

Abstract
Chronic hypoxia typically elicits pulmonary hypertension (PH) in mice with a male-dominant phenotype. There is an opposite-sex bias in human PH, with a higher prevalence in women, but greater survival (the “estrogen paradox”). We investigated the involvement of the STAT5a/b species, previously established to mediate sexual dimorphism in other contexts, in the sex bias in PH. Mice with heterozygous or homozygous deletions of the STAT5a/b locus in vascular smooth muscle cells (SMCs) were generated in crosses between STAT5a/bfl/fl and transgelin (SM22α)-Cre+/+ parents. Wild-type (wt ) males subjected to chronic hypoxia showed significant PH and pulmonary arterial remodeling, with wt females showing minimal changes (a male-dominant phenotype). However, in conditional STAT5+/− or STAT5−/− mice, hypoxic females showed the severest manifestations of PH (a female-dominant phenotype). Immunofluorescence studies on human lung sections showed that obliterative pulmonary arterial lesions in patients with idiopathic pulmonary arterial hypertension (IPAH) or hereditary pulmonary arterial hypertension (HPAH), both male and female, overall had reduced STAT5a/b, reduced PY-STAT5 and reduced endoplasmic reticulum (ER) GTPase atlastin-3 (ATL3). Studies of SMCs and endothelial cell (EC) lines derived from vessels isolated from lungs of male and female IPAH patients and controls revealed instances of coordinate reductions in STAT5a, STAT5b and ATL3 in IPAH-derived cells, including SMCs and ECs from the same patient. Taken together, these data provide the first definitive evidence for a contribution of STAT5a/b to the sex bias in PH in the hypoxic mouse and implicate reduced STAT5 in the pathogenesis of the human disease.
INTRODUCTION
There is an unusual “estrogen paradox” in the pathogenesis of pulmonary arterial hypertension (PAH) (1–5). The human disease idiopathic PAH (IPAH) has a two- to four-fold higher prevalence in postpubertal women than in men, although women with PAH survive longer than their male counterparts (1–5). This female bias in prevalence is observed even in kindreds of hereditary PAH (HPAH) in which both the male and female family members were identified to have the same mutation in bone morphogenetic protein receptor type 2 (BMPR2) (1–3,6–8). It has been suggested that mutations in cytochrome P450 CYP1B1, an enzyme involved in estrogen metabolism, may be a candidate cofactor cocontributing to the development of the overt disease (8). However, there is an opposite male-dominant sex bias in the customarily used rodent models of pulmonary hypertension (PH) (chronic hypoxia, monocrotaline) such that the male typically shows higher prevalence and higher severity than the female (2–5,9–12). Estrogenic compounds [estradiol-17β (E2), 2-methoxyestradiol (2-ME)] attenuated development of PH in these rodent models in the male, and ovariectomy of the female enhanced PH (4). However, in a converse example, female mice overexpressing the serotonin transporter (SERT) or the S100 calcium-binding protein S100A4/mts1, but not male mice, spontaneously developed PH, with modest increases in right ventricular pressure at 5 months of age (3,7). The mechanisms for these variations in sex dependence of this disease in humans and rodent models, and the underlying estrogen paradox, remain incompletely understood. A perplexing question has remained: what are the differences between a female mouse (or female rat) and a female human?
In addressing this question more broadly, we realized the existence of sex-bias mechanisms, in addition to E2 (and steroid hormones), that drive male versus female patterns of gene expression (13–24). Specifically, that male versus female patterns of growth hormone (GH) release (pulsatile versus continuous respectively) by the pituitary under hypothalamic control cause male versus female patterns of activation of signal transducer and activator of transcription (STAT)5a and STAT5b transcription factors has been well established in the last 15 years in other contexts (body growth, breast, liver) (13–28). Thus, the major transcription factors that mediate sex bias resulting from dynamic-patterned fluctuations of circulating GH are STAT5a and STAT5b (19,23). Downstream of the STAT5 transcription factors in sex-bias determinism lie master transcription factors such as B-cell lymphoma 6 (Bcl6) and cut-like homeobox 2 (CUX2), which, together with STAT5a/b, regulate the sex-biased expression of >400–500 genes (19,24,29–34). Indeed, it is already known that a difference between humans and rodents lies in circadian (diurnal versus nocturnal) and quantitative differences in the sex-biased patterns of circulating GH (13,18,35,36). However, these GH-STAT5–based mechanisms of sex bias have never been evaluated in vascular biology or in the pathogenesis of PH.
STAT5a and STAT5b are ~90-kDa proteins that are 96% related and have overlapping as well as distinct functions (26–28). The two genes lie adjacent to each other in the human and mouse genomes at the STAT5a/b locus (25). There is an extensive literature establishing the cytokine, GH and prolactin- and estradiol-17β (E2) responsiveness of STAT5a/b transcription factors through activation by Tyr-phosphorylation (PY) and/or Ser-phosphorylation (19,23,26,28). To investigate the role of STAT5 species in pulmonary vascular biology in vivo, especially in the sex bias observed in the hypoxic mouse model of PH, we developed novel mouse lines that have heterozygous or homozygous vascular smooth muscle–specific deletion of the STAT5a/bfl/fl locus using the SM22α-Cre+/+ approach (25,37). For the present investigation, we selected the male-dominant chronic hypobaric hypoxia model in the mouse (C57BL/6 background) to study the influence of sex on development of hemodynamic and histologic features of PH in these tissue-specific conditional STAT5a/b+/− and STAT5a/b−/− knockouts. We also investigated STAT5 in cells in sections of obliterative pulmonary arterial lesions and in vascular cells isolated from lungs of IPAH and HPAH patients of both sexes.
MATERIALS AND METHODS
Antibodies
Rabbit pAbs to STAT5a (sc-1081x, L-20), STAT5a+b (sc-835x, C-17), Tyr-694-P-STAT5 (PY-STAT5, sc-101806; reactive to both a and b), murine monoclonal antibodies (mAbs) to STAT5b (sc-1656, G-2), and goat pAb to reticulon-4 (RTN4; also called Nogo-B) (sc-11027, N-18) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), whereas rabbit pAb to atlastin-3 (ATL3; ab104262) was from Abcam (Cambridge, MA, USA). Mouse mAbs to GM130 (#610823) and Vti1a (#611220) were from BD Biosciences (San Jose, CA, USA). The mAb to α-smooth muscle actin (SMA) (A5228) and rabbit pAb to β-actin (A2066) were from Sigma-Aldrich (St. Louis, MO, USA). Alexa Fluor–tagged donkey secondary antibodies were from Life Technologies (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Generation of Mice with Smooth Muscle–Specific Deletion of STAT5a/b
Protocols for mouse breeding, animal care and experimental use of mice in the present studies were approved by the Institutional Animal Care and Use Committee of the New York Medical College. A breeder pair of STAT5a/bfl/fl mice carrying loxP inserts bracketing the entire STAT5 locus containing both a and b genes generated by Cui et al. (25) in C57BL/6 × 129J background mice was provided by Lothar Hennighausen (National Institutes of Health, Bethesda, MD, USA). Hennighausen and colleagues have previously produced hematopoietic- and endothelial-specific STAT5-null mice by mating these STAT5-floxed mice with Tie-2-Cre+/+ mice; however, such STAT5-null mice had hypochromic anemia and reduced postnatal survival (38). For the present experiments, STAT5a/bfl/fl mice were mated with the SM22-Cre+/+ mice expressing Cre re-combinase under the control of the mouse transgelin smooth muscle protein 22α promoter purchased from The Jackson Laboratory (Bar Harbor, ME, USA) [stock number 004746, Tg(Tagln-cre)1Her/J in C57BL/6:129SJL background] (37). Polymerase chain reaction analysis of the tail DNA using appropriate primers was used to determine the genotype (wt and floxed STAT5 alleles were from Cui et al. [25]; Cre alleles were from The Jackson Laboratory; primers are summarized in Supplementary Table S1). Examples of STAT5a/b wt or fl, and Cre-positive and -negative alleles are illustrated in Supplementary Figure S1. Cre-positive mice with the heterozygous STAT5a/bwt/fl or homozygous STAT5a/bfl/fl genotypes were viable and fertile and showed weight gain indistinguishable from the respective wt mice. Control mice with Cre-positive STAT5a/bwt/wt genotype used in the heterozygous deletion experiment were generated by crossing wt/wt C57BL/6 mice with SM22-Cre+/+ mice. Control mice with Cre-positive STAT5a/bwt/wt genotype used in the homozygous deletion experiment were littermates of the Cre-positive STAT5a/bfl/fl mice; both wt and homozygous knockout groups were offspring of the crossbreeding between STAT5a/bfl/wt, SM22-Cre+/− heterozygous mice; and thus, background matched completely.
Assessment of PH and Lung Vascular Remodeling in Mice after Chronic Hypoxia
Groups of Cre-positive male and female mice (wt/wt compared with wt/fl in the heterozygous deletion experiment [STAT5+/−] and wt/wt compared with fl/fl in the homozygous deletion experiment [STAT5−/−]) were used at approximately 8 wks of age in the respective chronic hypoxia experiments (n = 4–5 per group per variable). Mice were exposed to hypobaric hypoxia (0.5 atmospheres) for 7 wks; normoxia controls were left in ordinary room air (39,40).
The systemic blood pressure parameters of the respective groups of normoxic or hypoxic mice were measured during the last week of each experiment by using the tail-cuff method and a blood pressure monitor (Non-Invasive BP Monitor; Columbus Instruments, OH, USA). The systolic and diastolic blood pressures were determined using the Blood Pressure Monitor Software (NIBPE, version 1.23; Columbus Instruments).
Hemodynamic evaluation of the pulmonary circulation was carried out by using Millar catheterization beginning at the end of 7 wks in mice that were anesthetized by using the ketamine/xylazine cocktail (100 mg/kg ketamine, 10 mg/kg xylazine i.p.); three to four mice derived from different groups at random were evaluated per day until all evaluations were completed. A Millar catheter (SPR-839 for the experiment in Figure 3 or PVR-1030 for the experiment in Figure 4) was used for the measurement of heart rate and right ventricular systolic pressure (RVSP) (Millar Instruments, Houston, TX, USA). Numerical data for RVSP in hypoxic wt male mice were comparable to those previously reported by right ventricular (RV) catheterization (40). The animals were then euthanized, and the aorta and left lung were removed and formalin fixed (the right lung was snap frozen in liquid N2). The heart was dissected to obtain the ratio of right ventricle weight to that of the left ventricle plus septum (RV/LS+S) as a measurement of right ventricular hypertrophy (RVH). The left lung was formalin-fixed and paraffin-embedded for tissue sections (hematoxylin and eosin [H&E], van Giesson elastin and Masson trichrome stains, and quantitative immunofluorescence for SMA, STAT5a+b [C-17 pAb] or PY-STAT5 [Tyr-694-STAT5a+b] or without primary antibody). The extent of vascular remodeling in pulmonary arteries was quantitated by (a) medial wall thickness (as 2× wall thickness/outer vessel diameter in elastin-stained sections measured along two lines perpendicular to each other), (b) number of SMA-positive vessels (diameter <80 μm) per 10× field after immunofluorescence staining, and (c) extent of perivascular collagen deposition (blue after Masson trichrome staining) (39,40). Areal (two-dimensional) size of cells in the tunica media was evaluated by z-stack imaging on H&E-stained sections of arterial walls followed by three-dimensional reconstruction that then clearly displayed the intercellular boundaries.
Figure 3.

Conversion of the male-dominant PH response to chronic hypoxia in wt mice to a female-dominant response in heterozygous STAT5+/− mice. Respective groups (n = 5) of approximately 8-wk-old mice of either gender and indicated genotype were exposed to hypobaric hypoxia for 7 wks or kept in room air. At the conclusion of the experiment, various parameters of PH were assessed (RVSP in A, RV/LV+S weight in B, medial wall thickness [as % outer diameter] in C, number of SMA-positive vessels per 10× field in D, and extent of tunica adventitia/perivascular collagen deposition as evaluated by Trichrome staining [blue] expressed in AUs in E). Data in each chart summarize mean ± SE; *P < 0.05 using multiple-group ANOVA and pair-wise t test.
Figure 4.

Conversion of the male-dominant PH response to chronic hypoxia in wt mice to a female-dominant response in homozygous STAT5−/− mice. Respective groups (n = 4–5) of approximately 8-wk-old mice of either gender and indicated genotype were exposed to hypobaric hypoxia for 7 wks or kept in room air. At the conclusion of the experiment, various parameters of PH were assessed (RVSP in A, RV/LV+S weight in B, medial wall thickness [as % outer diameter] in C, number of SMA-positive vessels per 10× field in D, and extent of tunica adventitia/perivascular collagen deposition as evaluated by Trichrome staining [blue] expressed in AUs in E). Data in each chart summarize mean ± SE. *P < 0.05 using multiple-group ANOVA and pair-wise t test.
Immunofluorescence Analyses of Lung Sections from IPAH and HPAH Patients
Two separate sets of sections were used in the present study. The first set consisted of serial sections (5 μm) of formalin-fixed, paraffin-embedded lung tissue from patients with IPAH and from control individuals without PAH (n = 6 each, including males and females). These sections were provided by Rubin M Tuder (then at the Johns Hopkins University School of Medicine, Baltimore, MD, USA) without any personal identifiers in compliance with the guidelines of the Institutional Review Board at the Johns Hopkins University School of Medicine (the JHU set); these sections, as used in the present study, derive from our previous investigation of the Golgi tethers giantin and p115 in IPAH (42). Letter designations for individual patients and controls correspond to those in the article by Sehgal et al. (42). In Figure 6, control (Ctrl)-A and Ctrl-F were a 46-year-old female and a 25-year-old female, respectively; whereas IPAH-A, IPAH-B and IPAH-E were a 60-year-old male, a 59-year-old female and a 53-year-old female, respectively.
Figure 6.

Reduced STAT5 and ATL3 in obliterative lesions of human IPAH (JHU set). Representative formalin-fixed paraffin-embedded sections of lungs from human IPAH patients and controls (female: f; male: m) as previously reported in Sehgal et al. (42) (JHU set designations Ctrl-A, Ctrl-F, IPAH-A, IPAH-B and IPAH-E as in Sehgal et al. [42]) were immunostained for STAT5a (L-20 pAb) and for ATL3 (Abcam pAb). (A) STAT5a immunofluorescence; thick arrows point to normal pulmonary arteries in Ctrl-F; thin arrows point to obliterative lesions in IPAH patients. Scale bar = 45 μm. (B) Quantitation of average fluorescence pixel intensities for STAT5a in Ctrl vessels and IPAH lesions (n); mean ± SE. *P < 0.05 (t test). (C) Negative control using irrelevant primary pAb. (D) Double-label imaging validating that the obliterative lesions with reduced STAT5a represent cells that are SMA-positive. Scale bar = 40 μm. (E) ATL3 immunofluorescence; thick arrows point to normal pulmonary arteries in Ctrl-A with greatest fluorescence in the endothelium; thin arrows point to different obliterative lesions in an IPAH patient. Internal and external elastic laminas in this analysis were visualized by autofluorescence in green. Scale bar = 45 μm. (F) Quantitation of average fluorescence pixel intensities for ATL3 in Ctrl vessels and IPAH lesions (n); mean ± SE. *P < 0.05 (t test).
A second set of sections was provided by the Tissue Processing Core at the University of Colorado, Aurora, and the University of Alabama, Birmingham, through the auspices of the Pulmonary Hypertension Breakthrough Initiative (PHBI) and it consisted of serial sections of lung samples of male and female patients with IPAH and HPAH (total n = 16) and male and female controls (total n = 12), as enumerated in Supplementary Table S2 (the PHBI set). These were provided without any personal identifiers in compliance with the guidelines of the Institutional Review Board at the New York Medical College. There was no explicit attempt to include or exclude pediatric controls or patients. These human lung sections were evaluated by using H&E staining as well as indirect immunofluorescence for several proteins. Data collection from lung sections was completed before obtaining information from the PHBI about age, pulmonary artery (PA) pressures, pulmonary vascular resistance, functional status and treatment history of the respective patients. Thus, the conduct of this study was blind with respect to these parameters.
Immunofluorescence studies using these human lung sections were carried out for STAT5a (using the L-20 pAb), STAT5a+b (using the C-17 pAb), PY-STAT5, ATL3 and RTN4/Nogo-B, as described earlier (42,43) by using respective rabbit primary antibodies and donkey anti-rabbit Alexa Fluor 594–tagged pAbs (in red) as the secondary antibody. Controls included slides exposed to irrelevant primary pAbs and also double-label analysis by using anti-SMA in green (using Alexa Fluor 488–tagged pAbs as secondary). The STAT5b-specific mAb (G2) was not reactive on archived human lung sections.
Imaging was carried out by using a Zeiss AxioImager M2 motorized microscopy system equipped with a high-resolution RGB HRc AxioCam camera and AxioVision 4.8.1 software in a 1,388 × 1,040 pixel high-speed color capture mode (42–44). All data within each experiment were collected at identical imaging settings. Fluorescence intensity was quantitated by using the NIH ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/) with, typically, six representative areas per vessel wall or lesion image expressed as mean pixel intensity. Background subtraction within each image was derived from the bronchial airspace. Fluorescence intensities derived from individual images after background subtraction were normalized in terms of the strongest signal in each image (typically from red blood cells or bronchial epithelium), which was taken as 1.0.
Assessment of Proteins in Smooth Muscle Cells and Endothelial Cells Derived from Lungs of IPAH Patients and Controls
Early passage (up to P4) smooth muscle cell (SMC) and endothelial cell (EC) cultures derived from vessels in peripheral lung areas of IPAH patients (total n = 12 SMC lines; n = 5 EC lines) and controls (n = 9 SMC lines; n = 6 EC lines) of both sexes, as enumerated in Supplementary Table S2, were provided without any personal identifiers by the Cell Culture Core of the PHBI at the University of Pennsylvania School of Medicine. These cultures were provided in compliance with the guidelines of the Institutional Review Board at the New York Medical College. Number designations for individual patients correspond to those provided by the Cell Core Facility of the PHBI (Supplementary Table S2). As derived by PHBI, the SMC lines were >95% SMA-positive and the EC lines were >95% von Willebrand factor (vWF)-positive. The cells were grown for two passages at the New York Medical College by using the conditions recommended by the PHBI, and suitable cultures (in T-25 flasks or six-well plates) were used to prepare whole cell extracts for Western blot analyses or fixed (in cold 4% paraformaldehyde for 1 h), permeabilized (using digitonin at 50 μg/mL for 5 min) and used for immunofluorescence morphometry (41,45,46). Western blot data were normalized by using RTN4 as baseline and are expressed in arbitrary units (AUs).
Statistical Evaluations
These evaluations were performed using the two-tailed Student t test and single-factor analysis of variance (ANOVA) methods with (NCSS version 8, 2012; NCSS LLC, Kaysville, UT, USA). Multiple group comparisons were carried out by using ANOVA (Tukey-Kramer, Newman-Keuls and Tukey-Kramer tests). Cluster evaluations in the cell-culture hypertrophy experiments were carried out by using Hotelling’s T2 Report and k-means cluster analyses (NCSS version 8, 2012).
All supplementary materials are available online at www.molmed.org.
RESULTS
Development of Mice with Deletion of STAT5a/b in Vascular SMCs
Mouse lines with heterozygous and homozygous deletion of STAT5a/b in SMCs were generated by crossbreeding STAT5a/bfl/fl mice (13,15) with SM22α-Cre+/+ transgenic mice (see Materials and Methods, Supplementary Figure S1 and Supplementary Table S1) (37). Such mice were viable and fertile and showed weight gain commensurate with that in the wt mice. Verification of the deletion of STAT5a/b in arterial SMCs in the knockout mice was first carried out by characterizing STAT5a/b expression in the tunica media of the aorta. Both heterozygous (Supplementary Figures S2A, S2B) and homozygous (Supplementary Figures S3A, S3B) knockouts showed reduced STAT5 immunofluorescence in the aortic tunica media. Similarly, there was a marked reduction in transcripts for STAT5a and STAT5b in RNA isolated from the aorta of homozygous knockout mice (Supplementary Figure S3C and Table S3), and a marked reduction in STAT5 species by Western blotting in extracts of SMCs isolated from the aorta of homozygous knockout mice (Supplementary Figure S3D) as well as in extracts of aortas of heterozygous knockout mice (not shown). Despite this loss of STAT5 in the systemic arterial tree, neither of the homozygous nor heterozygous knockout mice of either sex showed a significant change in resting systolic, diastolic or mean blood pressure using the tail-cuff assay (Supplementary Figures S3E, S3F).
The consequences of the SM22α-Cre driven heterozygous or homozygous deletions of the STAT5a/b locus on STAT5 protein expression in the tunica media in the pulmonary arterial tree were evaluated using immunofluorescence methods (Figure 1 and Supplementary Figure S2C). A marked reduction in STAT5 immunofluorescence was observed in the tunica media of PA segments after the heterozygous (Supplementary Figure S2C) and homozygous deletions (Figures 1A, B).
Figure 1.

Tissue-specific reduction in STAT5a/b expression in the tunica media of segments of the main PA (A) as well as in a muscular (SMA-positive) PA segment within a lung section (B) in STAT5−/− mice. Immunofluorescence studies were carried out using the C-17 rabbit pAb (for STAT5a+b); black arrows point to the tunica media. Scale bars = 10 μm. The right side of each panel shows quantitation of STAT5 immunofluorescence over the tunica media normalized with respect to that over the tunica intima (A) or in terms of that observed in leukocytes in the same slide (B) (n = 6 images valuated per group; mean ± standard error (SE); *P < 0.05 (t test).
As has been previously established in other tissue and cell-type contexts (23, 47–49), there was little or no sex bias in the baseline expression of STAT5a/b proteins in aortic extracts derived from individual male or female wt mice (Figure 2A) or in extracts prepared from SMCs cultured from pools of aorta (n = 5/pool) derived from wt male or female mice (Figure 2B). However, SMC extracts derived from STAT5−/− mice were confirmed to show diminished STAT5a/b (Figure 2B). Similarly, there was little difference in STAT5 expression in the tunica media of PA segments in lung sections between wt male versus female mice as judged using quantitative immunofluorescence analyses (Figures 2C–E).
Figure 2.
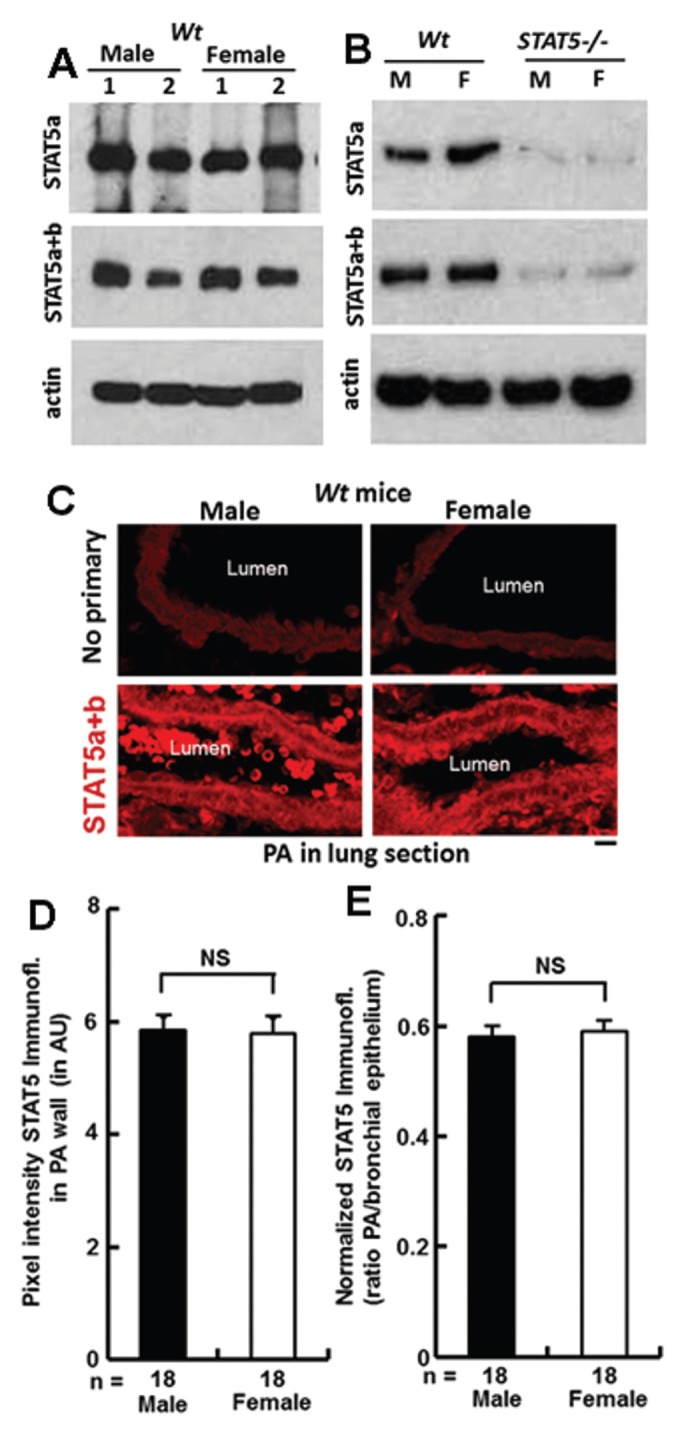
Expression of STAT5 species in different vascular tissues of male and female wt and STAT5−/− mice. (A) Aortic extracts from individual duplicate male and female wt mice were Western blotted for STAT5a (using L-20 pAb), STAT5a+b (using C-17 pAb) or β-actin. B: SMCs were cultured from aortic segments of pools of mice (n = 5 each) of the indicated genotype and sex. The cell extracts were then evaluated by Western blotting for STAT5 species as in A. (C–E) Quantitative evaluation of anti-STAT5a/b immunofluores-cence (C-17 pAb) in PA segments in lung sections from male and female wt mice (n = 2 mice per group with a total of 18 arterial segments quantitated) in two different ways: average pixel intensity over arterial walls after subtracting background in each individual slide over the bronchial air space (D) and normalized fluorescence intensity over arterial walls expressed as a ratio to that over the bronchial epithelium within the same individual slide (E). n = arterial segments in 2 different mice quantitated per group; mean ± SE; NS, not significant (t test).
Abrogation and Reversal of the Male-Dominant Sex Bias in PH Development in Hypoxic Heterozygous and Homozygous STAT5a/b SMC-Specific Knockout Mice
Groups (n = 5) of male and female wt or STAT5+/− mice were left in room air (normoxia) or exposed to chronic hypoxia for 7 wks and then evaluated for RVSP, RVH and lung vascular remodeling (Figure 3 and Supplementary Figure S4). There was little difference in baseline RVSP in groups of male or female wt or STAT5+/− mice kept under normoxia (Figure 3A). However, after exposure to hypoxia, male wt mice developed increased RVSP and RVH as well as pulmonary arterial remodeling with increased wall thickness. As expected, female wt hypoxic mice showed less hemodynamic and histologic changes of PH. Strikingly, female STAT5+/− mice developed PH and pulmonary arterial remodeling dramatically more severe than that in hypoxic wt females (Figures 3A–D and Supplementary Figure S4). Even among STAT5+/− mice, females showed more severe changes than males. Histologically, increased pulmonary arterial wall thickness in female hypoxic STAT5+/− mice included increased collagen deposition in the tunica adventitia/perivascular region (Figure 3E and Supplementary Figure S4B).
The abrogation of the male dominance in the development of hypoxic PH was observed to an even greater extent in homozygous STAT5−/− knockout mice (Figure 4 and Supplementary Figure S5). The data from this experiment again verified that development of hypoxic PH in wt mice displayed a male-dominant phenotype with respect to every parameter investigated (Figure 4 and Supplementary Figure S5). However, after STAT5−/− knockout, female mice developed the severest increases in RVSP and RVH, and arterial remodeling—even more than that observed in the male knockouts. Moreover, even among the male mice, the STAT5−/−males showed more severe disease than wt males (Figures 4A, C, D). Thus, both male and female STAT5−/− homozygous knockout mice had more severe disease than their wt counterparts, with female knockouts showing the severest disease of all. Histologically, the increased pulmonary arterial wall thickness in hypoxic female STAT5−/− mice was accompanied by increased collagen deposition in the tunica adventitia/perivascular region (Figure 4E and Supplementary Figure S5B).
At the cellular level, STAT5−/− knockout by itself did not lead to cell hypertrophy in the tunica media either in the male or female mouse (Figures 5A–C). After hypoxia, SMC hypertrophy was evident in male wt mice but not in the female (Figures 5A, C). In contrast, marked SMC hypertrophy was evident in the tunica media of both male and female STAT5−/− knockout mice after hypoxia (Figures 5A–C). The z-stack imaging and three-dimensional reconstruction helped in defining the intercellular boundaries between SMCs in the tunica media (two examples are shown in Figure 5B). Quantitation of the cellular hypertrophy in the tunica media of arterial segments in lungs of mice in the respective control and experimental groups is summarized in Figure 5C. Overall, SMC hypertrophy was a key cellular event in the pathogenesis of PH in this mouse model.
Figure 5.

SMC hypertrophy in the tunica media of pulmonary arterial segments of hypoxic STAT5−/− knockout mice. (A) H&E staining of lung sections of male and female wt normoxic and hypoxic mice and in male and female STAT5−/− normoxic and hypoxic mice from the experiment in Figure 4. Scale bars = 10 μm. (B) Three-dimensional representation of z-stack images of two illustrative examples of pulmonary arterial walls in the experiment summarized in A. (C) Quantitation of the areal size of cells (mean ± SE) in the tunica media of arterial walls (this is an SMA-positive cell type; data not shown) in the respective groups in A using the method in B; n > 200 cells derived from three mice from each group were enumerated; *P < 0.05 using multiple-group ANOVA and pair-wise t test.
Implications in Human PAH
The relevance of the preceding data showing development of marked PH in hypoxic mice with SMC-specific conditional deletion of STAT5a/b to the pathogenesis of PAH in humans was then investigated. It was already striking that vascular cell hypertrophy was a key observation in the arterial walls of hypoxic STAT5−/− knockout mice of both sexes (Figures 5A–C) as well as in the obliterative onion-skin lesions in IPAH (Figure 1 in Sehgal et al. [42]; Figure 15 in Lee et al. [45]). We have previously shown that acute siRNA-mediated knockdown of STAT5a/b in pulmonary vascular cells led to the development of a unique cystic endoplasmic reticulum (ER) and dilated and fragmented Golgi apparatus phenotype with deposition of RTN4 (an ER structural protein) and ATL3 (an ER/Golgi resident GTPase) at the cystzone boundaries, with one to three large juxta-nuclear cysts comprising dilation of the ER space between the inner and outer nuclear membranes resulting in a lunate distortion of the nuclei (45,46). Functionally, there was reduction of anterograde of member proteins including BMPR2 trafficking) (41). Thus, we investigated the expression of STAT5a/b, ATL3 and RTN4 in obliterative arterial lesions in human IPAH and HPAH by immunofluorescence analyses (42,43), as well as the presence of hypertrophic cells with enlarged ER and lunate distortion of nuclei (45,46) in such lesions (Figures 6, 7).
Figure 7.

Summary of data showing reduced STAT5 and ATL3 in obliterative vascular lesions in male and female IPAH and HPAH patients (PHBI set). (A) H&E staining of a PA segment in a male control (#6) and an obliterative lesion in a male patient with HPAH (#10) provided by PHBI (Supplementary Table S2). Scale bar = 30 μm. Insets show boxed areas at higher magnification. (B) Illustrative immunofluorescence analyses for STAT5a (L-20 pAb), STAT5a+b (C-17 pAb), PY-STAT5, ATL3 and RTN4/Nogo-B in PA segments from the same control male patient (top row; white arrows) and obliterative vascular lesion from the same HPAH patient (bottom row; white arrows) in A. Scale bar = 50 μm. (C) Quantitation of STAT5 immunofluorescence in tunica media of multiple arterial segments or obliterative vascular lesions in lung sections from control or PAH patients expressed in normalized arbitrary units (mean ± SE). Imaging data were collected from an average of five vessels/lesions per patient, and fluorescence intensities over vessels/lesions were expressed as a fraction of the highest intensity observed within each image (in epithelial cells or red blood cells). Patient number corresponds to the lab ID number listed in Supplementary Table S2; dashed line = overall group average; *P < 0.001 (t test). (D, E) Quantitation of PY-STAT5 and ATL3 immunofluorescence in normalized arbitrary fluorescence units in tunica media of multiple arterial segments or obliterative vascular lesions in lung sections from a subset of patients in B (n = average of 5 vessels/lesions per patient; mean ± SE); *P < 0.001 (t test).
Figures 6A and B illustrate a loss of STAT5a in the obliterative lesions in male and female IPAH patients (JHU set). Figure 6C shows an irrelevant antibody control, and Figure 6D shows a double-label analysis verifying that the obliterative lesions shown in Figure 6A contained SMA-positive cells. Figures 6E and F show a dramatic loss of ATL3 in illustrative obliterative lesions in an IPAH patient and the quantitation of this reduction.
These studies were extended to lung sections of sex-segregated patients with HPAH, IPAH and controls in the PHBI set (Supplementary Table S2) by using two different anti-STAT5 pAbs (STAT5a-specific L-20 and STAT5a+b-reactive C-17) as well as for PY-STAT5, ATL3 and RTN4. To begin with, evaluation of these sections using H&E staining readily showed the presence of hypertrophic cells with juxta-nuclear cysts and lunate distortion of the nucleus, reminiscent of the cell phenotype produced by acute siRNA-mediated knockdown of STAT5a/b. Figure 15 in Lee et al. (45) had shown an example of such cells in obliterative lesions of a female patient with IPAH; the present Figure 7A illustrates an example of cells with this phenotype in lesions in a male patient with HPAH. Serial sections from the set corresponding to the control and HPAH patient shown in Figure 7A, as well as from additional male and female IPAH and HPAH patients and controls, were then probed for STAT5a, STAT5a+b, PY-STAT5, ATL3 and RTN4 (Figures 7B–E). In the immunofluorescence data shown in Figure 7B, there was reduced STAT5, PY-STAT5 and ATL3 but increased RTN4 expression. Quantitative analyses for STAT5 expression in arterial tunica media of male and female controls, as well as in obliterative vascular lesions in male and female PAH patients in the PHBI set, showed a marked reduction of STAT5 in PAH patients of both sexes (Figure 7C and Supplementary Table S2). Whereas the individual STAT5 expression levels summarized in Figure 7C reveal a patient-specific heterogeneity in the extent of this reduction, overall, groups of male and female PAH patients both showed reduced STAT5 in the pulmonary vascular obliterative lesions. Tissue sections from a subset of the PAH patients were evaluated for PY-STAT5 and ATL3 (Figures 7D, E). The data obtained showed the reduction of PY-STAT5 and ATL3 in cells in obliterative vascular lesions in both male and female PAH patients.
Reduced STAT5 Correlates with Cell Hypertrophy in IPAH-Derived SMC Lines
The above immunofluorescence data derived from lung sections of PAH patients showing reductions in STAT5a, STAT5a+b and ATL3 in the same obliterative lesion raised the question whether cells derived from lung vessels of IPAH patients might also show coordinate reductions in these molecules. A related question derived from the H&E imaging data shown in Figure 7A was whether vascular cells derived from PAH patients were hypertrophic and to what extent. Thus, cell imaging studies and Western blot analyses of the primary cell lines provided by the PHBI (Supplementary Table S2) were carried out. For these studies each cell line was grown in triplicate wells of a six-well plate for imaging and simultaneously in a T-25 flask for preparing cell extracts. Eventually, 9 control SMC and 10 IPAH SMC lines were evaluable by phase contrast microscopy. By this inspection, all 9 control SMC lines consisted of small cells, whereas 4 of 11 IPAH SMC lines consisted of similar small cells. The remaining 7 of 11 IPAH SMC lines consisted of enlarged hypertrophic cells (see below). The EC lines did not show such overt size differences. In contrast to evidence of SMC hypertrophy in IPAH-derived primary cell lines, little size difference was evident among the control and IPAH-derived EC lines (data not shown).
Figure 8A is a compilation of one set of three controls and three IPAH-derived female SMC lines corresponding to the Western blot analysis of these cell extracts in Figure 8B. Morphometry data pertaining to cell size and Golgi apparatus size (and STAT5a levels by Western blots) for 10 such SMC cell lines (5 control and 5 IPAH-derived) are enumerated in Table 1. Overall, SMCs from the IPAH-derived group included lines that were hypertrophic when compared with those derived from controls. It is striking that these hypertrophic IPAH-derived SMCs also showed enlargement of the Golgi apparatus (Figure 8A), confirming our previous data derived from analyses of the Golgi size in cells in lung sections of control and IPAH patients (42).
Figure 8.

Relationship between cellular hypertrophy of IPAH-derived SMCs and STAT5 expression. (A) Representative SMC cell lines derived from three control and three IPAH patients were probed by triple-label immunofluorescence imaging for STAT5a, a Golgi marker (GM130 or Vti1a) and DAPI. The present study was blinded with respect to the age of the patients (indicated in parentheses). Scale bar = 20 μm; the same scale bar applies to control and IPAH images. The paraformaldehyde-fixed cells were permeabilized using digitonin; thus, the anti-STAT5a pAb does not penetrate into the nucleus (24). (B) Composites showing Western blot analyses of equal protein aliquots derived from extracts of cultures of the same SMC lines as in A and additional EC lines (Supplementary Table S2). Blots were probed for STAT5a (L-20 pAb), STAT5b (G-2 mAb), the ER-resident GTPase ATL3, the Golgi SNARE Vti1a, the ER structural protein RTN4 (Nogo-B) and β-actin. Boxed areas in B highlight individual examples with the triple loss of STAT5a, STAT5b and ATL3 (all of which were derived from IPAH patients). (C, D) Investigator-independent morphometric cluster analyses of cells in 10 SMC cell lines derived from control (n = 5; closed diamonds) and IPAH lungs (n = 5; open squares) for cell size (mean ± SE), for Golgi apparatus size (mean ± SE) and for STAT5a expression enumerated in Table 1; error bars not visible correspond to small numerical values, which are all tabulated in Table 1. The k-means cluster analysis optimized two clusters (the open squares cluster, which contained all the IPAH lines, and the closed diamonds cluster, which contained all the control lines). Statistical evaluation of distributions of IPAH (open squares) and Ctrl (closed diamonds) clusters in two-way and three-way comparisons using k-means cluster analyses gave P < 0.001 in all intercluster comparisons.
Table 1.
Morphometry of cells in control- and IPAH-derived SMC cell lines.
| Patient | SMC number | Cell size (μm2)a | Golgi size (μm2)a | STAT5a AUsb |
|---|---|---|---|---|
| Control | 54 | 423 ± 16 (84) | 32 ± 8 (47) | 0.95 |
| Control | 98 | 741 ± 42 (55) | 52 ± 4 (60) | 0.41 |
| Control | 102 | 648 ± 72 (24) | 107 ± 40 (72) | 0.90 |
| Control | 103 | 870 ± 71 (24) | 173 ± 42 (72) | 0.55 |
| Control | 104 | 629 ± 72 (22) | 55 ± 3 (121) | 0.46 |
| IPAH | 25 | 4,424 ± 360 (73) | 406 ± 94 (23) | 0.06 |
| IPAH | 36 | 13,040 ± 1,808 (18) | 1,150 ± 123 (16) | 0.17 |
| IPAH | 66 | 4,889 ± 773 (27) | 553 ± 75 (28) | 0.21 |
| IPAH | 74 | 8,860 ± 1,326 (11) | 566 ± 147 (16) | 0.17 |
| IPAH | 105 | 6,360 ± 1,327 (18) | 361 ± 89 (25) | 0.28 |
| All combined | ||||
| Control, mean ± SE | 662 ± 82 | 84 ± 29 | 0.65 ± 0.13 | |
| IPAH, mean ± SE | 7,515 ± 1,770 | 607 ± 158 | 0.18 ± 0.04 | |
| P | 0.0025 | 0.0066 | 0.0036 |
Data are mean ± SE (number of cells enumerated).
In normalized arbitrary units by Western blotting of cell extracts. Statistical testing used k-means cluster analysis.
An inspection of the Western blot shown in Figure 8B illustrates that as with the immunofluorescence data derived from IPAH lung sections (Figure 6), there were IPAH-derived SMC lines that showed reduced expression of STAT5a, STAT5b and ATL3. Moreover, reduced STAT5a/b and reduced ATL3 were also observed in both SMC and EC lines derived from the same IPAH patients (patients 36 and 105). Thus, there is a similarity (Figures 6, 7) in data derived from lung sections and from evaluation of cell lines (Figure 8).
The relationships between STAT5a content (by Westerns), cell hypertrophy and Golgi apparatus enlargement were tested in a subset of 10 SMC cell lines for which data regarding STAT5 protein levels as well as cell and organellar morphometry were evaluated (n = 5 controls and n = 5 IPAH-derived; Table 1). Investigator-independent cluster analyses were carried out by using all of the Western blot quantitation, cell size and Golgi apparatus size measurements for the 10 cell lines by using Hotelling’s T2 Report procedure incorporating two parameters at a time (NCSS version 8, 2012). With each pair of parameters, this process optimized two clusters: one cluster (open squares) that contained all the IPAH lines and another (solid diamonds) that contained all the control lines (Figures 8C, D). Statistical evaluation of distributions of IPAH and Ctrl clusters in two- and three-way comparisons using k-means cluster analyses gave P < 0.001 in all intercluster comparisons. Thus, low STAT5 expression clustered with cell hypertrophy and enlarged the Golgi apparatus in IPAH-derived SMC lines in a multiparameter pattern.
DISCUSSION
To focus first on the mouse data, the hypothesis that STAT5a/b may play a causal role in the pathogenesis of PH was tested in the hypoxic mouse model. The observations that heterozygous or homozygous deletions of the STAT5a/b locus in SMCs resulted in female mice that developed the severest PAH in response to chronic hypoxia abrogating the otherwise male-dominant phenotype in this model confirm a role for STAT5 in the pathogenesis of PH. This is the first time that STAT5 species have been definitively implicated in sex-dependent effects in any aspect of vascular biology. A role of STAT5 in sex-dependent and sex-independent mechanisms in the pathogenesis of hypoxic-PH was suggested by the observation that although the female STAT5−/− knockout mice developed the severest disease in response to chronic hypoxia, even the male knockout mice had more severe disease than the wt males (Figure 4) and displayed markedly increased SMC hypertrophy, but only in combination with hypoxia (Figure 5). STAT5−/− knockout by itself did not lead to SMC hypertrophy in the mouse pulmonary arterial wall (Figure 5).
To focus next on studies characterizing STAT5 expression in human disease–derived lung sections, there was reduced STAT5a/b, reduced PY-STAT5 and reduced ATL3 in cells in obliterative lesions in the majority of the human IPAH and HPAH patients evaluated, both male and female. While there were clear variations between individual patients (Figure 7), and even in lesions in the same patient (not shown), the overall trend was a reduction of STAT5a/b, PY-STAT5 and ATL3 in PAH in both sexes. A limitation in interpreting these data is that these lung tissue sections represent late-stage disease. The same limitation applies to the SMC and EC lines derived from patient lungs, with the additional limitation that cells in a derived line may not represent solely those that originate within the vascular lesion but also include nearby normal cells. Nevertheless, the discovery of a pattern of reduced STAT5a, STAT5b and ATL3 in IPAH-derived SMC and EC lines (in two instances the two cell types isolated from the same patient), as well as in lesions in lung sections, suggests an underlying biological significance. The clustering of low STAT5 expressing IPAH-derived lines into a hypertrophic cluster, in comparison to SMC lines derived from controls, is reminiscent of the marked SMC hypertrophy in PA observed in male and female hypoxic STAT5−/− mice. The biological significance of this clustering is further enhanced by our previous observation that mouse embryo fibroblasts derived global STAT5a/b−/− mice displayed transient cell hypertrophy, marked Golgi enlargement and fragmentation as well as a cystic ER change for 1–2 d after plating in cell culture (see Figures 11 and 12 in Lee et al. [45]). The mechanisms that lead to this cellular hypertrophy and accompanying altered organellar and growth properties remain to be elucidated.
Our focus on STAT5 in the pathogenesis of PAH (a schematic is shown in Figure 9) arose from the recognition in the last 3–4 years (41,43–46) that the subcellular phenotype produced by siRNA-mediated knockdown of STAT5a/b in human pulmonary ECs and SMCs (cystic ER/fragmented Golgi apparatus/lunate distortion of the nucleus) was similar to that reported in 1977 and 1979 by Smith and Heath in endothelial cells in the rat/hypoxia model and in cells in plexiform lesions in IPAH based on their electron micrographic studies (50,51). In a pilot study (45), we observed that cells in obliterative lesions in IPAH showed a loss of STAT5a accompanied by the cystic ER/Golgi enlargement/lunate nucleus and ER stress phenotype. These data led us to suggest that loss of STAT5a/b might contribute causally to the pathogenesis of PAH, through genomic and novel nongenomic mechanisms (for example, inhibition of BMPR2 trafficking to the plasma membrane and its signaling) discovered by us (41,43–46) (Figure 9). The present study represents a direct test of this hypothesis by using conditional STAT5a/b+/−and STAT5a/b−/− knockout mice.
Figure 9.

The STAT5a/b transcription factor hub in pulmonary hypertension.
Of the seven STAT transcription factors (STAT1, 2, 3, 4, 5a, 5b and 6), only STAT5a and STAT5b mediate sexual dimorphism (23,24,26,28). STAT5a/b are activated through Tyr- and/or Ser-phosphorylation by cytokines, growth factors, GH and prolactin and modulated by E2 (Figure 9). The work of Waxman and colleagues over the last decade has shown that mechanisms of sexual dimorphism converging on STAT5a and STAT5b (in mice) originate mainly from the sex-dependent dynamic patterns of release of GH from the pituitary, either in a pulsatile (male) or continuous (female) manner leading to pulsatile or continuous patterns of activation of STAT5 species (by Tyr-phosphorylation) (13–28,36) (Figure 9). This patterned low-level activation of PY-STAT5 species by GH in different tissues leads to the differential expression of 400–500 distinct genes in a male- or female-specific phenotype (Figure 9). As a baseline issue in our studies, vascular cells and tissues from male and female mice expressed similar levels of unphosphorylated STAT5a/b (U-STAT5a/b; Figure 2). Waxman and colleagues have emphasized that it is the quantitative differences in the dynamic GH-driven patterns of PY-STAT5 activation that determine the sex bias in downstream gene expression in their mouse studies (23,24). That both STAT5a/b+/− and STAT5a/b−/− knockout mice showed reversal of the sex bias in hypoxia-induced PH and its increased severity is consistent with the concept that small quantitative changes in the extent and pattern of STAT5a/b activation suffice to determine the underlying sex bias. The relationship between the extent of STAT5 reduction and disease severity remains an important open question.
In the context of PH in mouse and humans, STAT5a/b are affected by multiple signaling and miRNA pathways and, in turn, regulate other transcription factors, cell-cycle regulators and miRNAs already known to be important in the pathogenesis of this disease (Figure 9). The new observations from the present studies reflect an overlay of sex bias over these mechanisms driven, we suggest, by dynamic GH patterns in the male and female, quantitatively different between mouse and humans (Figure 9). Irrespective of the upstream mechanisms that regulate levels of STAT5a/b expression in cells (such as by regulating its transcriptional expression, mRNA degradation through miR-222 or proteasomal degradation; Figure 9), the downstream sex-bias mechanisms already identified by Waxman and colleagues include transcription factor hubs such as CUX2 and Bcl6 regulated by dynamic (pulsatile or continuous) activation of PY-STAT5a/b by GH in a sex-specific manner (17,19,37). In turn, these transcription factor hubs affect hundreds of target genes (including cytokines, BMPR2 and STAT5a expression and Smad signaling) and cell-cycle checkpoint genes and affect or are themselves affected by micro-RNAs already implicated in PAH (for example, miR-155 and miR-21) (29,33,34,52–56) (Figure 9). The new mouse model developed by us provides a novel technology platform for elucidating the molecular basis for how loss of STAT5 promotes pulmonary arterial SMC hypertrophy and increased PH. Sex-specific modulation of Bcl6/cytokines secondary to STAT5a/b loss represent attractive downstream candidate molecular mechanisms (17,19,32,52–56).
Most investigators thus far have considered mechanisms of sex bias in PH in rodent models (male predominant) and humans (female predominant) mainly in terms of differences due to sex hormones (1–8). The present study highlights a complementary mechanism, largely independent of steroid hormones, contributing to this sex bias in terms of male or female patterns of activation of PY-STAT5 by circulating GH. Whether exogenous estrogens might be protective in the hypoxic STAT5−/− male or female knockout mouse model of PH remains an open question.
In cystic fibrosis, there is a well-established “gender gap” such that women get the disease earlier and in more severe form (57,58). However, unlike the protective effect of E2 in rodent models of PH (22), exogenously administered E2 exacerbated the disease in the male cystic fibrosis knockout mouse accompanied by elevated Th17-associated proinflammatory mediators (59).
The combination of hypoxia with the inhibitor SU5416 has been used to derive a rodent model of PH that develops extensive pulmonary arterial remodeling (60,61). The inclusion of SU5416 in this model has customarily been presented as the addition of a “VEGF receptor 2 antagonist” (4,60,61). Intriguingly, in a recent review, Lahm et al. (4) point to their data in this model, suggesting little or no sex bias. We note that it is known that hypoxia activates PY-STAT5 (62,63) and that SU5416 also inhibits activation of PY-STAT5 (64,65). This result raises the possibility that an absence of sex bias in the hypoxia + SU5416 model may derive from the already known ability of SU5416 to inhibit PY-STAT5 activation (64,65). Thus, the SU5416-treated hypoxic rodent would be analogous to the STAT5−/− knockout hypoxic mouse. Additionally, polycythemia has been observed in hypoxic mice, and erythropoietin (Epo) and HIF-2 isoforms have been shown to participate in this process (66,67). Because it is also known that Epo signals through STAT5 in erythroid lineage cells (28) and that polycythemia can affect SMC reactivity by affecting NO bioavailability (68), future studies of hypoxia-induced polycythemia in our conditional STAT5a/b−/− mice might prove insightful.
In studies of human IPAH- and HPAH-derived lung sections, while there was patient-specific variation, these tissue materials showed reduced nonphosphorylated STAT5a/b and reduced PY-STAT5 in obliterative lesions in both male and female patients, together with reduced ATL3. Overall, this triple-loss phenotype was present in human vascular lesions equivalently in both groups of male and female patients. The absence of an evident sex bias in the loss of STAT5a/b may reflect a limitation of studies by using tissue sections from patients with late-stage disease (as available from PHBI) or may represent the intrinsic complexity and heterogeneity of this disease. A reduction in PY-STAT5 in both males and females would suggest similar downstream changes (Figure 9) in both sexes, at least in late-stage disease. Intriguingly, it has been estimated that dasatanib, a tyrosine kinase inhibitor also known to inhibit STAT5 signaling and downregulate STAT5 target gene expression (69), led to development of PH in approximately 0.45% of leukemia patients receiving this medication, with the identified cases occurring primarily in women (70).
CONCLUSION
To summarize, we developed a novel female-dominant mouse model of PH that can be used to understand the molecular pathogenesis of this disease. The mice with a smooth muscle–specific deletion of STAT5a/b converted the male-dominant hypoxia-induced PAH to a female-dominant phenotype. Although the knockout females developed the severest disease, disease severity was also higher in the knockout males compared with wt males. These data provide the first definitive evidence for the causal involvement of STAT5 in the pathogenesis of PAH. The data also draw attention to an alternative novel mechanism to understand the sex bias seen in human and rodent PH based on the GH-STAT5 sex-bias axis, with differences between humans and rodents suggested to arise from quantitative and circadian differences in circulating GH patterns in males and females in the respective species. We also provide evidence for a loss of STAT5 in cells in oblit-erative pulmonary vascular lesions in human IPAH and HPAH, in both men and women in late-stage disease. Candidate downstream molecular mechanisms leading to PAH pathogenesis may include disruption of transcription factor hubs regulated by STAT5/Bcl6 that affect cytokine biology, cell cycle progression and transition into mitosis. Thus, the data suggest new molecular targets that could be manipulated to modify the pathogenic cause of this devastating condition.
Supplemental Data
ACKNOWLEDGMENTS
We thank Lothar Hennighausen (National Institute of Diabetes, Digestive and Kidney Diseases, Bethesda, MD, USA) for a breeder pair of the STAT5a/bfl/fl mice and Rubin M Tuder (University of Colorado-Denver, Aurora, CO, USA) for the human lung sections (controls and IPAH). Additional lung sections, SMCs and ECs derived from lungs of male and female IPAH patients and controls were provided under the PHBI from the Baylor College of Medicine, The Cleveland Clinic Foundation, University of Alabama at Birmingham, Stanford University, Vanderbilt University, University of California at San Diego, Duke University, University of Michigan, University of Colorado–Denver, Allegheny General Hospital, and University of Pennsylvania. Funding for PHBI is provided by the Cardiovascular Medical Research and Education Fund (CMREF). This work was supported in part by National Heart, Lung, and Blood Institute Grants HL-087176 (to PB Sehgal), HL-114509 (to PB Sehgal) and HL-111469 (EJ Miller).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Austin ED, et al. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009;34:1093–9. doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin ED, et al. Gender, sex hormones and pulmonary hypertension. Pulm Circ. 2013;3:294–314. doi: 10.4103/2045-8932.114756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dempsie Y, MacLean MR. The influence of gender on the development of pulmonary arterial hypertension. Exp Physiol. 2013;98:1257–61. doi: 10.1113/expphysiol.2012.069120. [DOI] [PubMed] [Google Scholar]
- 4.Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307:L7–26. doi: 10.1152/ajplung.00337.2013. [DOI] [PubMed] [Google Scholar]
- 5.Umar S, Rabinovitch M, Eghbali M. Estrogen paradox in pulmonary hypertension: current controversies and future perspectives. Am J Respir Crit Care Med. 2012;186:125–31. doi: 10.1164/rccm.201201-0058PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm Circ. 2011;1:305–19. doi: 10.4103/2045-8932.87293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White K, et al. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res. 2011;90:373–82. doi: 10.1093/cvr/cvq408. [DOI] [PubMed] [Google Scholar]
- 8.White K, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–98. doi: 10.1161/CIRCULATIONAHA.111.062927. [DOI] [PubMed] [Google Scholar]
- 9.Rabinovitch M, Gamble WJ, Miettingen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and recovery. Am J Physiol Heart Circ Physiol. 1981;240:H62–72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- 10.Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol. 2001;280:L88–97. doi: 10.1152/ajplung.2001.280.1.L88. [DOI] [PubMed] [Google Scholar]
- 11.Smith P, Moosavi H, Winson M, Heath D. The influence of age and sex on the response of the right ventricle, pulmonary vasculature and carotid bodies to hypoxia in rats. J Path. 1974;112:11–8. doi: 10.1002/path.1711120104. [DOI] [PubMed] [Google Scholar]
- 12.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–91. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 13.Coutant R, Lahlou N, Bouvattier C, Bougneres P. Circulating leptin level and growth hormone response to stimulation in obese and normal children. Eur J Endocrin. 1998;139:591–7. doi: 10.1530/eje.0.1390591. [DOI] [PubMed] [Google Scholar]
- 14.Gebert CA, Park SH, Waxman DJ. Regulation of signal transducer and activator of transcription (STAT) 5b activation by the temporal pattern of growth hormone stimulation. Mol Endocrinol. 1997;11:400–14. doi: 10.1210/mend.11.4.9904. [DOI] [PubMed] [Google Scholar]
- 15.Holloway MG, et al. Loss of sexually dimorphic liver gene expression upon hepatocyte-specific deletion of Stat5a-Stat5b locus. Endocrinology. 2007;148:1977–86. doi: 10.1210/en.2006-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Low MJ, et al. Somatostatin is required for masculinization of growth hormone-regulated hepatic gene expression but not of somatic growth. J Clin Invest. 2001;107:1571–80. doi: 10.1172/JCI11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer RD, Laz EV, Su T, Waxman DJ. Male-specific hepatic Bcl6: growth hormone-induced block of transcription elongation in females and binding to target genes inversely coordinated with STAT5. Mol Endocrin. 2009;23:1914–26. doi: 10.1210/me.2009-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishida Y, Yoshioka M, St Amand J. Sexually dimorphic gene expression in the hypothalamus, pituitary gland, and cortex. Genomics. 2005;85:679–87. doi: 10.1016/j.ygeno.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Sugathan A, Waxman DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol. 2013;33:3594–610. doi: 10.1128/MCB.00280-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tannenbaum GS, Choi HK, Gurd W, Waxman DJ. Temporal relationship between the sexually dimorphic spontaneous GH secretory profiles and hepatic STAT5 activity. Endocrinology. 2001;142:4599–606. doi: 10.1210/endo.142.11.8480. [DOI] [PubMed] [Google Scholar]
- 21.Thangavel C, Shapiro BH. A molecular basis for the sexually dimorphic response to growth hormone. Endocrinology. 2007;148:2894–903. doi: 10.1210/en.2006-1333. [DOI] [PubMed] [Google Scholar]
- 22.Udy GB, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94:7239–44. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waxman DJ, Ram PA, Park SH, Choi HK. Intermittent plasma growth hormone triggers tyrosine phosphorylation and nuclear translocation of a liver-expressed, Stat 5-related DNA binding protein: Proposed role as an intracellular regulator of male-specific liver gene transcription. J Biol Chem. 1995;27:13262–70. doi: 10.1074/jbc.270.22.13262. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Laz EV, Waxman DJ. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol. 2012;32:880–96. doi: 10.1128/MCB.06312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui Y, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–47. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–21. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hosui A, et al. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-β and STAT3 activation. J Exp Med. 2009;206:819–31. doi: 10.1084/jem.20080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–14. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basso K, et al. Integrated biochemical and computational approach identifies BCL-6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115:975–84. doi: 10.1182/blood-2009-06-227017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Wang M, Guo M, Xie Y, Cong YS. miR-127 regulates cell proliferation and senescence by targeting Bcl6. PLoS One. 2013;8:e80266. doi: 10.1371/journal.pone.0080266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conforto TL, Zhang Y, Sherman J, Waxman DJ. Impact of CUX2 on the female mouse liver transcriptome: activation of female-biased genes and repression of male-biased genes. Mol Cell Biol. 2012;32:4611–27. doi: 10.1128/MCB.00886-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–92. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 33.Nazari-Jahantigh M, et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest. 2012;122:4190–202. doi: 10.1172/JCI61716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawant DV, Wu H, Kaplan MH, Dent AL. The Bcl6 target gene microRNA-21 promotes Th2 differentiation by a T cell intrinsic pathway. Mol Immunol. 2013;54:435–42. doi: 10.1016/j.molimm.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacLeod JN, Pampori NA, Shapiro BH. Sex differences in the ultradian pattern of plasma growth hormone concentrations in mice. J Endocrin. 1991;131:395–399. doi: 10.1677/joe.0.1310395. [DOI] [PubMed] [Google Scholar]
- 36.Muller EE, Locatelli V, Cocchi D. Neuroendocrine control of growth hormone secretion. Physiol Rev. 1999;79:511–607. doi: 10.1152/physrev.1999.79.2.511. [DOI] [PubMed] [Google Scholar]
- 37.El Bizri N, et al. SM22alpha-targeted deletion of bone morphogenetic protein receptor 1A in mice impairs cardiac and vascular development, and influences organogenesis. Development. 2008;135:2981–91. doi: 10.1242/dev.017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu BM, et al. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood. 2008;112:2071–80. doi: 10.1182/blood-2007-12-127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. Macrophage migration inhibitory factor mediates hypoxia-induced pulmonary hypertension. Mol Med. 2012;18:215–23. doi: 10.2119/molmed.2011.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogura S, et al. Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized LDL receptor. Am J Physiol Heart Circ Physiol. 2013;305:H155–62. doi: 10.1152/ajpheart.00169.2012. [DOI] [PubMed] [Google Scholar]
- 41.Yang YM, Lane KB, Sehgal PB. Subcellular mechanism in pulmonary arterial hypertension: combinatorial modalities that inhibit anterograde trafficking and cause bone morphogenetic protein receptor 2 mislocalization. Pulm Circ. 2013;3:533–50. doi: 10.1086/674336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sehgal PB, et al. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol Physiol. 2009;297:L729–37. doi: 10.1152/ajplung.00087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sehgal PB, Lee JE. Protein trafficking dysfunctions: role in the pathogenesis of pulmonary arterial hypertension. Pulm Circ. 2011;1:17–32. doi: 10.4103/2045-8932.78097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sehgal PB. Non-genomic STAT5-dependent effects at the endoplasmic reticulum and Golgi apparatus and STAT6-GFP in mitochondria. JAX-STAT. 2013;2:e24860. doi: 10.4161/jkst.24860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JE, et al. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol. 2012;302:C804–20. doi: 10.1152/ajpcell.00379.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JE, Yang YM, Yuan H, Sehgal PB. Definitive evidence using enucleated cytoplasts for a nongenomic basis for the cystic change in endoplasmic reticulum structure caused by STAT5a/b siRNAs. Am J Physiol Cell Physiol. 2013;304:C312–23. doi: 10.1152/ajpcell.00311.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi HK, Waxman DJ. Growth hormone, but not prolactin, maintains low-level activation of STAT5a and STAT5b in female rat liver. Endocrinology. 1999;140:5126–35. doi: 10.1210/endo.140.11.7106. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995;92:8831–5. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ortiz BL, Sanchez-Gomez M, Norstedt G. Comparison of STAT5 mRNA levels in GH-treated male and female rats analyzed by a solution hybridization assay. Growth Horm IGF Res. 2000;10:236–41. doi: 10.1054/ghir.2000.0159. [DOI] [PubMed] [Google Scholar]
- 50.Smith P, Heath D. Ultrastructure of hypoxic hypertensive pulmonary vascular disease. J Path. 1977;121:93–100. doi: 10.1002/path.1711210205. [DOI] [PubMed] [Google Scholar]
- 51.Smith P, Heath D. Electron microscopy of the plexiform lesion. Thorax. 1979;34:177–86. doi: 10.1136/thx.34.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao H, et al. Novel role for STAT-5B in the regulation of Hsp27-FGF-2 axis facilitating thrombin-induced vascular smooth muscle cell growth and motility. Circ Res. 2006;98:913–22. doi: 10.1161/01.RES.0000216954.55724.a2. [DOI] [PubMed] [Google Scholar]
- 53.Cocolakis E, et al. Smad signaling antagonizes STAT5-mediated gene transcription and mammary epithelial cell differentiation. J Biol Chem. 2008;283:1293–307. doi: 10.1074/jbc.M707492200. [DOI] [PubMed] [Google Scholar]
- 54.Pak SH, et al. Hypoxia upregulates Hsp90alpha expression via STAT5b in cancer cells. Int J Oncol. 2012;41:161–8. doi: 10.3892/ijo.2012.1450. [DOI] [PubMed] [Google Scholar]
- 55.Toney LM, et al. BCL-6 regulates chemokine gene transcription in macrophages. Nat Immunol. 2000;1:214–20. doi: 10.1038/79749. [DOI] [PubMed] [Google Scholar]
- 56.Wang D, et al. BCL6 represses Smad signaling in transforming growth factor-beta resistance. Can Res. 2008;68:783–9. doi: 10.1158/0008-5472.CAN-07-0008. [DOI] [PubMed] [Google Scholar]
- 57.Arrigo T, Rulli I, Sferlazzas C, DeLuca F. Pubertal development in cystic fibrosis: an overview. J Pediatr Endocrinol Metab Suppl. 2003;2:267–70. [PubMed] [Google Scholar]
- 58.Saint-Criq V, Harvey BJ. Estrogen and the cystic fibrosis gender gap. Steroids. 2014;81:4–8. doi: 10.1016/j.steroids.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Cela E, Gagnon S, Sweezey NB. Estrogen aggravates inflammation in Pseudomonas aeriginosa pneumonia in cystic fibrosis mice. Respir Res. 2010;11:166. doi: 10.1186/1465-9921-11-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ciuclan L, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–82. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- 61.Taraseviciene-Stewart L, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–38. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 62.Joung YH, et al. Hypoxia activates signal transducers and activators of transcription 5 (STAT5) and increases its binding activity to the GAS element in mammary epithelial cells. Exp Mol Med. 2003;35:350–7. doi: 10.1038/emm.2003.46. [DOI] [PubMed] [Google Scholar]
- 63.Joung YH, et al. Hypoxia activates the cyclin D1 promoter via the Jak2/STAT5b pathway in breast cancer cells. Exp Mol Med. 2005;37:353–64. doi: 10.1038/emm.2005.45. [DOI] [PubMed] [Google Scholar]
- 64.Loges S, et al. Downregulation of VEGF-A, STAT5 and AKT in acute myeloid leukemia blasts of patients treated with SU5416. Leuk Lymphoma. 2006;47:2601–9. doi: 10.1080/10428190600948253. [DOI] [PubMed] [Google Scholar]
- 65.Yee KWH, et al. SU5416 and SU5614 inhibit kinase activity of wild-type and mutant FLT3 receptor tyrosine kinase. Blood. 2002;100:2941–9. doi: 10.1182/blood-2002-02-0531. [DOI] [PubMed] [Google Scholar]
- 66.Harada T, et al. Decreased “ineffective erythropoiesis” preserves polycythemia in mice under long-term hypoxia. Clin Exp Med. 2014 doi: 10.1007/s1-238.014.0286.5. Epub Jun 13 2014. [DOI] [PubMed] [Google Scholar]
- 67.Tan Q, et al. Erythropoiesis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem. 2013;288:17134–44. doi: 10.1074/jbc.M112.444059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hasegawa J, et al. Altered pulmonary vascular reactivity in mice with excessive erythrocytosis. Am J Respir Crit Care Med. 2004;169:829–35. doi: 10.1164/rccm.200308-1154OC. [DOI] [PubMed] [Google Scholar]
- 69.Nam S, et al. Dasatanib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol Cancer Ther. 2007;6:1400–5. doi: 10.1158/1535-7163.MCT-06-0446. [DOI] [PubMed] [Google Scholar]
- 70.Montani D, et al. Pulmonary arterial hypertension in patients treated by dasatanib. Circulation. 2012;125:2128–37. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.