

Abstract
The study was designed to explore the role and possible mechanisms of hydrogen sulfide (H2S) in the regulation of myocardial collagen remodeling in spontaneously hypertensive rats (SHRs). We treated nine-week-old male SHRs and age- and sex-matched Wistar–Kyoto rats (WKYs) with NaHS (90 μmol/kg−1·day−1) for 9 wks. At 18 wks, plasma H2S, tail arterial pressure, morphology of the heart, myocardial ultrastructure and collagen volume fraction (CVF), myocardial expressions of collagen I and III protein and procollagen I and III mRNA, transforming growth factor-β1 (TGF-β1), TGF-β type I receptor (TβR-I), type II receptor (TβR-II), p-Smad2 and 3, matrix metalloproteinase (MMP)-13 and tissue inhibitors of MMP (TIMP)-1 proteins were determined. TGF-β1-stimulated cultured cardiac fibroblasts (CFs) were used to further study the mechanisms. The results showed that compared with WKYs, SHRs showed a reduced plasma H2S, elevated tail artery pressure and increased myocardial collagen, TGF-β1, TβR-II, p-Smad2 and p-Smad3 expressions. However, NaHS markedly decreased tail artery pressure and inhibited myocardial collagen, TGF-β1, TβR-II, p-Smad2 and p-Smad3 protein expressions, but H2S had no effect on the expressions of MMP-13 and TIMP-1. Hydralazine reduced blood pressure but had no effect on myocardial collagen, MMP-13 and TIMP-1 expressions and TGF-β1/Smad signaling pathway. H2S prevented activation of the TGF-β1/Smad signaling pathway and abnormal collagen synthesis in CFs. In conclusion, the results suggested that H2S could prevent myocardial collagen remodeling in SHR. The mechanism might be associated with inhibition of collagen synthesis via TGF-β1/Smad signaling pathway.
INTRODUCTION
Hypertension is one of the most common cardiovascular diseases, endangering human health and life. Cardiovascular remodeling is an important pathological change in the development of hypertension and also a factor leading to deterioration of the disease (1). Cardiac fibrosis is a major pathologic feature of hypertensive myocardial remodeling, including interstitial and perivascular fibrosis of intramyocardial coronary arteries. Myocardial collagen principally comprises collagen type I (80%) and type III (20%) (2). Deposition of collagen type I in the extracellular matrix is the most important factor in myocardial remodeling (3). Previous studies indicated that cardiac fibrosis was the result of an imbalance between the synthesis and degradation of collagen, characterized by excessive deposition of fibrillar collagen, disproportion of collagen types (increased I/III collagen ratio) and disorganized collagen arrangement (4,5). However, the mechanisms responsible for the abnormal metabolisms in hypertension have been unclear.
Transforming growth factor-β (TGF-β) is widely regarded as a key factor in the acceleration of the fibrotic process in organs (6). Three TGF-β isoforms, TGF-β1, TGF-β2 and TGF-β3, have been detected in mammal cells (7). TGF-β1, the major isoform of the TGF-β superfamily, is produced by myocardial fibroblasts, myofibroblasts and myocardial cells in the heart (8,9). Evidence indicates that TGF-β1 plays a crucial role in the myocardial remodeling process, particularly in cardiac fibrosis. Previous studies have shown that TGF-β1 can stimulate fibroblast-mediated collagen synthesis (10,11). In addition, TGF-β1 may inhibit degradation of collagen by suppressing the activity of matrix metalloproteinases and by inducing synthesis of protease inhibitors such as plasminogen activator inhibitor-1 and tissue inhibitor of matrix metalloproteinases (12–15).
While TGF-β1 action may involve multiple downstream signaling pathways and cross-talk, and the intracellular Smad pathway is thought to play a crucial role in mediating intracellular responses to TGF-β1 and related factors (16,17), activated TGF-β1 binds to a heteromeric complex of type I (TβR-I) and type II (TβR-II) receptors, which induces intracellular signals via phosphorylation of TβR-I-associated Smads (18,19). The receptor-activated Smads (R-Smads), Smad2 and Smad3, are translocated to the nucleus, where they regulate transcription, further modifying multiple cell functions including hypertrophy and proliferation (20).
Hydrogen sulfide (H2S) is a third endogenous gaseous transmitter following nitric oxide (NO) and carbon monoxide (CO), playing important roles in cardiovascular physiology and pathophysiology (21–29). Recent studies showed that the endogenous H2S pathway was downregulated in spontaneously hypertensive rats (SHRs), but exogenous administration of H2S to SHRs decreased blood pressure and lessened aortic structural remodeling (30). However, whether H2S affects excess accumulation of collagen in the myocardium and intramyocardial coronary arteries with arterial hypertension is unclear. The present study was, therefore, designed to explore the regulatory effect of H2S on myocardial collagen remodeling and its possible mechanisms in SHRs.
MATERIALS AND METHODS
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (31). The protocol was approved by the Committee on the Ethics of Animal Experiments of the Peking University (permit number: J200913). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Animals and Groups
Fourteen male Wistar–Kyoto rats (WKYs) (150–180 g) at the age of 9 wks were divided into two groups: a WKY control group (n = 7) and a WKY + NaHS group (n = 7). Twenty-one male SHRs (9 wks old) were divided into three groups: a SHR group (n = 7), a SHR + NaHS group (n = 7) and a SHR + hydralazine (Hyd) group (n = 7). All rats were purchased from Vital River (Beijing, China). They were housed in cages and fed a standard laboratory diet with fresh water. The animal cages were kept in a room with constant temperature (24°C ± 1°C) and relative humidity (65%–70%) and a 12-hr light–dark cycle.
For rats in the WKY + NaHS and SHR + NaHS groups, NaHS at a dose of 90 μmol/kg−1·day−1, dissolved in physiological (0.9%) saline, was injected intraperitoneally (IP). Hydralazine, a direct-acting smooth muscle relaxant, was injected IP at a dose of 10 mg/kg−1·day−1 into the SHR + Hyd rats. The same volume of physiological saline was injected into the rats in the WKY control group and the SHR control group. All rats were injected once daily for 9 wks. At 18 wks of age, their tail arterial blood pressure was measured.
Measurement of Tail Arterial Pressure
Tail systolic arterial pressure was measured by tail cuff plethysmography in all rats at 18 wks. Briefly, the systolic pressure in the rat’s tail was measured after heating for 10–15 min in a preheated box (RBP-1; China-Japan Friendship Hospital, Beijing China). The tail was wiped clean before the examination. During the measurement process, all of the rats were awake and quiet. Values were calculated as the average of three measurements.
Measurement of H2S Content in Plasma
At 18 wks, rat plasma H2S was measured using a sulfide-sensitive electrode (PXS-270, Shanghai, China). Before the experiment, the electrode was immersed into distilled water for 2 h. A volume of 0.5 mL of plasma was mixed with 0.5 mL of antioxidant buffer. The electrode was immersed in the mixture until a stable reading was obtained. The H2S concentration of each sample was calculated from a standard curve.
Measurement of Heart Morphological Parameters
At 18 wks, rats were anesthetized and weighed. The heart was excised, the atria and great vessels were removed, and the right ventricular free wall was carefully dissected from the left. The left ventricle weight (LVW) and the right ventricle weight (RVW) were recorded. After the measurements, hearts were frozen for paraffin sections and tissue homogenates.
Ultrastructural Analysis of Myocardium
Myocardial tissue was quickly cut into 1-mm3 pieces and placed immediately in 3% glutaraldehyde solution. Ultrathin sections were double stained with uranyl acetate and lead citrate. The myocardial ultrastructure was observed using a JEM1230 transmission electron microscope (JEOL Ltd., Tokyo, Japan). Two ultrathin sections were randomly selected from each rat and 10 microscopic fields per section were randomly selected for ultrastructural stereological analysis. The number of mitochondrion per unit area (N), mitochondrial mean diameter (mean D), volume density (Vv), numerical density (Nv) and mean volume (V) were calculated according to the method of Weibel (32) and Nouette-Gaulain (33). All stereological evaluations were performed using the Motic Digital Medical Image Analysis System (Motic Med 6.0; Motic Image Technology Co, Ltd, Beijing, China).
Myocardial Collagen Volume Fraction Assessed by Masson Trichrome Staining
After dewaxing with dimethylbenzene, sections of myocardial tissues were stained with Masson trichrome reagent. Results were observed under light microscope. Collagen volume fraction (CVF) was calculated as the sum of the connective tissue areas divided by the sum of all connective tissue and muscle area. An average of four microscopic fields was taken for the analysis for each animal.
Myocardial Collagen I and III Protein Expressions Evaluated by Enzyme-Linked Immunosorbent Assay (ELISA)
The hearts were homogenized and centrifuged at 3,000g. The liquid supernatants were obtained as homogenates for testing the concentrations of collagen I and III. Collagen I and III protein expressions were assayed using a double-antibody sandwich ELISA according to the manufacturer’s instructions (Rapidbio, West Hills, CA, USA).
Myocardial Procollagen I and III mRNA Expressions Evaluated by Quantitative Real-Time Polymerase Chain Reaction (PCR)
RNA from myocardial tissues of rats (n = 40) was extracted using TRIzol reagent (Thermo Fisher Scientific Inc., Waltham, MA USA) and reverse transcribed using an oligo d(T)18 primer and M-MLV reverse transcriptase. Quantitative real-time PCR was carried out using an ABI PRISM 7300 instrument (Applied Biosystems, Foster, CA, USA). Specific sequences of the primers and Taqma probes are listed in Table 1. The PCR condition for collagen I and collagen III was as follows: pre-denatured at 94°C for 5 min, 94°C for 30 s, 61°C for 30 s, 72°C for 45 s and 72°C for 6 min for 40 cycles. The amount of β-actin cDNA in the sample was used to calibrate the amount of sample needed for quantification.
Table 1.
The sequence of the primers and probes of collagen I, collagen III and β-actin.
| Collagen I | Forward primer | 5′-CTTGTTGCTGAGGGCAACAG |
| Reverse primer | 5′-GCAGGCGAGATGGCTTATTC | |
| TaqMan probe | 5′-ATTCACCTACACTGTCCTTGTCGATGGCTG | |
| Collagen III | Forward primer | 5′-GAAAAAACCCTGCTCGGAATT |
| Reverse primer | 5′-ATCCATCTTGCAGCCTTGGT | |
| TaqMan probe | 5′-AGAGACCTGAAATTCTGCCACCCTGAACTC | |
| β-actin | Forward primer | 5′-ACCCGCGAGTACAACCTTCTT |
| Reverse primer | 5′-TATCGTCATCCATGGCGAACT | |
| TaqMan probe | 5′-CCTCCGTCGCCGGTCCACAC |
Measurement of TGF-β1, TβR-I, TβR-II, p-Smad2, p-Smad3, Matrix Metalloproteinase (MMP)-13 and Tissue Inhibitors of MMP (TIMP)-1 Expressions in the Myocardium by Western Blotting
Myocardial tissues from all rats (n = 35) were homogenized and extracted in ice-cold protein lysis buffer containing 50 mmol/L Tris-Cl (pH 7.4), 150 mmol/L NaCl, 1 mmol/L ethylenediamine tetraacetic acid, 1% NP-40, 0.25% sodium deoxycholate, 1 mmol/L phenylmethylsulphonyl fluoride, and protease and phosphatase inhibitors. Equal amounts of proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrophoretically to nitrocellulose membrane according to the experimental protocol. The primary antibody dilutions were 1:100 for TGF-β1, 1:100 for TβR-I, 1:800 for TβR-II, 1:600 for p-Smad2, 1:600 for p-Smad3, 1:400 for MMP-13, 1:500 for TIMP-1 and 1:5000 for GAPDH antibodies. Secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used at a 1:10,000 dilution. The immunoreactions were visualized by electrochemiluminescence (ECL) and exposed to X-ray film (Kodak Scientific Imaging, New Haven, CT, USA).
Cell Culture and Experimental Treatments
Cardiac fibroblasts (CFs) were isolated from hearts of adult male Sprague Dawley rats (Vital River, Beijing, China) weighing 200–220 g. CFs were cultured with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 4 mmol/L l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C, with 5% CO2 and saturated humidity. The cells were divided into five treatment groups: a normal control group; a TGF-β1 group; a TGF-β1 + NaHS group; a TGF-β1 + TGF-β inhibitor SB431542 (SB) group; and a TGF-β1 + NaHS + SB group. The media was changed every 24 h and NaHS added again in the cells treated for 72 h.
Cell Viability Test
Cell viability was measured by cell counting kit (CCK-8, Dojindo, Kumamoto, Japan). Cells were cultured in an incubator with 5% carbon dioxide (CO2). Cells were cultured with Dulbecco’s modified Eagle medium (DMEM) mixed with 10% fetal bovine serum (FBS), 1% streptomycin, 1% penicillin and 1% glutamine. Before drug administration, cells were incubated with synchronization medium containing no FBS for 24 h. Cells were cultured in 96 wells with a final volume of 100 μL/well medium, incubated with different concentration of NaHS (Sigma-Aldrich, St. Louis, MO, USA) and TGF-β (Sigma-Aldrich) for 72 h, and culture medium with freshly prepared NaHS was changed every 12 h. Immediately after the incubation, the absorbance of sample was measured under a wavelength of 450 nm by Fluorescence Microplate Reader (FLx800, Biotek, Winooski, VT, USA).
Phosphorylation of TβR-I in CFs Evaluated by Immunoprecipitation
CFs were seeded in 100-mm dishes for immunoprecipitation. When the cells had grown to 70%–80% confluence, they were incubated in serum-free medium for 24 h. In the control group, they were incubated in DMEM with 0.5% FBS for 1 h. In the TGF-β1 group, the cells were stimulated with TGF-β1 at 10 ng/mL for 30 min. In the TGF-β1 + NaHS group, the cells were pretreated with NaHS at 2 × 10−4 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for the same duration. In the TGF-β1 + SB group, the cells were pre-treated with SB at 5 × 10−6 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for the same duration. In the TGF-β1 + NaHS + SB group, the cells were pretreated with NaHS at 2 × 10−4 mol/L and SB at 5 × 10−6 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for the same duration. The cells were then harvested and lysed. CFs lysates were incubated with the antibody against TβRI (Santa Cruz Biotechnology) before immunoprecipitation with protein A/G agarose beads (Thermo Fisher Scientific). The precipitated proteins were rexolved by 10% SDS-PAGE and then immunoblotted with antibody against phosphoserine (Abcam, Cambridge, MA, USA).
Determination of p-Smad2 and p-Smad3 Expressions in CFs by Western Blotting
CFs were seeded in 60-mm dishes for Western blotting. When the cells had grown to 70%–80% confluence, they were incubated in serum-free medium for 24 h. In the control group, they were incubated in DMEM with 0.5% FBS for 1 h. In the TGF-β1 group, the cells were stimulated with TGF-β1 at 10 ng/mL for 30 min. In the TGF-β1 + NaHS group, the cells were pretreated with NaHS at 2 × 10−4 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for the same duration. In the TGF-β1 + SB group, the cells were pre-treated with SB at 5 × 10−6 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for the same duration. In the TGF-β1 + NaHS + SB group, the cells were pretreated with NaHS at 2 × 10−4 mol/L and SB at 5 × 10−6 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for the same duration. The cells were then harvested and lysed as described previously. Equal amounts of proteins were separated by 10% SDS-PAGE and transferred electrophoretically to nitrocellulose membrane. The primary antibody dilutions were 1:400 for p-Smad2 (245/250/255), 1:400 for p-Smad2 (Ser465/467), 1:400 for p-Smad3 and 1:5000 for GAPDH antibodies. Secondary antibody (Santa Cruz Biotechnology) was used at a 1:10,000 dilution. The immunoreactions were visualized by ECL and exposed to X-ray film (Kodak Scientific Imaging, USA).
Collagen I and III Expression in CFs by Immunofluorescence and Confocal Microscopy
CFs were plated on glass coverslips. When the cells had reached 60%–70% confluence, they were incubated in DMEM containing 0.5% FBS for 24 h. The cells were then cultured in DMEM with 10% FBS. The cells of control group were incubated in DMEM with 10% FBS for 72 h. In the TGF-β1 group, the CFs were treated with TGF-β1 (Princeton Business Park, Rocky Hill, NJ, USA) at 10 ng/mL for 72 h. In the TGF-β1 + NaHS group, the cells were pretreated with NaHS (2 × 10−4 mol/L) for 30 min and then treated with TGF-β1 (10 ng/mL) for 72 h. In the TGF-β1 + SB group, the cells were pre-treated with SB at 5 × 10−6 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for 72 h. In the TGF-β1 + NaHS + SB group, the cells were pre-treated with NaHS at 2 × 10−4 mol/L and SB at 5 × 10−6 mol/L for 30 min and then treated with TGF-β1 (10 ng/mL) for 72 h. The cells in each group were fixed with 4% paraformaldehyde (0.01 mol/L PBS, pH 6.8), washed with PBS, and treated with 3% BSA and 0.3% Triton X-100 for 30 min at 37°C. The coverslips were then incubated with rabbit anti-collagen I primary antibody (1:100; Abcam) and rabbit anti-collagen III primary antibody (1:100; Abcam), respectively, in a humidified chamber at 4°C overnight. Next day, after washing, the coverslips were incubated with secondary antibody (1:100) (Santa Cruz Biotechnology) in the dark for 30 min at 37°C. Cellular nuclei were marked by propidium iodide (PI). The slides were viewed using a Fluoview laser scanning confocal microscope (Olympus, Tokyo, Japan). In the control group, 3% BSA was used instead of the first antibody.
Statistical Analysis
The data were analyzed using Excel and SPSS 13.0 statistical software. The results were expressed as the mean ± standard deviation (SD). Differences among groups were analyzed by one-way analysis of variance (ANOVA), and least significant difference (LSD) analysis was used to compare data between the two groups. A level of P < 0.05 was considered statistically significant.
RESULTS
Changes in Tail Arterial Pressure
Tail systolic arterial pressure was measured by tail cuff plethysmography in all rats at 18 wks. Compared with the WKY rats, tail arterial pressure significantly increased in the SHR group (P < 0.01) (Figure 1A). However, compared with the SHR group, tail arterial pressure decreased in both the SHR + NaHS group and the SHR + Hyd group (both P < 0.01) (see Figure 1A). There was no difference in tail arterial pressure between the WKY group and the WKY + NaHS group (P > 0.05) (see Figure 1A).
Figure 1.
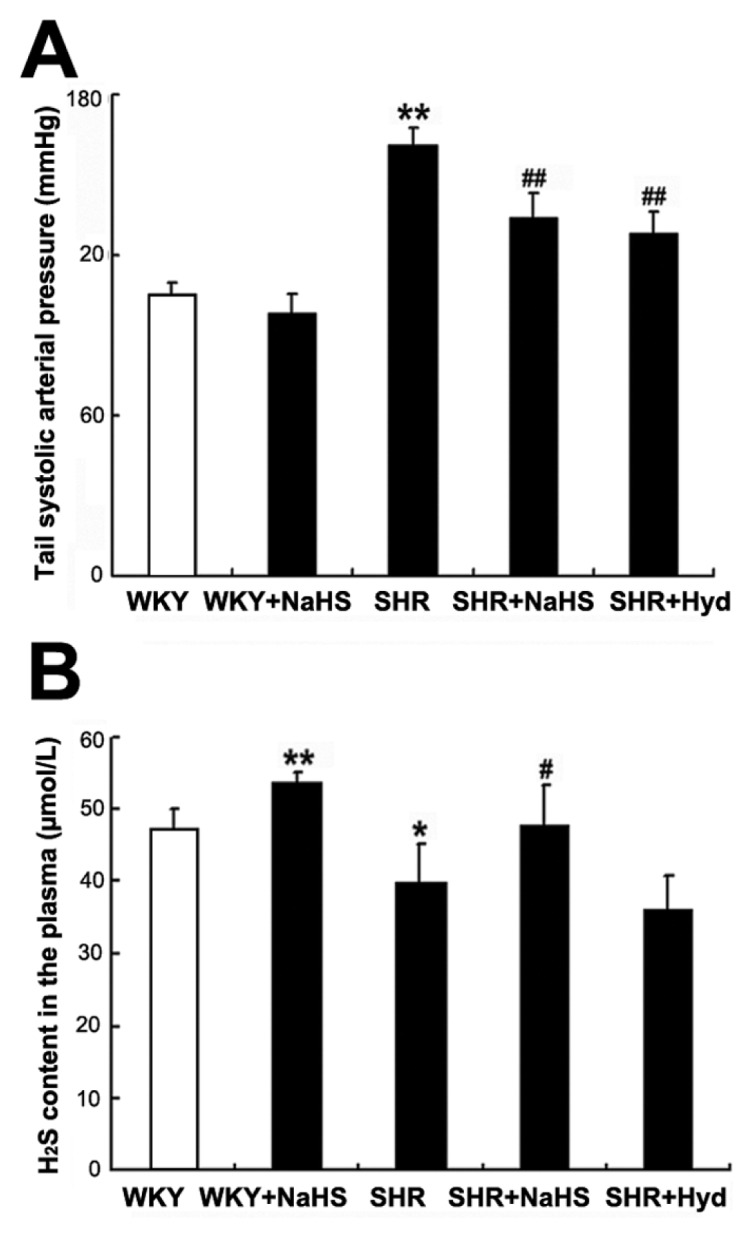
Changes in tail arterial pressure and H2S content in the plasma of rats. (A) Tail systolic arterial pressure for rats of each group (mmHg). (B) H2S content in the plasma (μmol/L). Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. *P < 0.05 compared with WKY group; #P < 0.05 compared with SHR group; **P < 0.01 compared with WKY group; ##P < 0.01 compared with SHR group.
H2S Content in Rat Plasma
Compared with the WKY group, the plasma H2S content was significantly greater in the WKY + NaHS group (P < 0.01) (Figure 1B), but markedly reduced in the SHR group (P < 0.05) (see Figure 1B). Compared with the SHR group, the plasma H2S content significantly raised in the SHR + NaHS group (P < 0.05) (see Figure 1B). However, there was no difference in plasma H2S content between the SHR group and the SHR + Hyd group (P > 0.05) (see Figure 1B).
Effect of H2S on Indices of Left-Ventricular Hypertrophy
As illustrated in Table 2, compared with the WKY group, SHRs showed a reduced BW, but increased LVW/BW (both P < 0.01) (Table 2). However, neither NaHS nor hydralazine exerted any significant effects on LVW, BW and LVW/BW in SHR control (both P > 0.05) (Table 2).
Table 2.
Effect of H2S on indexes of left-ventricular hypertrophy.a
| Groups | N | BW (g) | LVW (mg) | RVW (mg) | LVW/BW (mg/g) |
|---|---|---|---|---|---|
| WKY | 7 | 458 ± 19 | 973 ± 57 | 203 ± 12 | 2.125 ± 0.155 |
| WKY + NaHS | 7 | 451 ± 24 | 911 ± 65 | 216 ± 31 | 2.055 ± 0.158 |
| SHR | 7 | 329 ± 23b | 1036 ± 99 | 181 ± 19 | 3.218 ± 0.351b |
| SHR + NaHS | 7 | 321 ± 14 | 1016 ± 37 | 185 ± 15 | 3.269 ± 0.162 |
| SHR + Hyd | 7 | 312 ± 12 | 988 ± 77 | 181 ± 27 | 3.237 ± 0.119 |
BW, body weight; LVW, left ventricular weight; RVW, right ventricular weight; eLVW/BW, LVW-to-BW ratio.
Values are means ± SE.
P < 0.05 versus WKY group.
Effect of H2S on Myocardial Ultrastructure
In the WKY group, the myocardial cells were well aligned, muscle striations were clear, and the structure of the mitochondria and sarcoplasmic reticulum was normal. In the WKY + NaHS group, the ultrastructure was similar to that in the WKY group. In the SHR group, the cardiomyocytes were arranged erratically. The contour was deformed, the sarcoplasmic reticulum was significantly dilated, mitochondria were swollen, and the mitochondrial cristae were fractured and dissociated. In the SHR + NaHS group, the ultrastructure was similar to that in the WKY group. The cardiac muscle cells were well aligned, the sarcoplasmic reticulum was not dilated and only few mitochondria were slightly swollen. There were no obvious ultrastructural differences between the SHR group and the SHR + Hyd group (Figure 2).
Figure 2.

Myocardial ultrastructure of rats in each group. The black arrows indicate mitochondria. The red arrows indicate sarcoplasmic reticulum. WKY group: the cardiomyocytes lined up in order, muscle striations were clear, and the structure of mitochondria and sarcoplasmic reticulum was clear; WKY + NaHS group: the cardiomyocytes lined up in order, muscle striations were clear and the structure of mitochondria was normal; SHR group: the cardiomyocytes were arranged erratically, the contour was deformed, sarcoplasmic reticulum was significantly dilated, mitochondria were swollen and mitochondria cristae were fractured and dissolved; SHR + NaHS group: The cardiomyocytes lined up in order, sarcoplasmic reticulum did not dilate, and only few mitochondria were slightly swollen; SHR + Hyd group: sarcoplasmic reticulum was significantly dilated, mitochondria were swollen and mitochondria cristae were fractured and dissolved. Scale bar, 1 μm.
Stereological analysis revealed a reduced N and Nv, but increased mean D, Vv and V in the SHRs compared with the WKY group (both P < 0.05) (Table 3). Compared with the SHR group, SHR + NaHS group showed a significant reduction in mean D, Vv and V, and obvious increase in N and Nv (both P < 0.05) (Table 3). However, hydralazine had no effect on parameters of myocardial mitochondria (both P > 0.05) (Table 3).
Table 3.
Effect of NaHS on parameters of myocardial mitochondria.a
| Groups | N | Mean D (μm) | Vv (%) | Nv (μm−3) | V (μm3) |
|---|---|---|---|---|---|
| WKY | 23.50 ± 2.67 | 0.97 ± 0.08 | 37.78 ± 5.83 | 10.35 ± 2.17 | 3.84 ± 1.15 |
| WKY + NaHS | 24.13 ± 1.72 | 0.95 ± 0.04 | 38.67 ± 4.83 | 10.54 ± 1.20 | 3.72 ± 0.70 |
| SHR | 13.5 ± 2.88b | 1.17 ± 0.12b | 48.73 ± 4.43b | 4.07 ± 1.11b | 12.61 ± 2.89b |
| SHR + NaHS | 23.38 ± 2.50c | 0.88 ± 0.92c | 38.03 ± 7.24c | 10.24 ± 1.82c | 3.87 ± 1.14c |
| SHR + Hyd | 15.25 ± 2.50 | 1.13 ± 0.05 | 50.74 ± 3.34 | 4.61 ± 0.69 | 11.19 ± 1.6 |
N, number of mitochondria per unit area; mean D, mean diameter; Vv, volume density; Nv, numerical density; V, mean volume.
Values are means ± SE.
P < 0.05 versus WKY group.
P < 0.05 versus SHR group.
Myocardial Collagen Expression by Masson Trichrome Staining
Cardiac collagen deposition was assessed by Masson trichrome staining. The blue staining around the vascular and between the cardiomyocytes represents the presence of collagen. We observed that CVF was increased significantly in the SHR group compared with the WKY group (P < 0.01) (Figure 3). When SHRs were treated with NaHS, CVF was decreased significantly (P < 0.01) (see Figure 3).
Figure 3.

Collagen volume fraction (CVF) by Masson trichrome staining in myocardial tissues (blue color). (A) WKY group; (B) SHR group; (C) SHR + NaHS group; (D) Data of quantification of myocardial fibrosis. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. **P < 0.01 compared with WKY group; ##P < 0.01 compared with SHR group. Scale bar, 30 μm.
Expressions of Collagen I and III in Myocardium using ELISA
The expressions of myocardial collagen I and III proteins were increased in the SHRs compared with the WKYs (both P < 0.05) (Figures 4A, B). But, compared with the SHR group, myocardial collagen I and III protein expressions were decreased in the SHR + NaHS group (both P < 0.05) (see Figures 4A, B). However, there was no difference in collagen I or III protein expression between the SHR group and the SHR + Hyd group (both P > 0.05) (see Figures 4A, B).
Figure 4.

Expressions of collagen protein and procollagen mRNA in myocardium. (A) Collagen I protein content in myocardium of each group using ELISA; (B) Collagen III protein content in myocardium of each group using ELISA; (C) Procollagen I mRNA expression in myocardium of each group by real-time PCR; (D) Procollagen III protein mRNA expression in myocardium of each group by real-time PCR. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. *P < 0.05 compared with WKY group; #P < 0.05 compared with SHR group; **P < 0.01 compared with WKY group; ##P < 0.01 compared with SHR group.
mRNA Expressions of Myocardial Procollagen I and III by Real-Time PCR
Compared with the WKY group, the expressions of myocardial procollagen I and III mRNA were increased in the SHR group (procollagen I, P < 0.05; procollagen III, P < 0.01) (Figures 4C, D). However, when treated with NaHS, SHR showed reduced levels of myocardial procollagen I and procollagen III mRNA (procollagen I, P < 0.01; procollagen III, P < 0.05) (see Figures 4C, D). There was no difference in procollagen I or III mRNA expressions between the SHR group and the SHR + Hyd group (both P > 0.05) (see Figures 4C, D).
Effects of H2S on TGF-β1 Protein Expression in Myocardium by Western Blot
The TGF-β1 protein content of myocardium was significantly greater in the SHR group than in the WKY group (P < 0.05) (Figure 5A). However, in the SHR + NaHS group, the myocardial TGF-β1 content was significantly lower than that in the SHR group (P < 0.05) (see Figure 5A). TGF-β1 content did not differ between the SHR group and the SHR + Hyd group (P > 0.05) (see Figure 5A).
Figure 5.

Expressions of TGF-β1 and TGF-β receptor protein in myocardium by Western blot. (A) TGF-β1 protein content in myocardium of each group. Results are expressed as mean ± SD. Differences among groups were analyzed by Kruskal-Wallis Test. *P < 0.05 compared with WKY group; #P < 0.05 compared with SHR group. (B) TβR-I protein content in myocardium of each group by Western blot; (C) TβR-II protein content in myocardium of each group by Western blot. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. *P < 0.05 compared with WKY group; #P < 0.05 compared with SHR group.
Effects of H2S on TGF-β Receptor Expression in Myocardium by Western Blot
There was no significant difference in TβR-I protein content of myocardium among the five groups (Figure 5B). But TβR-II protein content of myo cardium from the SHR group was significantly greater than that from the WKY group (P < 0.05) (Figure 5C). In the SHR + NaHS group, TβR-II protein was significantly reduced compared with the SHR group (P < 0.05) (see Figure 5C). TβR-II protein expression did not differ between the SHR group and the SHR + Hyd group (P > 0.05) (see Figure 5C).
Myocardial p-Smad2 and p-Smad3 Determination by Western Blotting
Compared with the WKY group, the expressions of both p-Smad2 and p-Smad3 protein in myocardium were increased in the SHR group (both P < 0.01) (Figures 6A, B). Compared with the SHR group, the SHR + NaHS-treated rats showed lower levels of myocardial p-Smad2 and p-Smad3 protein expressions (p-Smad2, P < 0.05; p-Smad3, P < 0.01) (see Figures 6A, B), but expressions of p-Smad2 and p-Smad3 did not differ between the SHR and the SHR + Hyd group (P > 0.05) (see Figures 6A, B).
Figure 6.
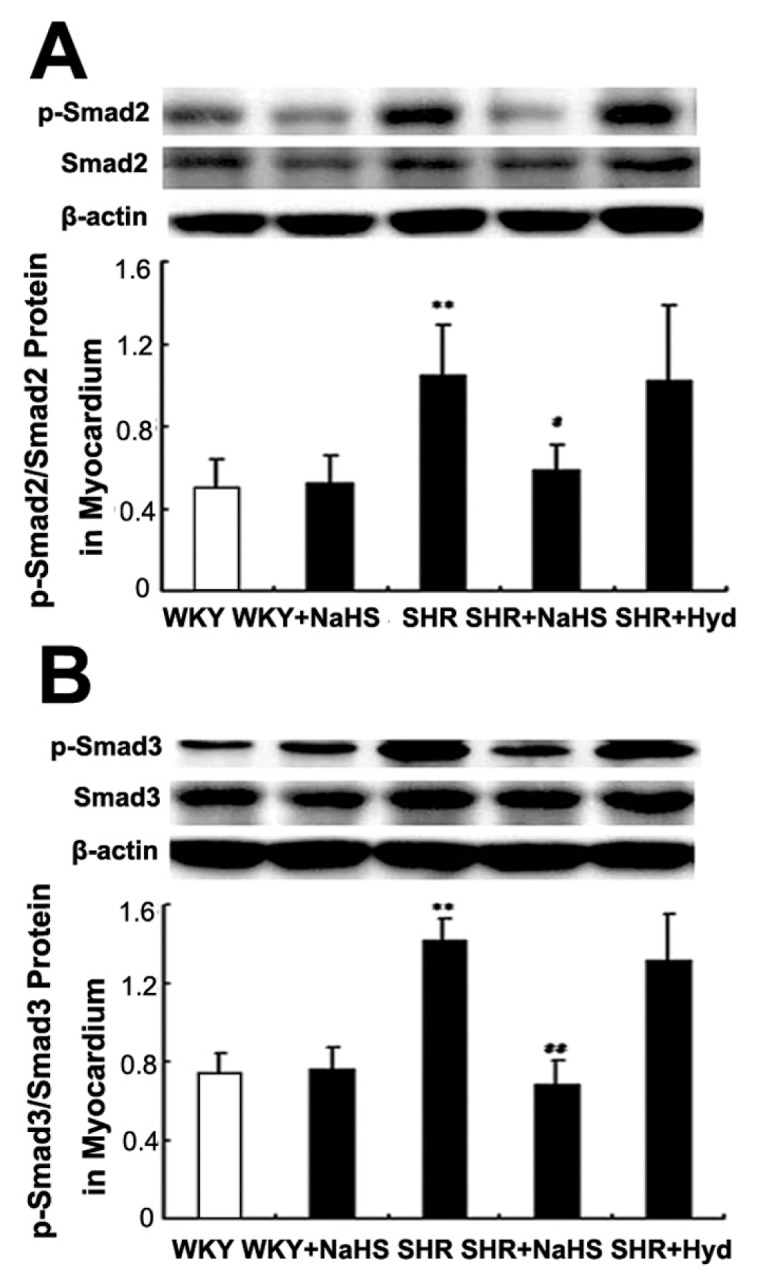
Expressions of p-Smad2 and p-Smad3 protein in myocardium by Western blot. (A) p-Smad2 protein content in myocardium of each group; (B) p-Smad3 protein content in myocardium of each group. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. **P < 0.01 compared with WKY group; ##P < 0.01 compared with SHR group; #P < 0.05 compared with SHR group.
Effects of H2S on MMP-13 and TIMP-1 Protein Expressions in Myocardium by Western Blotting
Compared with the WKY group, the expression of MMP-13 in myocardium was decreased in the SHR group (P < 0.01) (Figure 7A). In contrast, myocardial TIMP-1 content was significantly greater in the SHR group than in the WKY group (P < 0.01) (Figure 7B). However, compared with the SHR group, the expression of MMP-13 and TIMP-1 proteins did not differ either in the SHR + NaHS group or in the SHR + Hyd group (both P > 0.05) (see Figures 7A, B).
Figure 7.
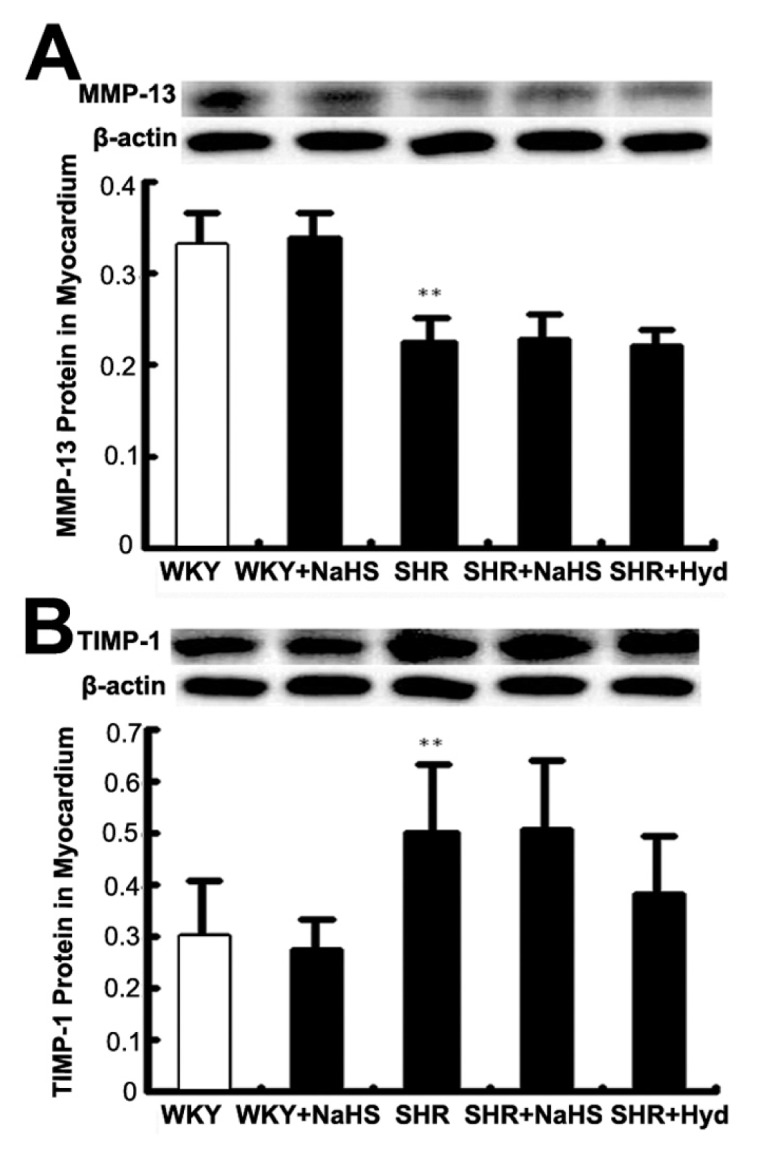
Expressions of MMP-13 and TIMP-1 protein in myocardium by Western blot. (A) MMP-13 protein content in myocardium of each group; (B) TIMP-1 protein content in myocardium of each group. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. **P < 0.01 compared with WKY group.
Cell Viability Treated with Different Concentrations of NaHS or TGF-β1
No significant difference of cell viability was found between vehicle cells and cells treated with 2 × 10−4 mol/L NaHS for 72 h. In addition, there was no significant difference among vehicle cells and cells treated with other concentrations of NaHS or TGF-β1 (P > 0.05) (Figure 8).
Figure 8.
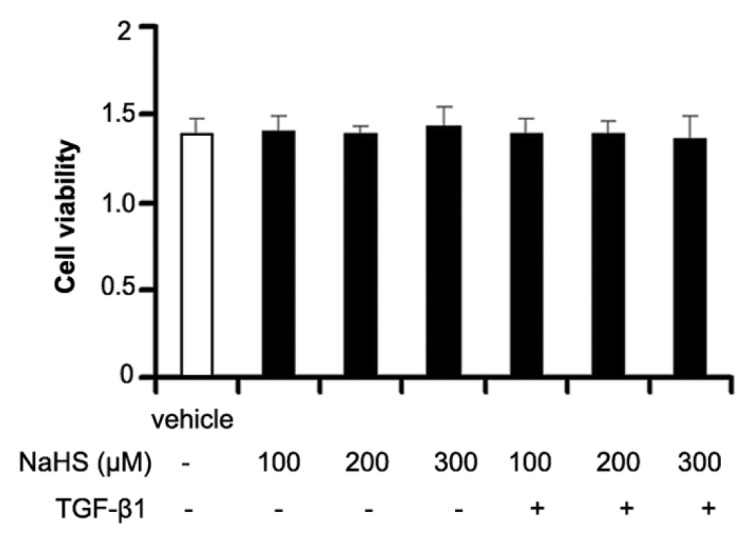
Cell viability of different concentration of NaHS or TGF-β1. There was no significant difference between vehicle cells and cells treated with different concentration of NaHS or TGF-β1.
Effect of H2S on Phosphorylation of TβR-I in CFs
The immunoprecipitation assay showed that the expression of p-TβR-I protein was significantly increased when CFs were treated with TGF-β1 at 10 ng/mL for 30 min (P < 0.01) (Figure 9). However, the expression of p-TβR-I protein induced by TGF-β1 was suppressed markedly after CFs were pretreated with NaHS at 2 × 10−4 mol/L for 30 min (P < 0.01) (see Figure 9). Furthermore, the TGF-β1 + SB treated cells also showed low levels of p-TβR-I protein expression compared with the TGF-β1 group (P < 0.01) (see Figure 9). There was no obvious difference in p-TβR-I protein expression between the TGF-β1 + SB group and the TGF-β1 + NaHS + SB group (P > 0.05) (see Figure 9).
Figure 9.
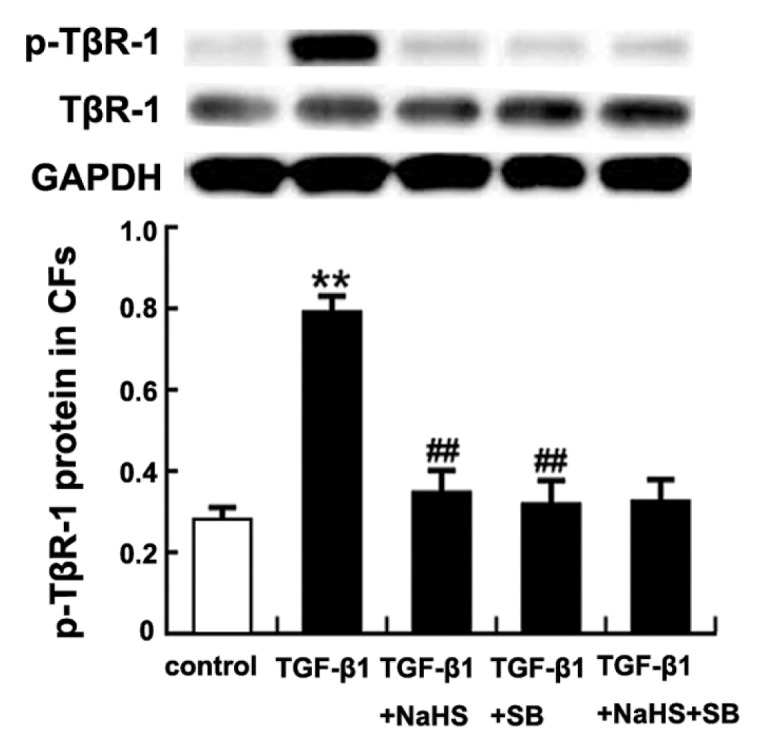
Effect of H2S on phosphorylation of TβR-I in CFs. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. **P < 0.01 compared with control group; ##P < 0.01 compared with TGF-β1 group.
Effects of H2S on p-Smad2 and p-Smad3 in CFs
Western blotting showed that the expressions of both p-Smad2 and p-Smad3 proteins were significantly increased when CFs were treated with TGF-β1 at 10 ng/mL for 30 min (both P < 0.01) (Figure 10). However, the expressions of p-Smad2 and p-Smad3 proteins induced by TGF-β1 were inhibited significantly when CFs were pretreated with NaHS at 2 × 10−4 mol/L for 30 min (p-Smad2 (Ser245/250/255), P < 0.05; p-Smad2 (Ser465/467), P < 0.01; p-Smad3, P < 0.01) (see Figure 10). Furthermore, the TGF-β1 + SB treated cells also showed lower levels of p-Smad2 and p-Smad3 protein expressions compared with the TGF-β1 group (both P < 0.01) (see Figure 10). There was no obvious difference in p-Smad2 and p-Smad3 protein expression between the TGF-β1 + SB group and the TGF-β1 + NaHS + SB group (both P > 0.05) (see Figure 10).
Figure 10.

Expressions of p-Smad2 and p-Smad3 protein in CFs by Western blot. (A) p-Smad2 (Ser245/250/255) protein content in CFs of each group; B: p-Smad2 (Ser465/467) protein content in CFs of each group; C: p-Smad3 protein content in CFs of each group. Results are expressed as mean ± SD. Differences among groups were analyzed by one-way ANOVA, and LSD analysis was used to compare data between the two groups. **P < 0.01 compared with control group; ##P < 0.01 compared with TGF-β group; #P < 0.05 compared with TGF-β1 group.
Effect of H2S on Collagen Type I and III Expressions in CFs
Confocal images showed that, compared with the control group, TGF-β1 induced accumulation of collagen type I and collagen type III in CFs. However, in CFs of the TGF-β1 + NaHS or TGF-β1 + SB group, where cells were pretreated with NaHS or SB, collagen type I and III expressions were both reduced to some extent. There were, however, no obvious difference in collagen type I and III expressions between the TGF-β1 + SB group and the TGF-β1 + NaHS + SB group (Figures 11, 12).
Figure 11.

Effect of H2S on collagen type I expression in CFs by confocal images. TGF-β1 induced accumulation of collagen type I in CFs. H2S inhibited collagen type I expression induced by TGF-β1 in CFs. PI: propidium iodide.
Figure 12.

Effect of H2S on collagen type III expression in CFs by confocal images. TGF-β1 induced accumulation of collagen type III in CFs. H2S inhibited collagen type III expression induced by TGF-β1 in CFs. PI: propidium iodide.
DISCUSSION
H2S is a recently identified endogenous gaseous transmitter that plays various roles in the cardiovascular system (34,35). In previous studies, we found that H2S attenuated hypertension and lessened aortic structural remodeling in hypertensive rats (30). In addition, Shi et al. reported that chronic NaHS treatment prevented hypertrophy of intramyocardial arterioles and ventricular fibrosis (36). However, whether H2S affects excess accumulation of collagen in the myocardium and what the underlying mechanisms are remain unclear. In the present study, we explored the regulatory effect of H2S on myocardial collagen remodeling and its possible mechanisms in SHRs.
The SHR model of genetic hypertension is analogous to human primary hypertension, including its genetic characteristics, pathogenesis and complications (myocardial remodeling), and is therefore widely used in experiments on primary hypertension and antihypertensive drugs (37). In our study, compared with WKYs, the blood pressure was significantly increased in 18-wk-old SHRs, indicating 18-wk-old SHRs successfully developed hypertension. We also recorded the indexes of left-ventricular hypertrophy. We found that, compared with the WKY group, SHRs showed an increased LVW/BW. Examination of the myocardial ultrastructure showed that the cardiomyocytes were arranged erratically, the sarcoplasmic reticulum was significantly dilated, the mitochondria were swollen, and the mitochondrial cristae were fractured and dissociated in SHRs. Stereological analysis of the mitochondrial revealed a reduced N and Nv, but increased mean D, Vv and V in the SHRs compared with the WKY group, suggesting that the myocardial mitochondria of SHRs were swelling and deformation. These observations implied that significant hypertension and myocardial ultrastructural remodeling occurred in the 18-wk-old SHRs.
We also found that, along with the increased blood pressure and myocardial ultrastructural remodeling, the plasma H2S content was markedly decreased in SHRs. Administration of H2S donor to SHRs significantly decreased the blood pressure. Yan et al. (30) reported that the downregulation of the gene expression of cystathionine γ-lyase (CSE), a H2S-generating enzyme, resulted in a suppression of H2S production, which might be responsible for the decreased H2S level in plasma in SRHs. In addition, in our study, myocardial ultrastructure showed that the cardiomyocytes were well aligned, the sarcoplasmic reticulum was not dilated and only a few mitochondria were slightly swollen in the SHR group. Mitochondrial stereological analysis showed a significant reduction in mean D, Vv and V and an obvious increase in N and Nv in the SHR + NaHS group compared with the SHR group, suggesting that NaHS treatment could improve the myocardial ultrastructural changes.
However, NaHS treatment (90 μmol/kg−1·day−1) for 9 wks did not induce a reduction in LVW and LVW/BW compared with SHR. The possible explanation is that myocardial remodeling is a complex pathologic process, including cardiomyocyte hypertrophy, an increase in perivascular and interstitial fibrosis, remodeling of the extracellular matrix and a reduced capillary-to-myocyte ratio (38). Cardiac collagen remodeling, the result of an imbalance between the synthesis and degradation of collagen, is just one of the abovementioned pathologic features of hypertensive myocardial remodeling. On the basis of the data we obtained in the present study, we could not exclude the possibility of the impact of H2S on other biological behaviors of cardiomyocytes with either stimulative or protective features in myocardial remodeling, in addition to the collagen remodeling. The above facts might explain why administration of H2S acted only on the collagen synthesis but had no effect on macroscopic hypertrophy in SHRs. With the further understanding of regulatory insights of H2S in myocardium, more detailed information about the biological behaviors of cardiomyocyte regulated by H2S would be obtained.
Cardiac collagen remodeling was represented by an excessive accumulation of collagen in the myocardium, increased concentration of cardiac collagen or increased CVF. To explore the effect of H2S on cardiac collagen remodeling in SHRs, we detected CVF and the myocardial collagen I and III protein. We found that CVF and myocardial collagen protein were significantly increased in SHRs, when compared with WKYs. However, CVF and myocardial collagen protein expressions were decreased significantly after treatment with NaHS. These results suggested that chronic NaHS treatment could improve myocardial collagen remodeling.
High blood pressure is an important causative factor in myocardial collagen remodeling. However, it is unclear whether the inhibitory effect of NaHS on myocardial collagen remodeling is due to the reduction of blood pressure, resulting in reduced afterload of the heart after NaHS treatment. In the present study, we found that hydralazine reduced blood pressure, but had no effect on the expressions of collagen I and III in myocardium, suggesting that the inhibitory effects of NaHS on myocardial collagen remodeling could not be solely ascribed to the BP-decreasing effect of NaHS treatment.
As is well known, the balance between collagen synthesis controlled mainly by procollagen gene transcription and collagen degradation adjusted by MMPs/TIMPs is crucial for vascular collagen metabolism. First, we examined if H2S could impact collagen synthesis by detecting procollagen mRNA changes. We found that myocardial procollagen I and III mRNA expressions were increased significantly in SHRs, when compared with WKYs. However, myocardial procollagen I and III mRNA expressions were decreased significantly after treatment with NaHS. These results suggested that chronic NaHS treatment could inhibit myocardial collagen synthesis.
It is well established that TGF-β1 plays a critical role in the progression of cardiac fibrosis. Previous studies have shown that TGF-β1 can stimulate collagen synthesis (10,11) and inhibit the degradation of collagen (12–15). To explore the possible mechanisms by which H2S regulated myocardial collagen synthesis, we determined the expression of TGF-β1 in myocardium by Western blotting. We found that, compared with WKYs, myocardial expression of TGF-β1 protein was raised significantly in SHRs. When H2S donor was administered, the myocardial TGF-β1 level was decreased. These results indicated that H2S regulated myocardial collagen synthesis, probably by inhibiting TGF-β1 protein expression.
The best-defined TGF-β signaling pathways are through Smad family members. Active TGF-β binds to a complex of TβR-I and TβR-II, resulting in phosphorylation of the Smad2 and Smad3 by TGF-β receptor I kinase. Activated Smad2 and Smad3 bind Smad 4 and becomes localized into the nucleus, where it activates transcription of procollagen (16). It has been shown in several experiments that TGF-β1 activates cardiac fibrosis predominantly through the TGF-β1/Smad signaling pathway. Bai et al. (20) found that TGF-β1 significantly increased protein synthesis in cardiac fibroblasts, partially through the Smad2-dependent pathway. Hao et al. (38) reported that TGF-β1 mRNA abundance and protein levels both were increased significantly in cardiac infarct scar, correlated with increased collagen type I expression and elevated Smad2, Smad3 and Smad4.
In the present study, to examine if H2S regulated myocardial collagen remodeling principally through the TGF-β/Smad signaling pathway, we detected the important elements in the TGF-β/Smad signaling pathway including protein expression of TβR-I and TβR-II, phosphorylation of Smad2 and Smad3 expressions in myocardial tissues in vivo. First, we found that NaHS inhibited myocardial TβR-II protein expression and phosphorylation of Smad2 and Smad3. Unfortunately, we did not find the impact of NaHS on myocardial TβR-I protein expression. To further explore how H2S blocked the signaling transduction from TβR-II to Smad2 and Smad3, we detected phosphorylation of TβR-I in CFs in vitro. In the in vitro experiment, we used 2 × 10−4 mol/L NaHS to treat the CFs for 72 h, in which the cell viability did not change as compared with vehicle cells. The data showed that TGF-β induced phosphorylation of TβR-I, Smad2 and Smad3 and collagen I and III expression. Moreover, H2S inhibited the phosphorylation of TβR-I protein, Smad2 and Smad3 and collagen I and III expression activated by TGF-β1 in CFs. Also, we found that the above mentioned inhibitory effect of H2S was abolished by Smad inhibitor SB-431542. The facts suggest that TGF-β and H2S might regulate the activation of Smad2 and Smad3 by targeting the phosphorylation of TGF-βRI instead of its protein expression.
MMPs comprise a family of Zn-dependent endopeptidases that can decompose the ECM and basement membrane. They participate in tissue remodeling, cell infiltration and tumor spread. At least 23 MMP family members have been characterized. In particular, MMP-2, MMP-9 and MMP-13 are thought to play key roles in tissue remodeling. MMP-2 degrades gelatin, type IV, V, VII, X and XI collagens, fibronectin, elastin and laminin. MMP-9 degrades gelatin, type IV, V, VII and X collagens, elastin, proteoglycan, entacin, fibronectin and laminin. Also called collagenase-3, MMP-13 degrades various substrates, including fibrillar-type I, II and III collagens, type IV, IX, X and XIV collagens, gelatin, tenascin-C, fibronectin, and proteoglycan core proteins (40). The activation of MMPs is inhibited by TIMPs, which form a 1:1 complex with MMPs. Four different TIMPs have been identified. TIMP-1 was an important inhibitor of MMP-1, -3, -9 and -13 (41).
Therefore, in the present study, we examined the effects of H2S on MMP-13 and TIMP-1 protein expression in myocardial tissue. We found that along with the increased TIMP-1 expression, the MMP-13 content was markedly decreased in SHRs, suggesting that alterations of the collagen degrading protease system were also involved in the development of myocardial collagen remodeling. But, we found that there was no difference in MMP-13 and TIMP-1 expressions between the SHR group and the SHR + NaHS group, not supporting the impact of H2S on collagen degradation.
This study had limitations. For example, we detected the blood pressure in 18-wk-old SHRs, instead of conducting a series of measurements at different time points of experiment. However, the parameters detected at the termination of the experiment were comparable among groups. Further studies are needed to observe the dynamic changes of the blood pressure and the myocardial structure.
CONCLUSION
The endogenous H2S pathway was downregulated during myocardial collagen remodeling in SHRs. Administering H2S donor to SHRs reduced blood pressure and alleviated the accumulation of collagen in interstitial and intramyocardial coronary arteries. The mechanisms might involve inhibition of the TGF-β1/Smad signaling pathway by H2S in cardiomyocytes. These findings also suggest that H2S might be a potential therapeutic approach for hypertension and myocardial collagen remodeling.
ACKNOWLEDGMENTS
This work was supported by the Major Basic Research Development Program of People’s Republic of China (2012CB517806, 2013CB933801 and 2011CB503904), National Natural Science Foundation of China (31130030, 81370154, 81100181 and 81121061), Beijing Natural Science Foundation (7122184 and 7121014) and The Open Project of Key Laboratory of Remodeling-related Cardiovascular Diseases, Ministry of Education (2014XXGB02).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Hackam DG, et al. The 2010 Canadian Hypertension Education Program recommendations for the management of hypertension: part 2-therapy. Can J Cardiol. 2010;26:249–58. doi: 10.1016/s0828-282x(10)70379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laviades C, Mayor G, Diez J. Treatment with lisinopriol normalizes serum concentrations of procollagen type III amino-terminal peptides in patients with essential hypertension. Am J Hypertens. 1994;7:52–8. doi: 10.1093/ajh/7.1.52. [DOI] [PubMed] [Google Scholar]
- 3.Seeland U, et al. Myocardial fibrosis in transforming growth factor beta(1)(TGF-beta(1)) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest. 2002;32:295–303. doi: 10.1046/j.1365-2362.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- 4.Tsuda T, et al. Post-ischemic myocardial fibrosis occurs independent of hemodynamic changes. Cardiovasc Res. 2003;59:926–33. doi: 10.1016/s0008-6363(03)00519-4. [DOI] [PubMed] [Google Scholar]
- 5.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–67. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 6.Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13:3056–62. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huh MI, et al. Distribution of TGF-beta isoforms and signaling intermediates in corneal fibrotic wound repair. J Cell Biochem. 2009;108:476–88. doi: 10.1002/jcb.22277. [DOI] [PubMed] [Google Scholar]
- 8.Deten A, Holzl A, Leicht M, Barth W, Zimmer HG. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. J Mol Cell Cardiol. 2001;33:1191–207. doi: 10.1006/jmcc.2001.1383. [DOI] [PubMed] [Google Scholar]
- 9.Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39:258–63. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- 10.Stow RC, Mallawaarachchi CM, Weissberg PL. Migration of adventitia myofibroblasts following vascular balloon injury: insights from in vivo gene transfer to rat carotid arteries. Cardiovasc Res. 2003;59:212–21. doi: 10.1016/s0008-6363(03)00292-x. [DOI] [PubMed] [Google Scholar]
- 11.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seeland U, et al. Myocardial fibrosis in transforming growth factor beta(1)(TGF-β1) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest. 2002;32:295–303. doi: 10.1046/j.1365-2362.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- 13.Jin L, et al. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther. 2006;318:288–95. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- 14.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Mauviel A. Transforming growth factor-beta: a key mediator of fibrosis. Methods Mol Med. 2005;117:69–80. doi: 10.1385/1-59259-940-0:069. [DOI] [PubMed] [Google Scholar]
- 16.Leask A. TGFβ, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–12. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 17.Li P, et al. Atrial natriuretic peptide inhibits transforming growth factor β–induced smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–92. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 19.Bertolino P, Deckers M, Lebrin F, Dijke P. Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest. 2005;128:585S–90S. doi: 10.1378/chest.128.6_suppl.585S. [DOI] [PubMed] [Google Scholar]
- 20.Lei B, et al. Effect of efonidipine on tgf-β1–induced cardiac fibrosis through smad2-dependent pathway in rat cardiac fibroblasts. J Pharmacol Sci. 2011;7:1–8. doi: 10.1254/jphs.11065fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter. FASEB J. 2002;16:1792–8. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 22.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–35. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 23.Hart JL. Role of sulfur-containing gaseous substances in the cardiovascular system. Front Biosci (Elite Ed) 2011;3:736–49. doi: 10.2741/e282. [DOI] [PubMed] [Google Scholar]
- 24.Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. JBiol Chem. 2010;285:21903–7. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2013;20:783–93. doi: 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura H, Shibuya N, Kimura Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal. 2012;17:45–57. doi: 10.1089/ars.2011.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baskar R, Bian J. Hydrogen sulfide gas has cell growth regulatory role. Eur J Pharmacol. 2011;656:5–9. doi: 10.1016/j.ejphar.2011.01.052. [DOI] [PubMed] [Google Scholar]
- 28.Liu YH, Yan CD, Bian JS. Hydrogen sulfide: a novel signaling molecule in the vascular system. J Cardiovasc Pharmacol. 2011;58:560–9. doi: 10.1097/FJC.0b013e31820eb7a1. [DOI] [PubMed] [Google Scholar]
- 29.Bian JS, et al. Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther. 2006;316:670–8. doi: 10.1124/jpet.105.092023. [DOI] [PubMed] [Google Scholar]
- 30.Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–7. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 31.Institute of Laboratory Animal Resources (U.S.), Committee on Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. Bethesda (MD): NIH; 1985. p. 83. Rev. 1985. (NIH publication; No. 85-23) [Google Scholar]
- 32.Weibel ER, Staubli W, Gnagi HR, Hess FA. Correlated morphometric and biochemical studies on the liver cell. I. Morphometric model, stereologic methods, and normal morphometric data for rat liver. J Cell Biol. 1969;42:68–91. doi: 10.1083/jcb.42.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nouette-Gaulain K, et al. Time course of differential mitochondrial energy metabolism adaptation to chronic hypoxia in right and left ventricles. Cardiovasc Res. 2005;66:132–40. doi: 10.1016/j.cardiores.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 34.Coletta C, Szabo C. Potential role of hydrogen sulfide in the pathogenesis of vascular dysfunction in septic shock. Curr Vasc Pharmacol. 2013;11:208–21. [PubMed] [Google Scholar]
- 35.Szabó C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–65. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi YX, et al. Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;293:H2093–100. doi: 10.1152/ajpheart.00088.2007. [DOI] [PubMed] [Google Scholar]
- 37.Pitcher TL, Wickens JR, Reynolds JN. Differences in striatal spiny neuron actinpotentials between the spontaneously hypertensive and Wistar-Kyoto rat strains. Neuroscience. 2007;146:135–42. doi: 10.1016/j.neuroscience.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Wang M, et al. Involvement of NADPH oxidase in age-associated cardiac remodeling. J Mol Cell Cardiol. 2010;48:765–72. doi: 10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hao J, et al. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol. 1999;31:667–78. doi: 10.1006/jmcc.1998.0902. [DOI] [PubMed] [Google Scholar]
- 40.Mori S, et al. Expression and roles of MMP-2, MMP-9, MMP-13, TIMP-1, and TIMP-2 in allergic nasal mucosa. Allergy Asthma Immunol Res. 2012;4:231–9. doi: 10.4168/aair.2012.4.4.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cevik C, et al. Rosuvastatin therapy does not affect serum MMP-13 or TIMP-1 levels in hypercholesterolemic patients. Tex Heart Inst J. 2011;38:229–33. [PMC free article] [PubMed] [Google Scholar]