− mutants the grnb mRNA levels were reduced by approx. 90% (S1 Fig.). The differences in residual mRNA levels might be attributed to the presence of a grna antisense RNA [14], which potentially prevents the efficient degradation of mutated grna by NMD. We generated monoclonal antibodies specifically detecting zebrafish Grna or Grnb protein to examine if the grna −/− and grnb −/− mutants are deficient in Grna and Grnb, respectively. Neither Grna nor Grnb was detectable in grna −/− and grnb −/− mutants (Fig. 1D, E), indicating that no protein is made in homozygous mutants.
Analysis of Grna and Grnb KOs for morphological phenotypes
Morphologically, the grna −/− and grnb −/− as well as the grna −/−;grnb −/− double mutant embryos and adults are indistinguishable from wildtype embryos (S2 Fig.). Incrosses of grna +/−, grnb +/,- and grna +/−;grnb +/− adults produce offspring of all genotypes in an expected Mendelian ratio. Also offspring from incrosses of grna −/−, grnb −/−, and grna −/−;grnb −/− adults had normal amounts of fertilized embryos and no morphological defects (data not shown).
Since GRN is a growth factor, we asked if the development is delayed in single and double homozygous grna and grnb mutant embryos. The developmental stage of the mutants was assessed by counting somites at a time point when the controls embryos reached the 13–15 somite stage. No developmental delay was detected in grna −/−, grnb −/−, and grna −/−;grnb −/− mutants when compared to wildtype embryos (S3 Fig.). As there are four Grn in zebrafish, we speculated, that the paralogues could compensate for each other by upregulation. Therefore, the grnb and grna mRNA expression levels and protein levels were determined in Grna and Grnb KO larvae, respectively. Additionally, the grn1/grn2 mRNA expression levels were analysed in grna −/−;grnb −/− larvae. No compensatory upregulation of the grna or grnb paralogue on mRNA and protein level was detected in grna −/− or grnb −/− mutants, respectively (S4 Fig. A-B,D-E). Moreover, the grna −/−;grnb −/− mutants did not show any transcriptional upregulation of grn1/grn2 (S4 Fig. 4C).
In summary, homozygous Grna and Grnb single and double KO mutants were successfully generated. They have wildtype morphology, are viable and fertile and are not developmentally delayed. Moreover, lack of Grna or Grnb in zebrafish does not result in compensatory transcriptional or translational upregulation of Grnb or Grna, respectively.
Analysis of SpMN in Grna and Grnb KOs
SpMN axons from Grna and Grnb KD embryos using MOs were reported to be truncated and hyperbranched [18, 19, 23]. We therefore stained SpMN axons of single and double Grna and Grnb KO embryos at 28hpf with the znp1 antibody. We previously experienced that variations between different clutches can result in slight differences in the SpMN axon length. Therefore, we only compared embryos from the same clutch to avoid inter-clutch variation and analysed the five SpMN axons anterior to the end of the yolk extension (Fig. 2A) [24, 25]. First, wildtype, heterozygous and homozygous embryos from matings of single and double heterozygous Grna and Grnb KOs were compared. Extended branching was not observed in any of the genotypes analysed (Fig. 2B). Measurements of the SpMN axon length, which is defined here as the length of the SpMN axon from the exit point of the spinal cord to the tip of the growth cone, revealed that the length is not significantly altered in any genotype analysed (Fig. 2C-E). When translation inhibition MOs are used for KD experiments maternal and zygotic mRNA is depleted whereas in KO animals from heterozygous females maternal mRNA could potentially preclude a phenotype. To exclude that maternal mRNA precludes SpMN axon outgrowth phenotypes, siblings from matings of homozygous females and heterozygous males were analysed. Also in maternal zygotic KOs of Grna, Grnb, or both, we did not detect any extended branching or reduced SpMN axon outgrowth (S5 Fig.).
Fig 2. No SpMN axonopathy in Grna and Grnb single and double KOs.

A: Schematic illustration of a zebrafish embryo at 28hpf (lateral view) and a detail of the region above the end of the yolk extension imaged for the analysis of SpMN axons (lateral view). B: In Grna and Grnb single and double KOs the SpMN axons show no extended branching. Whole-mount immunofluorescence staining of 28hpf embryos with znp1 antibody. The 5 SpMN axons above the end of the yolk extension are shown. Images taken by spinning disk confocal microscopy. Anterior to the left. Lateral view. Orthogonal projections. Scale bar: 100μm. C-E: Quantification of the SpMN axon length in homozygous and heterozygous Grna and Grnb single and double KOs and wildtype siblings. The SpMN axon length of the 5 SpMN axons (1–5) above the end of the yolk extension is measured from the exit point of the spinal cord to the tip of the growth cone. C: Homozygous and heterozygous Grna KOs and wildtype siblings. n = 30. D: Homozygous and heterozygous Grnb KOs and wildtype siblings. n = 30. E: Homozygous and heterozygous Grna and Grnb KOs and wildtype siblings. n = 25. S.E.M. Two-way ANOVA. Bonferroni post-test. All non-significant (n.s.).
In summary, SpMN axons and their length are indistinguishable in single and double Grna and Grnb stable genetic KO, in contrast to previously published reports from Grna and Grnb KD experiments [18, 19, 23].
Analysis of MPCs in Grna and Grnb KOs
A reduced number of MPCs was reported in Grna KD embryos [20]. A similar reduction, although less strong, was also reported for Grnb KD [20]. We performed immunofluorescence staining with the Pax7 antibody in wildtype and grna −/−;grnb −/− embryos at 24hpf and quantified the number of Pax7-positive cells in the four somites anterior to the end of the yolk extension (Fig. 3A). At this stage of development Pax7 labels the nuclei of MPCs, which are located at the surface of the trunk musculature [26] but also some xanthophores [27]. Xanthophores can be distinguished from Pax7-positive MPCs by their elongated shape, a more intense staining as well as the dorsal localization [27]. Images of Pax7-stained wildtype and grna −/−;grnb −/− embryos do not reveal any differences. In both, wildtype and grna −/−;grnb −/− embryos, nuclei are stained with the Pax7 antibody in different intensities and a few of them show an elongated shape (Fig. 3B). Quantification of the number of Pax7-positive nuclei in the most lateral part of the four somites anterior to the end of the yolk extension, independent of their staining intensity, revealed that the number of Pax7-positive cells is not significantly decreased in all four somites analysed of grna −/−;grnb −/− mutants when compared to wildtype embryos (Fig. 3C). Taken together, there is no significant reduction in Pax7-positive MPCs in homozygous Grna;Grnb KO embryos.
Fig 3. The number of MPCs is equal.
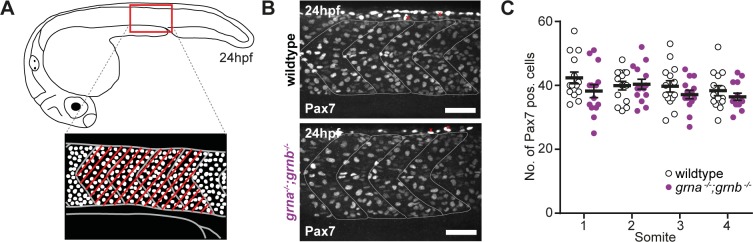
A: Schematic illustration of a zebrafish embryo at 24hpf (lateral view) and a detail of the region above the end of the yolk extension imaged for the analysis of the MPCs (lateral view). The four somites, which were considered for the quantification of MPCs are marked with red lines. B: Immunofluorescence staining with Pax7 at 24hpf in grna −/−;grnb −/− mutants and wildtype embryos. In each image two xanthophores are exemplarily marked with a red x. Images taken by spinning disk confocal microscopy. Anterior to the left. Lateral view. Orthogonal projections. Scale bar: 50μm. C: Quantification of Pax7-positive cells in the 4 somites (1–4) above the end of the yolk extension in grna −/−;grnb −/− mutants and wildtype embryos. Only Pax7-positive cells at the surface of the somites were counted. n = 15. S.E.M. Mann-Whitney test (two-tailed). All n.s. S1: p = 0.0854. S2: p = 0.7977. S3: p = 0.3489. S4: p = 0.2337.
Analysis of Grna and Grnb KOs for disease-related pathology
FTLD-TDP/GRN patients [28–30] and homozygous Grn KO mice [8–13] develop micro- and astrogliosis. Moreover, grna is expressed in microglia and has even been used as a microglia marker [31, 32]. We therefore asked, if grna −/−;grnb −/− mutants develop brain gliosis. To estimate the number of microglia in the larval brain at 3dpf we stained with neutral red, a dye that accumulates as red fluorescence in acidic vesicles of microglia and macrophages [33], and quantified the number of neutral red positive particles in the brain (Fig. 4A). This analysis did not reveal any differences in the amount of neutral red particles in the brain. We conclude that the number of microglia is not increased in the brain of grna −/−;grnb −/− mutants when compared to wildtype at 3dpf (Fig. 4B). Microglia in the larval brain are highly motile and phagocytic and mature to ramified microglia with small cell bodies from 5dpf onward [34]. Therefore, it is not feasible to categorize microglia in the larval zebrafish brain in active and resting microglia. A time lapse analysis of amoeboid microglia, using a transgenic line that labels a subset of microglia, revealed that the distance moved and the persistence of processes is identical in grna −/−;grnb −/− and wildtype larvae at 3dpf (Fig. 4C-E) indicating that grna −/−;grnb −/− mutants seem to lack a morphological microglial phenotype at larval stages.
Fig 4. No microgliosis and neurodegeneration in grna −/−;grnb −/− mutants.

A: Schematic illustration of a zebrafish larvae at 3dpf (lateral view) and a detail of the region (red line), dorsal view, imaged for the analysis of neutral red positive particles (B). The dashed red line marks the area that was imaged in the time lapse recordings of microglia (C). B: The number of neutral red positive particle in the region illustrated in A (Z-stack) is unchanged in wildtype and grna −/−;grnb −/− mutants. n = 15. S.E.M. Mann-Whitney test (two-tailed). p = 0.2884. C-E: Microglia in Tg(apoeb:lynEGFP) grna −/−;grnb −/− mutants and wildtype larvae at 3dpf are indistinguishable. C: Still images of the time lapse recordings in the optic tectum recorded by spinning disk confocal microscopy. Two microglia cells marked in each genotype by a white and yellow arrow. Dorsal view. Anterior to the left. n = 3. Scale bar: 50μm. Recording time: 60min. 1frame/min. D: The distance microglia move within one hour in grna −/−;grnb −/− mutants and wildtype larvae. Quantification of n = 3x5 randomly selected microglia from the time lapse recordings shown in C. S.E.M. Mann-Whitney test (two-tailed). p = 0.0671. E: Processes in the grna −/−;grnb −/− mutants and wildtype larvae persist for same durations. Quantification of n = 3x5 randomly selected processes from the time lapse recordings shown in C. S.E.M. Mann-Whitney test (two-tailed). p = 0.8296. F: Schematic illustration of a zebrafish larvae at 5dpf (lateral view) and a detail of the region, dorsal view, imaged for the analysis of acridine orange (AO) positive cells. G: The number of acridine orange positive cells in the region illustrated in C (Z-stack) is unchanged in wildtype and grna −/−;grnb −/− mutants. n = 15. S.E.M. Mann-Whitney test (two-tailed). p = 0.69.
Smith and colleagues linked GRN loss of function to NCL [4]. Furthermore, Grn KO mice develop NCL-related pathological and biochemical alterations [5, 8, 10, 11, 35]. We therefore analysed the grna −/−;grnb −/− mutants for NCL-related behavioural, pathological, and biochemical alterations. Due to massive neurodegeneration one of the clinical symptoms described in the NCL/GRN patients is epilepsy [4]. We first examined if cell death is increased in the larval brain of grna −/−;grnb −/− mutants at 5dpf by acridine orange staining, a vital dye that is frequently used in zebrafish to estimate the amount of cell death [25]. The number of acridine orange positive particles was quantified in the area of the larval brain marked by a square in the illustration (Fig. 4F). This quantification revealed that there is no increase in acridine orange positive particles in the grna −/−;grnb −/− mutants when compared to age-matched wildtype embryos (Fig. 4G). Despite the lack of neurodegeneration in the grna −/−;grnb −/− mutants, we tracked the swim path in grna −/−;grnb −/− mutants, potentially revealing seizure-like swim behaviour, which has been described in a CLN2-NCL disease model zebrafish [36]. We analysed the swimming behaviour of 5dpf grna −/−;grnb −/− mutants and age matched wildtype larvae for 5min after an adaptation period of 30min in the recording device and plotted the swim path. Moreover, we extracted the total distance moved, the mean velocity and the percentage of time spent for movements with a velocity above 2mm/s from the data sets acquired. Seizure-like swim behaviour in zebrafish has been described as quick, circling spinning [36]. As a positive control we applied pentylenetetrazole (PTZ), a drug that was previously demonstrated to induce seizures in zebrafish [37]. In contrast to wildtype siblings and grna −/−;grnb −/− mutants, the PTZ-treated larvae are much more active, which is reflected in the total distance moved (Fig. 5A,B). The mean velocity as well as the percentage of time spent for movements faster than 2mm/s is comparable in wildtype and grna −/−;grnb −/− mutants but significantly increased in the PTZ-treated wildtype larvae (Fig. 5C,D). This data indicate that swimming of 5dpf grna −/−;grnb −/− mutants is very similar to that of wildtype larvae and devoid of seizure-like behaviour as seen upon PTZ treatment.
Fig 5. grna −/−;grnb −/− mutants swim like wildtype.

A: The swim path of wildtype, grna −/−;grnb −/− mutants, DMSO-treated, and PTZ-treated larvae is shown. PTZ treatment was used as a positive control. 5dpf. Movements < 2mm/s: black lines. Movements 2–6mm/s: green lines. Movements > 6mm/s: red lines. Recording time: 5min. B: The total distance moved within 5min in wildtype, grna −/−;grnb −/− mutants, DMSO-treated, and PTZ-treated larvae is shown. Wt-grna −/−;grnb −/−: p = 0.2386. Wt-DMSO: p = 0.0534. Wt-PTZ: ***p = 0.0002. DMSO-PTZ: **p = 0.004. C: A graph of the mean velocity of wildtype, grna −/−;grnb −/− mutants, DMSO-treated, and PTZ-treated larvae is shown. Time frame: 5min. Wt-grna −/−;grnb −/−: p = 0.5657. Wt-DMSO: p = 0.8081. Wt-PTZ: *p = 0.0137. DMSO-PTZ: **p = 0.0014. D: Percentage of time spent for movements with a velocity above 2mm/s in wildtype, grna −/−;grnb −/− mutants, DMSO-treated, and PTZ-treated larvae is plotted. Time frame: 5min. Wt-grna −/−;grnb −/−: p = 0.2585. Wt-DMSO: p = 0.0668. Wt-PTZ: **p = 0.0016. DMSO-PTZ: **p = 0.0037. B-D: n = 18. S.E.M. Mann-Whitney test (two-tailed).
To address, if the lysosomal function is altered as in NCL patients lacking GRN, we determined the expression of Ctsd on the transcriptional and translational level. Grn KO mice have elevated Ctsd mRNA expression and Ctsd protein levels [5, 11, 38]. Moreover, FTLD-TDP/GRN patients have increased CTSD levels in the frontal cortex [5]. In contrast to mice and human, the ctsd mRNA expression levels were unchanged in samples derived from 5dpf grna −/−;grnb −/− mutants and adult grna −/−;grnb −/− brains of up to 22-month-old zebrafish, when compared to the corresponding wildtype controls (Fig. 6A). Ctsd immuno-blots of 5dpf larvae and adult brain samples showed no difference on the Ctsd protein level between grna −/−;grnb −/− mutants and age-matched wildtype controls (Fig. 6B,C).
Fig 6. No disease-related biochemical alterations in grna −/−;grnb −/− mutants.

A-C: ctsd mRNA expression and Ctsd protein levels in grna −/−;grnb −/− mutants. A: ctsd mRNA expression at 5dpf and in 7mpf as well as 22mpf brain samples derived from grna −/−;grnb −/− mutants compared to wildtype controls. 5dpf: p = 1.0. 7mpf: p = 0.4. 22mpf: p = 0.7. B: Western blots showing Ctsd and α-tubulin in 5dpf whole lysis samples as well as 7mpf and 22mpf brain samples from grna −/−;grnb −/− mutants and wildtype controls. C: Quantification of Ctsd from the Western blots shown in B normalized to α-tubulin. 5dpf: n = 4. 7mpf and 22mpf: n = 3. 5dpf: p = 0.8857. 7mpf: p = 1.0. 22mpf: p = 0.7. D: stat3 mRNA levels in 5dpf grna −/−;grnb −/− mutants and 7mpf as well as 22mpf grna −/−;grnb −/− mutant brains compared to wildtype control samples. 5dpf: p = 0.1. 7mpf: p = 0.4. 22mpf: p = 0.4. E: flnca mRNA levels in grna −/−;grnb −/− larvae at 5dpf and 7mpf as well as 22mpf grna −/−;grnb −/− brain samples in comparison to wildtype samples. 5dpf: p = 0.4. 7mpf: p = 0.7. 22mpf: p = 0.7. F: grna −/−;grnb −/− larvae of 5dpf and 7mpf and 22mpf grna −/−;grnb −/− brain samples are analysed for flncb mRNA expression levels and compared to wildtype. 5dpf: p = 0.1. 7mpf: p = 0.4. 22mpf: p = 0.4. A,D-E: Normalized to actb1 and tbp. qPCR. 5dpf: n = 4. 7mpf and 22mpf: n = 3. A,C-E: S.E.M. Mann-Whitney test (two-tailed).
A quantitative proteomic analysis of the zebrafish TDP-43 orthologous (tardbp −/−;tardbpl −/−) identified Flnca and Stat3 to be upregulated upon loss of Tardbp;Tardbpl [24]. Interestingly TDP-43 aggregates are a hallmark of FTLD-TDP/GRN and also in brain samples from FTLD patients FLNC mRNA expression levels are significantly increased [24]. In contrast, qPCR experiments revealed a slight but not statistically significant decrease in stat3 expression in samples derived from grna −/−;grnb −/− mutants, when compared to wildtype controls (Fig. 6D). qPCR analysis of the two FLNC orthologous in zebrafish, flnca and flncb, revealed that flnca mRNA expression levels are not altered in grna −/−;grnb −/− mutants and wildtype controls (Fig. 6E). mRNA expression of flncb appears to be decreased in 5dpf grna −/−;grnb −/− mutants when compared to wildtype control larvae, but this is not statistically significant. flncb is expressed at comparable levels in 7mpf grna −/−;grnb −/− and control brains and slightly but not significantly increased at 22mpf in grna −/−;grnb −/− brains when compared to control brains (Fig. 6F).
In summary, neuropathology as well as biochemical alterations reported in Grn-deficient humans and mice and the TDP-43 loss of function zebrafish were not observed in grna −/−;grnb −/− mutant zebrafish.
Discussion
Grna and Grnb mutants are complete loss of function mutants
Among the four zebrafish granulins, grna and grnb were chosen for the generation of KO mutants since they have a domain structure reminiscent of mammalian GRN. grna and grnb are both orthologous to human GRN with potentially redundant functions [14, 17]. Interestingly, we isolated a grna transcript with 12 granulin domains instead of the 10 granulin domains previously described [14]. Also in contrast to previous publications, Grna and Grnb [18, 20] were migrating at a higher molecular weight of approx. 210–230kDa and approx. 100–120kDa in denaturing SDS-PAGE.
The homozygous Grna and Grnb single and double KOs are viable and show no obvious morphological phenotype and fertility differences. Moreover, the lack of Grna and Grnb does not slow development.
No FTLD-TDP/GRN- and NCL/GRN-related phenotypes in grna −/−;grnb −/− mutants
Among the most consistent neuropathological findings in Grn KO mice are increased micro- and astrogliosis and enhanced lipofuscinosis [1] both neuropathological hallmarks of FTLD-TDP/GRN and NCL/GRN patients, respectively [4, 28–30]. In zebrafish larvae amoeboid microglia, which are highly motile and phagocytic, are present already at 3dpf and phagocytise cells and debris during brain development in a healthy environment [33, 34, 39]. These larval microglia, which can respond to inflammatory stimuli [40], are indistinguishable in a motion analysis performed with microglia of Grna;Grnb KO and wildtype larvae. A phenotypic categorization of amoeboid microglia according to the terms active and resting, commonly used for the analysis of microglia in the adult central nervous system [41] does not apply in larval zebrafish [34]. As the number of microglia in the Grna;Grnb KO larvae is also not increased we concluded that the grna −/−;grnb −/− mutants are devoid of microgliosis. “Astrocytes” are different in zebrafish compared to mouse. Zebrafish radial ependymoglia located at the ventricle fulfil functions attributed to parenchymal astrocytes in mammals [41], indicating that the cell types involved in inflammation in zebrafish are different than in mammals. Even though Grn KO mice develop micro- and astrogliosis they do not suffer from neurodegeneration suggesting that micro- and astrogliosis is not a result of the neurodegeneration in patients. To our knowledge, there is so far no zebrafish model for gliosis in the absence of injury. Therefore, it remains to be shown if this pathology can occur in zebrafish at all. In wildtype zebrafish the existence of autofluorescent neurolipofuscin is controversially discussed [42, 43]. Our attempts to demonstrate neurolipofuscin in aged wildtype brains using PAS stain supports the absence of lipofuscin deposits in aged zebrafish [42, 44].
In contrast to a CLN2 zebrafish NCL disease model associated with mutations in tripeptidyl peptidase 1 (TPP1) [36], grna −/−;grnb −/− mutants are devoid of increased cell death in the brain and do not display movement phenotypes reminiscent of seizures, a clinical hallmark of NCL pathology [36]. Neurodegeneration was only reported in one aged Grn KO mouse strain [8] but not in others [9, 11] consequently seizures are also not likely to occur in Grn KO mice. Possibly, seizures are specific for the clinical representation in humans due to the massive neurodegeneration observed in the NCL/GRN patients [4].
Biochemical alterations observed in FTLD-TDP/GRN and Grn KO mice [5, 11, 24] were not detectable in grna −/−;grnb −/− mutant zebrafish. The lysosomal protease Ctsd, which is itself linked to NCL by disease causing loss of function mutations [45], is slightly different in zebrafish compared to mammalian CTSD. Zebrafish Ctsd is mono-glycosylated and matures into a single-chain protein, whereas the di-glycosylated human CTSD proprotein is cleaved into a light and heavy chain [46, 47]. However, both the proprotein and the heavy chain are increased in Grn KO mice [5], whereas zebrafish Ctsd is unchanged. In Tardbp/Tardbpl-deficient zebrafish Stat3 was increased, whereas there was a trend to slightly but not significantly decreased stat3 expression in grna −/−;grnb −/− mutants. Whether STAT3 is increase in FTLD-TDP/GRN and Grn KO mice is not examined so far. STAT3 is involved in many processes, including neuroinflammation [48]. Therefore, it is possible, that a STAT3 increase occurs as a result of micro- and astrogliosis observed in FTLD-TDP/GRN and Grn KO mice [8–13]. If this is the case it would explain why stat3 is not increased in grna −/−;grnb −/− mutant zebrafish as they are devoid of microgliosis. The flnca and flncb expression levels are not significantly increased in grna −/−;grnb −/− mutants. It is possible, that the grna −/−;grnb −/− mutant zebrafish were too young when they were analysed, as the pathology and biochemical alterations do also manifest themselves over time in the Grn KO mice [1, 5]. Furthermore, heterozygous Grn KO mice did not develop micro- and astrogliosis and lack biochemical signatures described in the homozygous Grn KO mice [8, 35].
Potential reasons for the absence of phenotypes in grna −/−;grnb −/− mutants
We hypothesize that additional challenges are needed in the grna −/−;grnb −/− mutants to obtain disease-related phenotypes. In line with this hypothesis is the observation that the disease onset and manifestation in FTLD-TDP/GRN patients is quite variable [49, 50] supporting the need of a second hit such as oxidative stress, inflammation, or injury. The second or multiple hit theory was also proposed for TDP-43 and Fused-In-Sarcoma (FUS) pathogenesis, two proteins that are also linked to FTLD [51]. In contrast to mice used in biomedical research, zebrafish inbred lines are not available. Outbred lines enrich genetic variability and make the animals more resistant and robust, potentially precluding phenotypes. In contrast to mammals zebrafish also have a tremendous regenerative capacity [52]. This could possibly lead to a quick replacement of cells that are not fully functional because of a lysosomal dysfunction and thereby escape detection.
We also cannot exclude that the short Grn1 and Grn2 proteins sharing a similar domain structure might compensate the loss of Grna and Grnb. However, qPCR analysis of the shorter grn1/grn2 did not reveal an upregulation of grn1/grn2 on the transcriptional level in the grna −/−;grnb −/− mutants. Still, we cannot exclude a compensatory upregulation of Grn1/Grn2 in the Grna;Grnb KOs on the translational level. Additional KO of Grn1 and Grn2 can experimentally address this possibility in the future. It is also possible, that the zebrafish Granulins have, despite similar structures, diverse function and are independently regulated. In mice it was shown that Grn and the granulin peptides have opposing functions [53], which might be the case in zebrafish as well.
KD versus KO phenotypes in zebrafish
Previous KD studies in zebrafish embryos using Grna or Grnb translation inhibition MO resulted in SpMN axonopathies and a reduced number of MPCs [18–20]. In our Grna and Grnb single and double KOs we did not observe extended branching or truncation of SpMN axons despite careful quantifications and controls. Contradictory results obtained from morphants and mutants regarding SpMN axon outgrowth phenotypes have been described previously for other genes related to neurodegenerative diseases, e.g. Tardbp [24, 54] and Fragile X mental retardation 1 (Fmr1) [55, 56]. Similarly, the number of Pax7-positive MPCs in the grna −/−;grnb −/− embryos was not reduced as described after Grna or Grnb KD [20]. We speculate that the reduced number of MPCs in the morphants is due to toxic effects of the MO since the KD embryos have also morphological defects [20], which we do not observe in the KO embryos. To our knowledge, none of the studies in Grn KO mice showed muscle or motor neuron axons phenotype. Most importantly, GRN is not linked to any muscle disease and only published to be a minor risk factor for ALS [57, 58] and therefore not expected to affect SpMN axon outgrowth or muscle development but rather exclusively lead to NCL or FTLD-related phenotypes when mutated. These discrepancies raise concerns about the validity of the MO-induced phenotypes and suggest that MO-induced (neuro)toxicity is responsible for the SpMN axon outgrowth and MPCs phenotypes observed in morphants. With the ease of generating mutations in the zebrafish genome by the novel genome editing tools such as CRISPR/Cas9 we now have additional tools to circumvent the KD technologies [59].
Conclusion
In summary, we successfully generated Grna and Grnb KO mutants by targeted genome editing using ZFNs. The Grna and Grnb single and double KO mutants did not develop phenotypes previously published in KD studies [18–20, 23], highlighting the importance of generating stable genetic mutations. Moreover, the grna −/−;grnb −/− mutants are also devoid of FTLD-TDP/GRN or NCL/GRN related neuropathology and biochemical alterations reported for Grn KO mice [1, 5, 11, 24]. Since zebrafish granulins, especially grna, are upregulated after traumatic injuries [31, 60, 61] or upon infection [62, 63] it is likely that the zebrafish Granulins are involved in the inflammatory response as well as in regeneration in line with their enrichment in haematopoietic tissues [14, 31, 64]. Moreover, it was demonstrated that the inflammatory response in Grn KO mice is exaggerated after an injury [65, 66]. Further clarification of the role of the zebrafish Granulins in injury, inflammation, and regeneration remains to be addressed in more specialized functional assays and might provide further support for the hypothesis that GRN mutation carriers need to be exposed to a second hit to develop FTLD-TDP/GRN.
Materials and Methods
Zebrafish
Zebrafish embryos were kept at 28.5°C and staged according to Kimmel et al. [67]. For all experiments, unless stated otherwise, the wild-type line AB was used. Animal experiments were performed in accordance with the animal protection standards of the Ludwig-Maximilians University Munich and were approved by the government of Upper Bavaria (Regierung von Oberbayern, Munich, Germany, GZ:55.2-1-54-2532-127-10). The transgenic line Tg(apoeb:lynEGFP) was used in this study [39]. Embryo, larvae, and adult zebrafish were euthanized by an overdose of Tricaine (300 mg/l, Pharmaq Ltd) [68].
ZFN and identification of induced genomic lesions
CompoZr Custom zinc finger nucleases (ZFN) (Sigma-Aldrich) were designed for the grna (ENSDARG00000004954, Zv8) and the grnb (ENSDARG00000025081, Zv8) locus. ZF nucleotide recognition sequences are highlighted in uppercase:
grna ZFN: TTTGCTCGCAGTGCCCCAataatGAAGTCTGTGAAGCAGGC
grnb ZFN: TACCACCTGCTGCCAGatgcctgATGGGGGCTGGGGCT
ZFN-induced genomic mutations were identified by PCR amplification around the ZFN target site and subsequent restriction endonuclease digest. Samples were derived from euthanized 1dpf or 2dpf embryos or adult fin biopsies from anesthetized adult zebrafish (1xTricaine, 80 mg/l, Pharmaq Ltd). Oligonucleotides used are displayed in 5’-3’ orientation: P_grna ZFN forward TTCAGTCATTGTTTCAGAGGTCA, P_grna ZFN reverse TTCCTCTGATCCACTTTCTACCA, P_grnb ZFN forward AATGACACAAGACGTCCTCATAAA, P_grnb ZFN reverse AAAAATAATAACCACAGCGCAACT. 5μl of the 17μl PCR reactions were then digested at 37°C for approx. 3h with the following enzymes: grna ZFN PCR: Eco91I (Fermentas), grnb ZFN PCR: XcmI (New England Biolabs)
Whole mount immuno-fluorescence staining
Whole mount immuno-fluorescence staining was performed as previously described by Schmid and colleagues [24]. For znp1 staining 28hpf embryos and for Pax7 staining 24hpf embryos were anesthetized in E3 1xTricaine (80 mg/l, Pharmaq Ltd) and fixed in 4% PFA.
Neutral red staining
Neutral Red (Sigma-Aldrich) staining was performed at 3dpf to estimate the number of microglia as described by Herbomel and colleagues [33]. Larvae were anesthetized in E3 1xTricaine (80 mg/l, Pharmaq Ltd). After in vivo recording larvae were euthanized by an overdose of Tricaine (300 mg/l, Pharmaq Ltd) [68].
Acridine orange staining
Acridine orange (Sigma-Aldrich) staining was performed at 5dpf as described by Paquet and colleagues [25]. Larvae were anesthetized in E3 1xTricaine (80 mg/l, Pharmaq Ltd). After in vivo recording larvae were euthanized by an overdose of Tricaine (300 mg/l, Pharmaq Ltd) [68].
PTZ treatment and locomotion analysis
Locomotion analysis was performed in a ZebraBox Revolution (ViewPoint). Data was recorded by a digital camera with high resolution 1024x768 at 30 frames/s and analysed by ZebraLab tracking software version 3,22,3,9 (View Point). 5dpf larvae were placed into the 24-well plates (one larvae/well) in 800μl embryo medium (E3), 800μl 1%DMSO (Merck) E3, or 800μl 15mM PTZ (Sigma-Aldrich) 1%DMSO E3. The plate was transferred to the ZebraBox and the following tracking was performed: 30min adaptation, 5min tracking. Inactivity was defined as movements with a velocity below 2mm/s and large movements included all with a velocity above 6mm/s. After recording larvae were euthanized by an overdose of Tricaine (300 mg/l) (Pharmaq Ltd) [68].
Cloning
For grna cDNA (ENSDART00000137973, Zv9) cloning 5dpf AB cDNA and for grnb cDNA (ENSDART00000105686, Zv9) cloning 4dpf AB cDNA was used as a template. Oligonucleotides used are displayed in 5’-3’ orientation: P_grna forward CTGCTCAAAAAATGTTGAGACTG, P_grna reverse GCTCTAGAGCTTATAGAGTTAGGGCTCGTTTC, P_grna+MYC reverse GCTCTAGAGCTTATTCATTCAAGTCCTCTTCAGAAATGAGCTTTTGCTCCATCCCTAGAGTTAGGGCTCGTTTC, P_grnb forward ATGGTGCGTGCAGCTTTCAT, P_grnb reverse TTAGAGAGAATTATTCCACCACGT, P_grnb+MYC reverse TTATTCATTCAAGTCCTCTTCAGAAATGAGCTTTTGCTCCATGAGAGAATTATTCCACCACGT. All PCR products were cloned as described by Schmid and colleagues [24].
mRNA injections
mRNA for microinjections was synthesized in vitro using the MessageMAX T7 mRNA transcription kit (Epicentre) and the mMESSAGE mMACHINE T7 kit (Ambion) according to the manufacturer’s instruction. 2–4pl of 0.4μg/μl ZFN mRNA or 0.2μg/μl grnb mRNA (ENSDART00000105686, Zv9) were injected into the yolk. Translation of the mRNAs was confirmed using Western blotting.
qPCR
Samples for qPCR derived from 5dpf larvae that were anesthetized in E3 1xTricaine (80 mg/l, Pharmaq Ltd) and put in liquid nitrogen or from adult zebrafish biopsies taken from 7mpf or 22mpf zebrafish euthanized by an overdose of Tricaine (300 mg/l, Pharmaq Ltd) [68]. The RNeasy kit (Qiagen) was used with on column DNase I (Qiagen) treatment for total RNA isolation. cDNA synthesis was performed with M-MLV reverse transcriptase (Invitrogen) and Random Primer Mix (NEB), followed by a RNase H (Invitrogen) digest. qPCR was performed using SsoFast Evagreen Supermix (BioRad) with standard protocols. Oligonucleotides used are displayed in 5’-3’ orientation:
P_actb1 forward GATCTTCACTCCCCTTGTTCA (ENSDARE00000990736, Zv9), P_actb1 reverse AAAACCGGCTTTGCACATAC (ENSDARE00001086149, Zv9), P_ctsd forward GAAATACAACCTGGGCTTCC (ENSDARE00001100358, Zv9), P_ctsd reverse GAAGGTCTGGACAGGAGTGC (ENSDARE00000580787, Zv9), P_flnca forward CCTTCGTGGGTCAGAAGAAC (ENSDARE00000111992, Zv9), P_flnca reverse GGAGTTCTAGGACCGTGGAC (ENSDARE00000408366, Zv9), P_flncb forward GGCCCTACAAAGTGGACATC (ENSDARE00000522696, Zv9), P_flncb reverse CTTCAAACCAGGCCCATAAG (ENSDARE00000736832, Zv9), P_grn1/grn2 forward CCACCAGAACCTTCCAGAAA (ENSDARE00000899372, and ENSDARE00000953342, Zv9), P_grn1/grn2 reverse TGGCCAGTTCTAGTCTTGCAG (ENSDARE00000948245, ENSDARE00001069257, and ENSDARE00000948034, Zv9) P_grna qPCR forward ACCACATGGGGATGTTGC (ENSDARE00000060406, Zv9), P_grna qPCR reverse CCAAGTCTCCGGCTGAAATA (ENSDARE00000985214, Zv9), P_grnb qPCR forward GTCGCAGGAAGCCATTAGAG (ENSDARE00000188550, Zv9), P_grnb qPCR reverse CAGCATGTTGTATCTTCTGGACA (ENSDARE00000468841, Zv9), P_stat3 forward CCAGCTCAAAATCAAAGTGTG (ENSDARE00000257597, Zv9), P_stat3 reverse TTGGATTCCTCCATGTTCATC (ENSDARE00000257592, Zv9), P_tbp forward TCAGCATGGAGCAGAACAAC (ENSDARE00000320694, Zv9), P_tbp reverse CCCATACGGCATCATAGGAC (ENSDARE00000327919, Zv9). All samples were run in triplicates and normalized to the housekeeping genes TATA-binding protein (tbp) and actin, beta 1 (actb1). Relative mRNA abundance was calculated using the ΔΔCt method.
Antibodies
α-Tubulin, monoclonal, mouse, Sigma-Aldrich Cat# T6199, RRID:AB_477583, Western blotting (WB) 1:10000; Calnexin, polyclonal, rabbit, Enzo Life Sciences Cat# SPA-860D, RRID:AB_2069021, WB 1:10000; Cathepsin D, raised against rat Cathepsin D, polyclonal, rabbit, WB 1:8000 [47]; Pax7, monoclonal, mouse, Developmental Studies Hybridoma Bank Cat# pax7, RRID:AB_528428, immuno-fluorescence (IF) 1:100; znp-1, mouse, Developmental Studies Hybridoma Bank Cat# znp-1, RRID:AB_531910, IF 1:100; Anti-mouse IgG, HRP conjugated (conj.), Promega Cat# W4021, RRID:AB_430834, WB 1:5000; Anti-rabbit IgG, HRP conj., Promega Cat# W4011, RRID:AB_430833, WB 1:10000; goat anti-mouse IgG (H+L) antibody, Alexa Fluor 488, conjugated, Life Technologies Cat# A11001, RRID:AB_10566289, IF 1:500
The following antibodies were generated by the Institute of Molecular Immunology, Helmholtz Centre Munich by standard procedures: Grna 12E7-111 (Grna epitope: KQKKPETQRTTTRPTGTTS, NP_001001949.2), WB 1:1, monoclonal rat IgG2b; Grnb 11F4-11 (Grnb epitope: CTKSSSSTWWNNSL, NP_997903.1), WB 1:1, monoclonal rat IgG2c; Anti-rat IgG2b, HRP conj., WB 1:2000; Anti-rat IgG2c, HRP conj., WB 1:32000. All supernatants of hybridoma pools were first screened ELISA with the respective peptides and subsequently by Western Blotting with respective tagged grn proteins.
Western blotting
Western Blotting was performed as previously demonstrated by in Schmid and colleagues [24]. Samples for Western Blotting were derived from 1.5dpf embryos or 3dpf or 5dpf larvae that were anesthetized in E3 1xTricaine (80 mg/l, Pharmaq Ltd) and subsequently put in liquid nitrogen or from adult zebrafish biopsies taken from 7mpf or 22mpf zebrafish euthanized by an overdose of Tricaine (300 mg/l, Pharmaq Ltd) [68]. Quantifications were performed with ImageJ.
Image acquisition and in vivo imaging
Images were taken with a Cell Observer CSU-X1 Yokogawa Spinning Disk, AxioCam MRm and Evolve 512 (Zeiss). For in vivo imaging 1.5dpf embryos or 3dpf/5dpf larvae were anesthetized in E3 1xTricaine (80 mg/l) (Pharmaq Ltd) and were mounted in 1.5% agarose (Invitrogen) dissolved in E3 1xTricaine. Embedded embryos were covered with E3 1xTricaine. Brightness and contrast were adjusted using Zen blue (Zeiss) and ImageJ. Image stitching was performed with the ImageJ plugin Image stitching [69] and microglia were tracked with the ImageJ plugin mTrackJ [70].
Statistics
The software Graph Pad Prism 6 (Graph Pad Software) was used for statistical analyses of the data. The statistical tests used are indicated in the figure legend.
Supporting Information
A: mRNA levels of grna in 5dpf old Grna KOs and wildtype. B: grnb mRNA levels in 5dpf Grnb KOs and age-matched wildtype controls. Normalized to actb1 and tbp. qPCR. S.E.M. Mann-Whitney test (one-tailed). *p < 0.05. n = 3. n = 1 grna mde54c−/− and grnb mde357a−/−.
(TIF)
Images of 1.5dpf old wildtype, Grna and Grnb single and double KOs. Anterior to the left. Lateral view. Single images were taken on a spinning disk microscope using transmitted light and were stitched using the Image Stitching plugin of ImageJ.
(TIF)
A: The number of somites in grna −/− mutants compared to wildtype. B: Analysis of the number of somites in grnb −/− mutants and wildtype. C: The number of somites in grna −/−;grnb −/− mutants and wildtype. SD. Mann-Whitney test (two-tailed). All n.s. n > = 30.
(TIF)
A: mRNA levels of grnb in wildtype and Grna KOs. B: grna mRNA levels in wildtype and Grnb KOs. C: grn1/grn2 mRNA in Grna;Grnb KOs compared to wildtype. Normalized to actb1 and tbp. qPCR experiments with 5dpf old larvae. S.E.M. Mann-Whitney test (two-tailed). A, B: n = 3, n = 1 grna mde54c−/− and grnb mde357a−/−. C: n = 4. D: Grnb in wildtype and grna −/− mutants at 3dpf. α-tubulin serves as a loading control. E: Grna in adult kidney samples from grnb −/− mutants and wildtype. α-tubulin serves as a loading control.
(TIF)
A-C: Quantification of the SpMN axon length in homozygous and heterozygous Grna and Grnb single and double KOs that are devoid of maternal mRNA. The SpMN axon length is measured from the exit point of the spinal cord to the growth cone. Spinal motor neuron axon length of the 5 SpMN axon (1–5) above the end of the yolk extension is determined. A: Homozygous and heterozygous Grna KO siblings. n = 30. B: Homozygous and heterozygous Grnb KO siblings. n = 30. D: Homozygous and heterozygous Grna and Grnb double KO siblings. n = 30. S.E.M. Two-way ANOVA. Bonferroni post-test. all n.s.
(TIF)
Acknowledgments
We thank Alex Hruscha for support in experimental design and input as well as establishing the movement tracking and Laura Hasenkamp for establishing the PTZ treatment in our laboratory. We thank Katharina Weininger, Mathias Teucke, Alexandra Rechenberg, Rielana Wichert, and Michael Graf for technical support, Anja Capell, Fargol Mazaheri, Frauke van Bebber, Sophie Rothhämel, and Yu Ohki for critically reading the manuscript and Daniela Würzinger and Roberto RojasRojas for taking care of the fish. Moreover, we thank Ciro Isidoro and Carlo Follo from Dipartimento di Scienze della Salute, Università del Piemonte Orientale 'Amedeo Avogadro', Laboratorio di Patologia Molecolare (Novara, Italy) for providing us the Ctsd antibody, Francesca Peri, EMBL (Heidelberg, Germany) for the Tg(apoeb:lynEGFP) and Kim Wager for experimental assistance during the visit of B.So. in the laboratory of Claire Russell.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Sonderforschungsbereich 596, International Max Planck Research School for Molecular and Cellular Life Sciences (B. So.), the Elite Network of Bavaria (B. So.), a Short Term Scientific Mission COST Action BM0804 (B.So.), the Batten Disease Family Association (C.R.), the European Research Council under the European Union’s Seventh Framework Program FP7/2007–2013)/ ERC Grant Agreement No. 321366-Amyloid (advanced grant to C.H.), the general legacy of Mrs. Ammer (to the Ludwig-Maximilians University/ C.H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kleinberger G, Capell A, Haass C, Van Broeckhoven C. Mechanisms of granulin deficiency: lessons from cellular and animal models. Mol Neurobiol. 2013;47(1):337–360. 10.1007/s12035-012-8380-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. 10.1038/nature05016 . [DOI] [PubMed] [Google Scholar]
- 3. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. 10.1038/nature05017 . [DOI] [PubMed] [Google Scholar]
- 4. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90(6):1102–1107. 10.1016/j.ajhg.2012.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014;127(6):845–860. 10.1007/s00401-014-1262-6 . [DOI] [PubMed] [Google Scholar]
- 6. Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114(1):49–54. 10.1007/s00401-007-0223-8 . [DOI] [PubMed] [Google Scholar]
- 7. van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet neurology. 2008;7(10):965–974. 10.1016/S1474-4422(08)70194-7 [DOI] [PubMed] [Google Scholar]
- 8. Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177(1):311–324. 10.2353/ajpath.2010.090915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. 2012;45(1):395–408. 10.1016/j.nbd.2011.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, et al. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 2012;45(2):711–722. 10.1016/j.nbd.2011.10.016 . [DOI] [PubMed] [Google Scholar]
- 11. Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, et al. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol. 2012;228(1):67–76. 10.1002/path.4043 . [DOI] [PubMed] [Google Scholar]
- 12. Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207(1):117–128. 10.1084/jem.20091568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010;24(12):4639–4647. 10.1096/fj.10-161471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cadieux B, Chitramuthu BP, Baranowski D, Bennett HP. The zebrafish progranulin gene family and antisense transcripts. BMC Genomics. 2005;6:156 10.1186/1471-2164-6-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132(Pt 3):583–591. 10.1093/brain/awn352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gijselinck I, van der Zee J, Engelborghs S, Goossens D, Peeters K, Mattheijssens M, et al. Progranulin locus deletion in frontotemporal dementia. Human mutation. 2008;29(1):53–58. 10.1002/humu.20651 . [DOI] [PubMed] [Google Scholar]
- 17. Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283(3):1744–1753. 10.1074/jbc.M705115200 . [DOI] [PubMed] [Google Scholar]
- 18. Chitramuthu BP, Baranowski DC, Kay DG, Bateman A, Bennett HP. Progranulin modulates zebrafish motoneuron development in vivo and rescues truncation defects associated with knockdown of Survival motor neuron 1. Mol Neurodegener. 2010;5:41 10.1186/1750-1326-5-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laird AS, Van Hoecke A, De Muynck L, Timmers M, Van Den Bosch L, Van Damme P, et al. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS ONE. 2010;5(10):e13368 10.1371/journal.pone.0013368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y-H, Chen H-Y, Li Y-W, Wu S-Y, Wangta-Liu, Lin G-H, et al. Progranulin regulates zebrafish muscle growth and regeneration through maintaining the pool of myogenic progenitor cells. Scientific reports. 2013;3:1176 10.1038/srep01176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48(7):999–1009. . [DOI] [PubMed] [Google Scholar]
- 22. Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nature reviews. Molecular cell biology. 2007;8(2):113–126. 10.1038/nrm2104 . [DOI] [PubMed] [Google Scholar]
- 23.De Muynck L, Herdewyn S, Beel S, Scheveneels W, Van Den Bosch L, Robberecht W, et al. The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiology of aging. 2013. [DOI] [PubMed]
- 24. Schmid B, Hruscha A, Hogl S, Banzhaf-Strathmann J, Strecker K, van der Zee J, et al. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc Natl Acad Sci U S A. 2013;110(13):4986–4991. 10.1073/pnas.1218311110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119(5):1382–1395. 10.1172/JCI37537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patterson SE, Mook LB, Devoto SH. Growth in the larval zebrafish pectoral fin and trunk musculature. Dev Dyn. 2008;237(2):307–315. 10.1002/dvdy.21400 . [DOI] [PubMed] [Google Scholar]
- 27. Seger C, Hargrave M, Wang X, Chai RJ, Elworthy S, Ingham PW. Analysis of Pax7 expressing myogenic cells in zebrafish muscle development, injury, and models of disease. Dev Dyn. 2011;240(11):2440–2451. 10.1002/dvdy.22745 . [DOI] [PubMed] [Google Scholar]
- 28. Chen-Plotkin AS, Xiao J, Geser F, Martinez-Lage M, Grossman M, Unger T, et al. Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 2010;119(1):111–122. 10.1007/s00401-009-0576-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66(2):142–151. 10.1097/nen.0b013e31803020cf . [DOI] [PubMed] [Google Scholar]
- 30. Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129(Pt 11):3081–3090. 10.1093/brain/awl271 . [DOI] [PubMed] [Google Scholar]
- 31. Craig SE, Calinescu AA, Hitchcock PF. Identification of the molecular signatures integral to regenerating photoreceptors in the retina of the zebra fish. J Ocul Biol Dis Infor. 2008;1(2–4):73–84. 10.1007/s12177-008-9011-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang T, Cui J, Li L, Hitchcock PF, Li Y. The role of microglia in the neurogenesis of zebrafish retina. Biochem Biophys Res Commun. 2012;421(2):214–220. 10.1016/j.bbrc.2012.03.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Herbomel P, Thisse B, Thisse C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238(2):274–288. [DOI] [PubMed] [Google Scholar]
- 34. Svahn AJ, Graeber MB, Ellett F, Lieschke GJ, Rinkwitz S, Bennett MR, et al. Development of ramified microglia from early macrophages in the zebrafish optic tectum. Developmental neurobiology. 2013;73(1):60–71. 10.1002/dneu.22039 . [DOI] [PubMed] [Google Scholar]
- 35. Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013;33(12):5352–5361. 10.1523/JNEUROSCI.6103-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mahmood F, Fu S, Cooke J, Wilson SW, Cooper JD, Russell C. A zebrafish model of CLN2 disease is deficient in tripeptidyl peptidase 1 and displays progressive neurodegeneration accompanied by a reduction in proliferation. Brain. 2013;136(Pt 5):1488–1507. 10.1093/brain/awt043 [DOI] [PubMed] [Google Scholar]
- 37. Baraban SC, Taylor MR, Castro PA, Baier H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience. 2005;131(3):759–768. 10.1016/j.neuroscience.2004.11.031 . [DOI] [PubMed] [Google Scholar]
- 38. Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience. 2013;250(C):8–19. 10.1016/j.neuroscience.2013.06.049 . [DOI] [PubMed] [Google Scholar]
- 39. Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133(5):916–927. 10.1016/j.cell.2008.04.037 . [DOI] [PubMed] [Google Scholar]
- 40. Sieger D, Moritz C, Ziegenhals T, Prykhozhij S, Peri F. Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev Cell. 2012;22(6):1138–1148. 10.1016/j.devcel.2012.04.012 . [DOI] [PubMed] [Google Scholar]
- 41. Baumgart EV, Barbosa JS, Bally-Cuif L, Gotz M, Ninkovic J. Stab wound injury of the zebrafish telencephalon: a model for comparative analysis of reactive gliosis. Glia. 2012;60(3):343–357. 10.1002/glia.22269 . [DOI] [PubMed] [Google Scholar]
- 42. Kishi S, Uchiyama J, Baughman AM, Goto T, Lin MC, Tsai SB. The zebrafish as a vertebrate model of functional aging and very gradual senescence. Exp. Gerontol. 2003;38(7):777–786. [DOI] [PubMed] [Google Scholar]
- 43. Rosa CE, Kuradomi RY, Almeida DV, Lannes CF, Figueiredo Mde A, Dytz AG, et al. GH overexpression modifies muscle expression of anti-oxidant enzymes and increases spinal curvature of old zebrafish. Exp Gerontol. 2010;45(6):449–456. 10.1016/j.exger.2010.03.012 . [DOI] [PubMed] [Google Scholar]
- 44. Kishi S, Bayliss PE, Uchiyama J, Koshimizu E, Qi J, Nanjappa P, et al. The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet. 2008;4(8):e1000152 10.1371/journal.pgen.1000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129(Pt 6):1438–1445. 10.1093/brain/awl107 . [DOI] [PubMed] [Google Scholar]
- 46. Follo C, Ozzano M, Montalenti C, Ekkapongpisit M, Isidoro C. Similarities and differences in the biogenesis, processing and lysosomal targeting between zebrafish and human pro-Cathepsin D: functional implications. Int J Biochem Cell Biol. 2013;45(2):273–282. 10.1016/j.biocel.2012.10.010 [DOI] [PubMed] [Google Scholar]
- 47. Follo C, Ozzano M, Mugoni V, Castino R, Santoro M, Isidoro C. Knock-down of cathepsin D affects the retinal pigment epithelium, impairs swim-bladder ontogenesis and causes premature death in zebrafish. PLoS One. 2011;6(7):e21908 10.1371/journal.pone.0021908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nicolas CS, Amici M, Bortolotto ZA, Doherty A, Csaba Z, Fafouri A, et al. The role of JAK-STAT signaling within the CNS. JAKSTAT. 2013;2(1):e22925 10.4161/jkst.22925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghidoni R, Paterlini A, Albertini V, Binetti G, Benussi L. Losing protein in the brain: the case of progranulin. Brain Res. 2012;1476:172–182. 10.1016/j.brainres.2012.01.075 . [DOI] [PubMed] [Google Scholar]
- 50. Bonvicini C, Milanesi E, Pilotto A, Cattane N, Premi E, Archetti S, et al. Understanding phenotype variability in frontotemporal lobar degeneration due to granulin mutation. Neurobiology of aging. 2014;35(5):1206–1211. 10.1016/j.neurobiolaging.2013.10.097 [DOI] [PubMed] [Google Scholar]
- 51. Dormann D, Haass C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011;34(7):339–348. 10.1016/j.tins.2011.05.002 . [DOI] [PubMed] [Google Scholar]
- 52. Gemberling M, Bailey TJ, Hyde DR, Poss KD. The zebrafish as a model for complex tissue regeneration. Trends Genet. 2013;29(11):611–620. 10.1016/j.tig.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111(6):867–878. . [DOI] [PubMed] [Google Scholar]
- 54. Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19(4):671–683. 10.1093/hmg/ddp534 . [DOI] [PubMed] [Google Scholar]
- 55. Tucker B, Richards RI, Lardelli M. Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum Mol Genet. 2006;15(23):3446–3458. 10.1093/hmg/ddl422 . [DOI] [PubMed] [Google Scholar]
- 56. den Broeder MJ, van der Linde H, Brouwer JR, Oostra BA, Willemsen R, Ketting RF. Generation and characterization of FMR1 knockout zebrafish. PLoS One. 2009;4(11):e7910 10.1371/journal.pone.0007910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Del Bo R, Corti S, Santoro D, Ghione I, Fenoglio C, Ghezzi S, et al. No major progranulin genetic variability contribution to disease etiopathogenesis in an ALS Italian cohort. Neurobiology of aging. 2011;32(6):1157–1158. 10.1016/j.neurobiolaging.2009.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PW, et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 2008;71(4):253–259. 10.1212/01.wnl.0000289191.54852.75 . [DOI] [PubMed] [Google Scholar]
- 59. Schmid B, Haass C. Genomic editing opens new avenues for zebrafish as a model for neurodegeneration. J Neurochem. 2013;127(4):461–470. 10.1111/jnc.12460 [DOI] [PubMed] [Google Scholar]
- 60. Lien CL, Schebesta M, Makino S, Weber GJ, Keating MT. Gene expression analysis of zebrafish heart regeneration. PLoS Biol. 2006;4(8):e260 10.1371/journal.pbio.0040260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mathew LK, Sengupta S, Franzosa JA, Perry J, La Du J, Andreasen EA, et al. Comparative expression profiling reveals an essential role for raldh2 in epimorphic regeneration. J Biol Chem. 2009;284(48):33642–33653. 10.1074/jbc.M109.011668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Meijer AH, Verbeek FJ, Salas-Vidal E, Corredor-Adámez M, Bussman J, van der Sar AM, et al. Transcriptome profiling of adult zebrafish at the late stage of chronic tuberculosis due to Mycobacterium marinum infection. Mol. Immunol. 2005;42(10):1185–1203. [DOI] [PubMed] [Google Scholar]
- 63. Borst LB, Patterson SK, Lanka S, Suyemoto MM, Maddox CW. Zebrafish (Danio rerio) as a Screen for Attenuation of Lancefield Group C Streptococci and a Model for Streptococcal Pathogenesis. Veterinary Pathology Online. 2013;50(3):457–467. 10.1177/0300985811424731 [DOI] [PubMed] [Google Scholar]
- 64. Zakrzewska A, Cui C, Stockhammer OW, Benard EL, Spaink HP, Meijer AH. Macrophage-specific gene functions in Spi1-directed innate immunity. Blood. 2010;116(3):e1–11. 10.1182/blood-2010-01-262873 . [DOI] [PubMed] [Google Scholar]
- 65. Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M. Exacerbated inflammatory responses related to activated microglia after traumatic brain injury in progranulin-deficient mice. Neuroscience. 2013;231:49–60. 10.1016/j.neuroscience.2012.11.032 . [DOI] [PubMed] [Google Scholar]
- 66. Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122(11):3955–3959. 10.1172/JCI63113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. 10.1002/aja.1002030302 . [DOI] [PubMed] [Google Scholar]
- 68. Matthews M, Varga ZM. Anesthesia and euthanasia in zebrafish. ILAR journal / National Research Council, Institute of Laboratory Animal Resources. 2012;53(2):192–204. 10.1093/ilar.53.2.192 . [DOI] [PubMed] [Google Scholar]
- 69. Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25(11):1463–1465. 10.1093/bioinformatics/btp184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. 10.1016/B978-0-12-391857-4.00009-4 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: mRNA levels of grna in 5dpf old Grna KOs and wildtype. B: grnb mRNA levels in 5dpf Grnb KOs and age-matched wildtype controls. Normalized to actb1 and tbp. qPCR. S.E.M. Mann-Whitney test (one-tailed). *p < 0.05. n = 3. n = 1 grna mde54c−/− and grnb mde357a−/−.
(TIF)
Images of 1.5dpf old wildtype, Grna and Grnb single and double KOs. Anterior to the left. Lateral view. Single images were taken on a spinning disk microscope using transmitted light and were stitched using the Image Stitching plugin of ImageJ.
(TIF)
A: The number of somites in grna −/− mutants compared to wildtype. B: Analysis of the number of somites in grnb −/− mutants and wildtype. C: The number of somites in grna −/−;grnb −/− mutants and wildtype. SD. Mann-Whitney test (two-tailed). All n.s. n > = 30.
(TIF)
A: mRNA levels of grnb in wildtype and Grna KOs. B: grna mRNA levels in wildtype and Grnb KOs. C: grn1/grn2 mRNA in Grna;Grnb KOs compared to wildtype. Normalized to actb1 and tbp. qPCR experiments with 5dpf old larvae. S.E.M. Mann-Whitney test (two-tailed). A, B: n = 3, n = 1 grna mde54c−/− and grnb mde357a−/−. C: n = 4. D: Grnb in wildtype and grna −/− mutants at 3dpf. α-tubulin serves as a loading control. E: Grna in adult kidney samples from grnb −/− mutants and wildtype. α-tubulin serves as a loading control.
(TIF)
A-C: Quantification of the SpMN axon length in homozygous and heterozygous Grna and Grnb single and double KOs that are devoid of maternal mRNA. The SpMN axon length is measured from the exit point of the spinal cord to the growth cone. Spinal motor neuron axon length of the 5 SpMN axon (1–5) above the end of the yolk extension is determined. A: Homozygous and heterozygous Grna KO siblings. n = 30. B: Homozygous and heterozygous Grnb KO siblings. n = 30. D: Homozygous and heterozygous Grna and Grnb double KO siblings. n = 30. S.E.M. Two-way ANOVA. Bonferroni post-test. all n.s.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.