

Abstract
Protein-based vaccines offer a number of important advantages over organism-based vaccines but generally elicit poor CD8+ T cell responses. We have previously demonstrated that pH-responsive, endosomolytic polymers can enhance protein antigen delivery to major histocompatibility complex class I (MHC-I) antigen presentation pathways thereby augmenting CD8+ T cell responses following immunization. Here, we describe a new family of nanocarriers for protein antigen delivery assembled using architecturally distinct pH-responsive polymers. Reversible addition-fragmentation chain transfer (RAFT) polymerization was used to synthesize linear, hyperbranched, and core-crosslinked copolymers of 2-(N,N-diethylamino)ethyl methacrylate (DEAEMA) and butyl methacrylate (BMA) that were subsequently chain extended with a hydrophilic N,N-dimethylacrylamide (DMA) segment copolymerized with thiol-reactive pyridyl disulfide (PDS) groups. In aqueous solution, polymer chains assembled into 25 nm micellar nanoparticles and enabled efficient and reducible conjugation of a thiolated protein antigen, ovalbumin. Polymers demonstrated pH-dependent membrane-destabilizing activity in an erythrocyte lysis assay, with the hyperbranched and cross-linked polymer architectures exhibiting significantly higher hemolysis at pH ≤ 7.0 than the linear diblock. Antigen delivery with the hyperbranched and cross-linked polymer architecture enhanced in vitro MHC-I antigen presentation relative to free antigen, whereas the linear construct did not have a discernible effect. The hyperbranched system elicited a four- to fivefold increase in MHC-I presentation relative to the cross-linked architecture, demonstrating the superior capacity of the hyperbranched architecture in enhancing MHC-I presentation. This work demonstrates that the architecture of pH-responsive, endosomolytic polymers can have dramatic effects on intracellular antigen delivery, and offers a promising strategy for enhancing CD8+ T cell responses to protein-based vaccines.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9697-1) contains supplementary material, which is available to authorized users.
KEY WORDS: MHC-I antigen presentation, pH-responsive nanoparticle, polymer architecture, RAFT polymerization, vaccine
INTRODUCTION
Activation of CD8+ cytotoxic T cell (CTL) responses is widely considered to be an essential component for effective vaccination against many intracellular pathogens and cancers (1,2). However, the vast majority of currently approved vaccine formulations primarily elicit humoral immune responses with minimal activation of cellular immunity (3). Recombinant viral vectors have been engineered that elicit strong CTL responses (4,5), but anti-vector immunity can dramatically compromise efficacy and safety concerns remain (6). By contrast, recombinant protein-based vaccines provide antigen specificity and generally favorable safety profiles (7,8), but at the expense of immunogenicity and cellular immunity (9,10).
The ongoing development of fully synthetic and chemically defined nanoparticles has afforded an opportunity to design antigen nanocarriers that efficiently promote antigen presentation on class I major histocompatibility complex (MHC) by dendritic cells (11–17). Considerable research has focused on the development of nanoparticles in the viral size range (∼20–200 nm) that enhance class I cross-presentation of exogenous protein antigens. These include liposomes (18–20), immune-stimulating complexes (ISCOMs) (21), inorganic nanoparticles (15,22), and polymer-based nanoparticles such as polymersomes (23,24), dendrimers (25), and micelles (11,12,26). Our group has focused on the development of reversible addition-fragmentation chain transfer (RAFT)-synthesized block copolymer carriers that alter intracellular trafficking pathways and enhance cytosolic delivery of proteins (11,12,27). These micellar carriers have been shown to enhance class I antigen presentation in vitro and elicit CD8+ T cell responses in vivo (11,12).
The assembly of nanocarriers using polymers synthesized by RAFT polymerization offers a number of important advantages for antigen delivery. In addition to practical advantages such as low cost and scalable manufacture, RAFT polymerization allows for the modular incorporation of diverse monomers into a single well-defined polymer, yielding multifunctional carriers with tunable chemical properties (28–35). Additionally, RAFT enables the synthesis of interesting carrier architectures that may potentially enhance carrier efficacy (36,37). There have been several reports studying how the dimensionality of synthetic vaccine carriers relate to immune stimulation (38–41), but relatively few studying how polymeric architectures could enhance antigen delivery. Here, we have explored how the structural geometry of segments directing intracellular trafficking activities and pH-induced nanocarrier structural transitions relate to class I antigen presentation. A new class of hyperbranched and core-crosslinked antigen nanocarriers were constructed and compared to the linear diblock architectures. The pH-responsive segments were based on a recently described composition of 2-(N,N-diethylamino)ethyl methacrylate (DEAEMA) and butyl methacrylate (BMA) originally designed for delivery of plasmid DNA and mRNA (42,43). The DEAEMA-co-BMA polymers of linear diblock, hyperbranched, and cross-linked architecture were synthesized by RAFT polymerization and chain extended with a hydrophilic segment composed of N,N-dimethylacrylamide (DMA) and pyridyl disulfide (PDS) groups to enable antigen conjugation via a disulfide bond (11,12). While both the hyperbranched and cross-linked architectures are branched, in the cross-linked architecture the branched points are introduced by copolymerization of a cross-linker (44), providing a microgel structure. By contrast, in the hyperbranched structure the RAFT R-group provides a branching point from which a dendritic structure is derived. In aqueous solution, the polymers self-assemble into ca. 25-nm diameter nanoparticles, yielding comparably sized antigen nanocarriers composed of architecturally distinct polymer chains (Fig. 1).
Fig. 1.
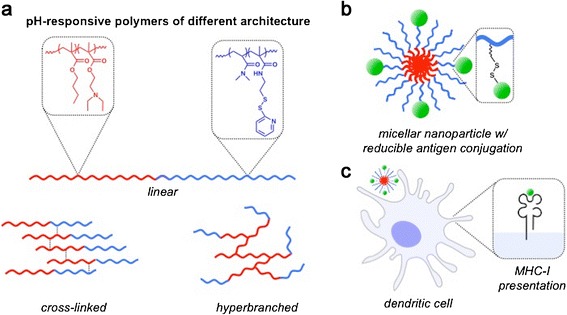
pH-responsive nanoparticles assembled using polymer chains of different architectures were utilized as carriers for delivery of protein antigen into the MHC-I antigen processing pathway. a pH-responsive diblock copolymers were synthesized with linear, core-crosslinked, or hyperbranched architectures. The pH-responsive, endosomolytic component (red) is a copolymer of butyl methacrylate (BMA) and 2-(N,N-diethylamino)ethyl methacrylate (DEAEMA) which was chain extended with a copolymer of dimethylacrylamide (DMA) doped with a pyridyl disulfide functionalized monomer (PDSMA) for antigen conjugation. b Polymer chains of all architectures assembled into nanoparticles under aqueous conditions at neutral pH, PDS groups were used for conjugation of protein antigen via disulfide exchange. c Nanocarriers composed of architecturally distinct polymer chains were evaluated for their ability to enhance MHC-I antigen presentation by dendritic cells
MATERIALS AND METHODS
Materials
All solvents were of analytical reagent grade unless otherwise stated. 2,2′-Azobis[2-methylpropionitrile] (AIBN) was purchased from Acros and purified by recrystallization twice from methanol prior to use. 1,1′-Azobis(cyclohexanecarbonitrile) (ABCC, supplied by DuPont as Vazo 88) was purified by recrystallization twice from methanol prior to use. All deuterated solvents were obtained from Cambridge Isotope Laboratories. The trithiocarbonate chain transfer agent (CTA) 4-cyano-4-(((ethylthio)carbonothioyl)thio)pentanoic acid (ECT) (45) and pyridyl disulfide methacrylamide (PDSMA) (46) were synthesized as previously reported. The monomers BMA, DEAEMA, DMA, and ethylene glycol dimethylacrylate (EGDMA) were purchased from Aldrich and pretreated using neutral alumina (for removal of hydroquinone and monomethyl ether hydroquinone polymerization inhibitors) prior to flash distillation. All solvents were obtained from Merck KGaA, and anisole was obtained from BDH Chemicals Ltd.; all were used without further purification. Bond-Breaker TCEP solution, Traut’s reagent (2-iminothiolane-HCl), and Ellman’s reagent (5,5′-dithio-bis-[2-nitrobenzoic acid], DTNB) were obtained from Thermo Fisher Scientific.
NMR Characterization
NMR solvent (MeOD-d4) was purchased from Cambridge Isotope Laboratories and used as received. 1H NMR spectra were recorded at 400 MHz using a Bruker Avance 400 MHz NMR spectrometer (Billerica, MA, USA). Chemical shifts (δH) are reported in parts per million (ppm). Spectra were taken as follows: number of scans = 32 and D1 relaxation delay = 23 s.
Gel Permeation Chromatography
Gel permeation chromatography (GPC) was performed on a Shimadzu system equipped with a CMB-20A controller system, an SIL-20A HT autosampler, an LC-20AT tandem pump system, a DGU-20A degasser unit, a CTO-20AC column oven, an RDI-10A refractive index detector, and 4× Waters Styragel columns (HT2, HT3, HT4, and HT5, each 300 mm × 7.8 mm2, providing an effective molar mass range of 100–4 × 106). N,N-Dimethylacetamide (DMAc) (containing 2.1 g L−1 lithium chloride (LiCl)) was used as an eluent with a flow rate of 1 mL/min at 80°C. Number (Mn) and weight-average (Mw) molar masses were evaluated using Shimadzu LC Solution software. The GPC columns were calibrated with low dispersity polystyrene (PSt) standards (Polymer Laboratories) ranging from 3100 to 650,000 g mol−1, and molar masses are reported as PSt equivalents. A 3rd-order polynomial was used to fit the log Mpvs. time calibration curve, which was linear across the molar mass ranges.
Synthesis of Inimer RAFT Agent
The synthesis of the inimer RAFT agent was adapted from the method described by Wei et al. (47) Briefly, ECT (1.0 g, 3.8 mmol) and hydroxyethyl methacrylate (HEMA) (0.74 g, 5.7 mmol) were dissolved in dry dichloromethane (DCM) (10 mL), followed by addition of 4-(dimethylamino)pyridine (DMAP) (0.47 g, 3.8 mmol). After the dissolution of DMAP, N,N′-diisopropylcarbodiimide (DIC) (0.96 g, 7.6 mmol) was added at 0°C and stirred for 30 min. The reaction was allowed to warm up to room temperature and further stirred for 3 h. DCM was removed from the reaction mixture which was subsequently dissolved in ethyl acetate, the insoluble DIC urea by-products were removed by filtration, and the solution was dried on a rotary evaporator to afford the crude product. The crude product was further purified via elution through a silica gel column (eluent:pentane to ethyl acetate:pentane, 1:4 v/v) to afford the inimer RAFT agent as a yellow oil upon drying (1.16 g, 80% yield).
1H NMR (400 MHz, CDCl3) is as follows: δ 1.35 (t, 3H, CH3–CH2), 1.87 (s, 3H, C(CH3)(CN)), 1.94 (s, 3H, CH3–C=C), 2.45 (m, 2H, CH2(CO)O), 2.65 (t, 2H, C(CH3)(CN)–CH2), 3.33 (m, 2H, –CH2–S), 4.35 (s, 4H, –OCH2CH2O–), 5.60 (s, 1H, C=C–Hb), and 6.13 (s, 1H, C=C–Ha). 13C NMR (125 MHz, CDCl3) is as follows: δ 12.9, 18.4, 25.0, 29.8, 31.5, 33.9, 46.4, 62.3, 62.8, 119.1, 126.3, 136.0, 167.2, 171.4, and 216.8. HRMS (EI) calculated for C15H21O4NS3 [M]+ is 375.0627 and found to be 375.0629.
Synthesis of Linear RAFT Copolymer of DEAEMA and BMA
RAFT copolymerization of DEAEMA and BMA was conducted with ECT and ABCC as the chain transfer agent and initiator, respectively, in dioxane at 90°C. The initial monomer ([M]o) to CTA ([CTA]o) to initiator ([I]o) ratio was 100:1:0.05, respectively. Individual polymerization solutions from the stock solution were transferred to nitrogen-flushed glass ampoules, freeze-evacuate-thaw degassed to constant high vacuum (8–9 × 10−3 mbar) and flame sealed. After this time, the polymerization ampoules were transferred to a preheated oil bath at 90°C and allowed to polymerize for the prescribed time period. Following polymerization, the individual polymerization ampoules were quenched by immersion in liquid nitrogen and then opened and evaluated via1H NMR in CD3OD and GPC (DMAc eluent).
A representative procedure for the synthesis of a poly(DEAEMA-co-BMA) macroCTA (Mn = 10,700 g/mol, Ð = 1.16) used to prepare the corresponding poly[(DEAEMA-co-BMA)-b-(DMA-co-PDSMA)] copolymers used in these studies is a prepared stock solution comprising of DEAEMA (3.89 g, 21 mmol), BMA (1.99 g, 14 mmol), ECT (92 mg, 0.35 mmol), dioxane (5.88 g), and ABCC (4.3 mg, 0.017 mmol). Individual polymerization solutions from the stock solution were transferred to nitrogen-flushed glass ampoules, freeze-evacuate-thaw degassed to constant high vacuum (8–9 × 10−3 mbar) and flame sealed. After this time, the polymerization ampoules were transferred to a preheated oil bath at 90°C and allowed to polymerize for the prescribed time period. Following polymerization, the individual polymerization ampoules were quenched by immersion in liquid nitrogen. The polymers were precipitated from the polymerization mixture into pentane and cooled to −10°C, the solvent was decanted, and the polymer oil was resuspended in fresh pentane. After three precipitations, the polymer was isolated via rotary evaporation followed by drying under high vacuum. For the 7-h sample, the final molecular weight, Ð, and composition of the poly(DEAEMA-co-BMA) macroCTA were 10,700 g/mol (Mntheoretical = 11,350), 1.16, and 61:39 DEAEMA:BMA (60:40 feed), respectively. Monomer conversions were determined via1H NMR spectroscopy by diluting the polymerization solutions in CD3OD and comparing the DEAEMA vinyl resonances at 6.08 ppm to the BMA vinyl resonances at 6.05 ppm relative to an anisole spike (6.88 ppm). Copolymer composition was determined by comparison of the DEAEMA aliphatic amine resonances at 2.5–3.0 ppm to the combined ester resonances between 3.9 and 4.3 ppm.
Inimer RAFT-Controlled Synthesis of Branched Copolymer of DEAEMA and BMA
Dendritically branched poly(DEAEMA-co-BMA) macroCTA was synthesized as for the linear copolymer described above, under the control and copolymerization of the inimer RAFT agent. The initial monomer ([M]o) to CTA ([CTA]o) to initiator ([I]o) ratio was 100:1:0.05, respectively. A representative procedure for the synthesis of branched poly(DEAEMA-co-BMA) is provided in the Supporting Information.
RAFT Synthesis of EGDMA-Cross-Linked Copolymer of DEAEMA and BMA
Cross-linked poly(DEAEMA-co-BMA) macroCTA was synthesized in the same manner as the linear copolymer described above with the addition of a divinyl cross-linker, EGDMA. The initial monomer ([M]o) to CTA ([CTA]o) to initiator ([I]o) ratio was 100:1:0.05, respectively. The RAFT to EGDMA ratio was 1:0.7. A representative procedure for the synthesis of poly(dimethylaminoethyl methacrylate (DMAEMA)-co-BMA-co-EGDMA) is provided in the Supporting Information.
RAFT Block Extension Copolymerization of DMA and PDSMA
To a solution of the poly(DEAEMA-co-BMA) or poly(DEAEMA-co-BMA-co-EGDMA) macroCTA in dioxane, DMA, PDSMA, and AIBN ([M]o:[CTA]o:[I]o = 100 or 200:1:0.047) were added. The polymerization solution was then transferred to nitrogen-flushed glass ampoules, freeze-evacuate-thaw degassed to constant high vacuum (8–9 × 10−3 mbar) and flame sealed. After this time, the polymerization ampoules were transferred to a preheated oil bath at 60°C and allowed to polymerize for 9 h. The polymer was precipitated from the polymerization mixture into pentane, filtered, re-dissolved in acetone, and re-precipitated into cooled pentane, repeated three times. After this time, the polymer was isolated via filtration and air-dried, followed by drying under high vacuum. Monomer conversions were determined via1H NMR spectroscopy by diluting the polymerization solutions in CD3OD and comparing the DMA vinyl resonances at 6.17 ppm to the PDSMA vinyl resonances at 5.37 ppm relative to an anisole spike (6.88 ppm). The theoretical copolymer composition was determined via comonomer conversion. Representative procedures for addition of the DMA-co-PDSMA blocks for each of the architectures are provided in the Supporting Information.
Micelle Formulation and Antigen Conjugation
Aqueous polymer solutions were prepared by first dissolving a dry copolymer into ethanol at 50 mg/mL followed by rapid dilution into 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered glucose (HBG; 20 mM HEPES, 5% glucose, pH 7.4) to a final concentration of 10 mg/mL. Ovalbumin (ova; EndoGrade®, Hyglos GmbH) was conjugated to the PDS groups on polymer micelles via a disulfide exchange reaction. Thiol groups were incorporated onto ova using a 22-M excess of 2-iminothiolane (Traut’s reagent) as previously described (11). Non-reacted 2-iminothiolane was removed using a Zeba desalting column (0.5 mL, 7K MWCO; Thermo Fisher Scientific) equilibrated with HBG. The average number of thiol groups per ova was determined using Ellman’s reagent (Thermo Fisher Scientific) according to manufacturer’s instructions. For all studies, four to five thiols per ova were introduced. In some instances, ova was labeled with Alexa Fluor 488® TFP (Invitrogen) prior to thiolation with approximately one dye/protein according to manufacturer’s instructions. Thiolated ova was subsequently reacted with polymer micelles at a polymer concentration of 7.5 mg/mL. The extent of conjugation was determined via nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of conjugates prepared with fluorescently labeled ova (137 V; 4–20% Tris-glycine; PROTEAN TGC precast gel, Bio-Rad). To demonstrate the reducibility of the disulfide bond between polymer and protein, conjugates were incubated with 20 mM tris(2-carboxyethyl)phosphine (TCEP; Pierce) for 1 h at room temperature. Gels were imaged using a Storm 860 Molecular Imager (GMI, Inc.) to determine protein shifts, and ImageQuant TL software was used to quantify the extent of conjugation.
Dynamic Light Scattering
Dynamic light scattering (DLS) measurements were conducted using a Malvern Zetasizer Nano ZS (Worcestershire, UK) at a constant scattering angle of 173° as described previously (11). Briefly, particle sizes of copolymer micelles and protein-polymer conjugates were determined by DLS at RT in HBG (pH 7.4) at 1 mg/mL polymer. In some cases, DLS measurements were performed with copolymers (0.5 mg/mL) incubated with 10 mM sodium phosphate buffer (supplemented with 150 mM NaCl) in the pH range of the endosomal processing pathway (7.4, 7.0, 6.6, 6.2, and 5.8). Mean diameters are reported as the number average ± standard deviation from three or more independent measurements.
Erythrocyte Lysis Assay
Polymer disruption of lipid bilayer membranes was assessed via a red blood cell hemolysis assay as previously described (48). Briefly, polymers were incubated for 1 h at 37°C in the presence of human erythrocytes at the indicated concentration in 100 mM sodium phosphate buffer (supplemented with 150 mM NaCl) in the pH range of the endosomal processing pathway (7.4, 7.0, 6.6, 6.2, and 5.8). Extent of cell lysis (i.e., hemolytic activity) was determined spectrophotometrically by measuring the amount of hemoglobin released (Abs 541 nm) and normalized to a 100% lysis control (1% Triton X-100). Samples were run in quadruplicate.
Cell Lines
The mouse dendritic cell line DC2.4 (H-2Kb positive) was kindly provided by Kenneth Rock (University of Massachusetts Medical School) and cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 2 mM l-glutamine, 100 U/mL penicillin/100 μg/mL streptomycin (Gibco), 55 μM 2-mercaptoethanol (Gibco), 1× nonessential amino acids (CellGro), and 10 mM HEPES (Invitrogen). Cells were passaged at ∼60–70% confluency using 0.25% trypsin-EDTA (Gibco). B3Z T cells, a lacZ-inducible T cell hybridoma specific for the SIINFEKL-H-2Kb complex, were a generous gift from Nilabh Shastri (UC Berkeley) and cultured in RPMI 1640 (Gibco) supplemented with 10% FBS, 100 U/mL penicillin/100 μg/mL streptomycin (CellGro), 50 μM 2-mercaptoethanol (Gibco), and 1 mM sodium pyruvate (Gibco). Both cell types were grown in a humidified atmosphere with 5% CO2 at 37°C.
In Vitro Dendritic Cell Uptake
Intracellular uptake of ovalbumin was evaluated by flow cytometry using Alexa Fluor 488®-labeled ovalbumin. DC2.4 cells were plated at 75 k cells/well in 24-well plates and allowed to adhere overnight. Cells were subsequently incubated with formulations containing fluorescently labeled ova for 5 h, rinsed twice with Dulbecco’s phosphate-buffered saline (DPBS), trypsinized (0.25%, 5 min), pelleted by centrifugation, and resuspended in DPBS containing 2% FBS. Flow cytometry was performed on a FACSCanto II (BD) and analyzed using FlowJo software (Tree Star Inc.).
In Vitro MHC-I Antigen Presentation Assay
The ability of polymeric nanoparticles to enhance MHC class I antigen presentation was assessed by in vitro antigen presentation assay using a DC2.4 cell as the antigen-presenting cell. This assay utilizes a specialized lacZ B3Z T cell hybridoma that produces β-galactosidase upon recognition of the immunodominant ovalbumin class I epitope SIINFEKL presented on MHC class I H-2Kb on DC2.4 cells. DC2.4 cells were plated at 5 × 104 cells/well in U-bottom 96-well cell culture plates and grown overnight. The following day, ova-containing formulations were added to the final indicated concentration (7.7–245 μg/mL) and incubated with DC2.4 cells for 5 h at 37°C in a 5% CO2 incubator. Cells were then carefully rinsed three times with DPBS, and 1 × 105 B3Z T cells were added to each well and co-cultured for 20 h in RPMI 1640 supplemented with 10% FBS, 2 mM l-glutamine, 55 μM beta-mercaptoethanol, 1 mM pyruvate, and 100 U/mL penicillin/100 μg/mL streptomycin. Cells were then pelleted via centrifugation (7 min, ∼500 rcf), media were carefully aspirated, and 150 μL of CPRG/lysis buffer (0.15 M chlorophenol red-β-d-galactopyranoside (Calbiochem), 0.1% Triton X-100, 9 mM MgCl, 100 μM mercaptoethanol) was added. Plates were incubated at 37°C in the dark for 20 h, and the absorbance of released chlorophenol red was measured at 570 nm using a Tecan Safire 2 plate reader.
RESULTS
Polymer Synthesis and Characterization
DEAEMA-co-BMA polymers were synthesized with linear, hyperbranched, or core-crosslinked architectures according to Scheme 1. All poly(DEAEMA-co-BMA) macroCTAs (mCTAs) were prepared with a DEAEMA to BMA feed ratio of 60:40 mol %. This feed ratio was selected based on our previous work that showed that this composition was most active at early and late endosomal pH values (i.e., pH 6.8–5.8) (43). Monomer conversion as a function of reaction time for each architecture is provided in the Supporting Information (Tables S1, S3, and S5). The evolution of polymer molecular weight and Ð with monomer conversion is shown in Fig. 2 along with representative GPC chromatograms of DEAEMA-co-BMA macroCTAs used in this work. The progression of Mn with conversion was found to behave as expected (43) with some deviation from calculated Mn at lower number average of molar masses. This can be accounted for, as the characteristics for this copolymer would be different to that of polystyrene, which was used for the calibration of GPC with refractive index detection. Polymer chain-length dispersity (Ð) was low (<1.18) over the course of the polymerization (Fig. 2a) with minimal tailing at higher retention times. Furthermore, the kinetics of the copolymerization (Table S1, Fig. S1) were found to behave predictably for the linear system with the ECT RAFT agent.
Scheme 1.
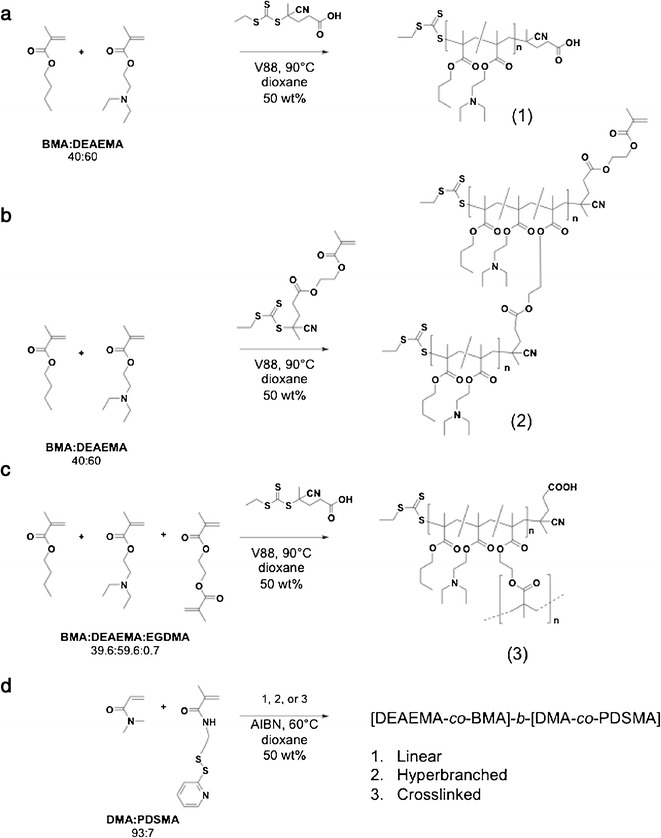
Synthesis of [BMA-co-DEAEMA]-b-[DMA-co-PDSMA] polymers of linear (a), hyperbranched (b), or core-crosslinked (c) architecture. Block extension of these architectures by DMA and PDSMA (d)
Fig. 2.
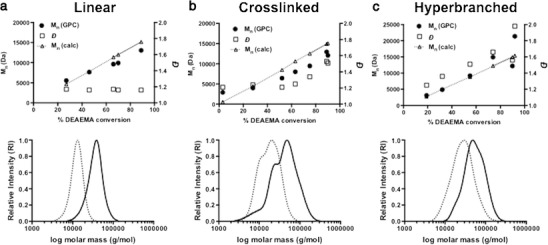
Evolution plot of polydispersity (Ð) and molar mass progression (M n (GPC)) with DEAEMA conversion vs. calculated molar mass (M n (calc)) for DEAEMA-co-BMA polymers (top) of linear (a), cross-linked (b), and hyperbranched (c) architecture. Representative molar mass distributions (bottom) of DEAEMA-co-BMA mCTA (dotted line) and [DEAEMA-co-BMA]-b-[DMA-co-PDSMA] diblock copolymer (solid line) of linear (a), hyperbranched (b), and cross-linked (c) architectures
The cross-linked and hyperbranched architectures exhibited higher Ðs, and their GPC traces displayed some shoulders or multimodality (Fig. 2b, c), both of which are common for polymers synthesized using inimer RAFT agents and divinyl monomers (37,49,50). The DEAEMA/BMA copolymerization with the divinyl monomer EGDMA controlled by the ECT RAFT agent and the DEAEMA/BMA copolymerization with the inimer RAFT agent were found to behave differently compared to the linear polymer architecture. Mn was observed to deviate from the calculated Mn, particularly at higher conversion (>80%) (Fig. 2b, c). For the cross-linked system, a EGDMA:CTA ratio of 0.7:1 was chosen to prevent macrogel formation during the later block extension (with DMA/PDSMA) which could occur due to the presence of pendant methacrylate groups. Molar mass dispersities also increased from 1.25 (at 60% conversion) to 1.53 (at 90% conversion) for the cross-linked architecture due to divinyl cross-linking of EGDMA becoming more significant as the polymerization proceeded. The development of a high molar mass shoulder was also more apparent with conversion (Fig. S5, Table S3), which is a characteristic of cross-linking (51,52). The inimer-hyperbranched system on the other hand showed molar mass dispersity broadening at earlier conversions (∼25%) and progressed to a final dispersity of 1.56 at high conversion, along with characteristic growth of high molecular weight shoulders in the GPC traces (Fig. S11, Table S6).
DEAEMA-co-BMA polymers of each architecture were then used as mCTAs to generate chain-extended copolymers comprising DMA (93%) and PDSMA (7%). This DMA-co-PDSMA block provided a hydrophilic segment that facilitates aqueous solubility and micelle formation while also providing reactive pyridyl disulfide (PDS) handles for conjugation of thiolated antigen. All DEAEMA-co-BMA mCTAs were successfully chain extended with DMA-co-PDSMA, as indicated in their GPC chromatographs by both a clear shift in molecular weight distributions (Fig. 2) and the appearance of characteristic DMA and PDSMA peaks in 1H NMR spectra (Figs. S4, S10, and S16). The specific evolution plots showing Mn and Ð, with respect to monomer conversion for DMA-co-PDSMA chain extensions from each of the mCTA architectures, are provided in the Supporting Information (Figs. S2, S6, S8, S12, and S14). The DEAEMA-co-BMA mCTAs were chain extended with DMA-co-PDSMA blocks of different lengths. By controlling the relative length of the second block, carriers with a relatively high (50–60%) or low (30–40%) pH-responsive content could be synthesized to allow the effect of endosomolytic mass fraction on antigen delivery to be investigated. DMA-co-PDSMA block extension of linear DEAEMA-co-BMA macroRAFT showed the linear progression of Mn with conversion and dispersities to just below 1.3 (Fig. S2, Table S2). A clear shift in the molar mass distribution to shorter retention times was observed, without the appearance of a significant low molecular weight tail. Also evident was a lack of any high molecular weight asymmetry in the GPC peak that could result from polymer-polymer coupling or conjugation reactions (Figs. 2a and S3). 1H NMR analysis of the copolymer showed successful removal of any residual comonomer, apparent from the lack of vinyl resonances (Fig. S4). Also apparent was the appearance of a large resonance between 2.5 and 3.0 ppm, which was associated with the N,N-dimethyl groups of DMA and the appearance of the aromatic signals of the pyridyl pendant group of PDSMA between 7 and 8.5 ppm. The block extensions for both the divinyl systems (Figs. S6 and S8) and the hyperbranched systems (Figs. S12 and S14) showed kinetic anomalies for Mn progression with conversion. Some of these can be explained by hydrodynamic volume differences of these architectures compared to linear PS standards as measured via GPC. The progression of monomer conversion with time, however, indicated nonideal polymerization kinetics, particularly for the cross-linked system. These systems may therefore require further investigation and optimisation, with respect to this aspect. Nonetheless, successful block extension for both the divinyl cross-linked system (Fig. 2b) and inimer-hyperbranched system (Fig. 2c) was evident from a shift in the molar mass distribution as determined by GPC. This was also evidenced in the 1H NMR spectra with the appearance of N,N-dimethyl resonance between 2.5 and 3.0 ppm and the appearance of the pyridyl pendant group resonances between 7 and 8.5 ppm (Figs. S10 and S16). A summary of the properties of all polymers used in subsequent investigations is provided in Table I.
Table I.
Properties of pH-Responsive Copolymers for Protein Antigen Delivery
| Architecture | M n (1st block) | Ð (1st block) | M n (2nd block) | M n (overall) | Ð (overall) | PDS/chain | wt% (1st block)a |
|---|---|---|---|---|---|---|---|
| High pH-responsive polymer content (50–60 wt%) | |||||||
| Linear | 10,500 | 1.15 | 11,500 | 22,000 | 1.30 | 3.6 | 0.50 |
| Hyperbranched | 12,900 | 1.55 | 30,100 | 43,000 | 1.39 | 6.1 | 0.61 |
| Cross-linked | 14,700 | 1.36 | 12,000 | 26,700 | 1.83 | 4.7 | 0.61 |
| Low pH-responsive polymer content (30–40 wt%) | |||||||
| Linear | 11,300 | 1.16 | 19,200 | 30,500 | 1.22 | 13.6 | 0.28 |
| Hyperbranched | 19,100 | 1.42 | 21,100 | 40,200 | 1.69 | 12.5 | 0.43 |
| Cross-linked | 14,700 | 1.36 | 32,500 | 47,200 | 2.78 | 15.4 | 0.30 |
aDetermined via 1H NMR
Micelle Formulation and pH-Responsive Behavior
Rapid dilution of all polymers from ethanol into HEPES-buffered glucose (HBG; pH 7.4) resulted in nanoparticle formation with conserved sizes of ca. 25 nm in diameter as measured by DLS (Fig. 3a). Particle size was measured across a pH range of 7.4–5.8 in order to mimic the physiological trafficking of carriers from the extracellular environment and into acidic endosomal/lysosomal compartments of antigen-presenting cells. In accordance with our previous work, a pH-triggered phase transition was observed, shifting the equilibrium from a micelle (∼25 nm) to unimeric polymer chains (∼5–10 nm) with decreasing pH (Fig. 3b). Interestingly, while similar particle sizes were observed at pH 7.4 and 7.0, the steepness of the transition in particle size between pH 7.0 and 5.8 was significantly different for the hyperbranched polymer compared to the linear diblock and cross-linked polymers that displayed similar transition profiles. The hyperbranched polymer also displayed a transition that was shifted to lower pH values with a midpoint of approximately pH 6.4 compared to approximately 6.8 for the linear diblock and cross-linked polymers.
Fig. 3.
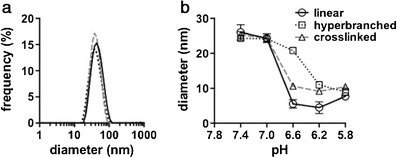
Dynamic light scattering (DLS) characterization of pH-responsive diblock copolymers (high pH-responsive polymer content; see Table I) in aqueous solution. a Representative size distribution (number average diameter) of particles at pH 7.4 in HEPES-buffered glucose. b Particle size measurements of copolymers as a function of pH (0.5 mg/mL in 10 mM sodium phosphate buffer with 150 mM NaCl)
A red blood cell hemolysis assay was used to evaluate the pH-dependent membrane-destabilizing activity of polymers (Fig. 4). The hyperbranched and cross-linked architectures displayed significantly more potent hemolytic activity at all pH values below 7.0, with the hyperbranched material demonstrating slightly higher activity at a fixed total polymer concentration of 1.25 μg/mL (Fig. 4a). To account for differences in the composition and molecular weights of the polymers, concentrations were normalized to an equivalent mass concentration of the pH-responsive component (1.25 μg/mL). Again, the hyperbranched and cross-linked polymers demonstrated heightened activity relative to the linear diblock (Fig. 4b), suggesting that the increased hemolytic activity relative to the linear construct is indeed an architectural effect. Analogous carriers were also synthesized with a higher percentage of the DMA-co-PDSMA hydrophilic block, and similar trends were observed amongst the architectures (Fig. 4c). Notably, extension of the hydrophilic segment resulted in decreased hemolytic activity even when concentrations were normalized to an equivalent amount of the pH-responsive component (1.25 μg/mL), suggesting that the increased hydrophilic chain length may interfere sterically with polymer-membrane interactions.
Fig. 4.
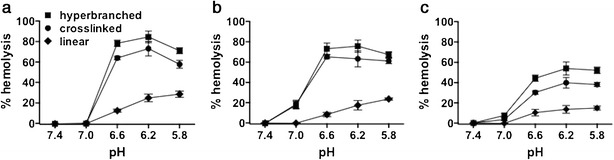
Erythrocyte lysis assay demonstrating pH-dependent membrane-destabilizing activity of copolymers with linear, cross-linked, and hyperbranched architectures. a Total polymer concentration fixed at 1.25 μg/mL. b Polymer concentration normalized to 1.25 μg/mL of the pH-responsive element (DEAEMA-co-BMA). c Hemolytic activity of polymers with higher mass fraction of hydrophilic DMA-co-PDSMA block (lower pH-responsive content), normalized to 1.25 μg/mL of the pH-responsive element. Data represent mean ± SD, n = 4
Antigen Conjugation
Conjugation of thiol-bearing antigens to PDS group on polymeric carriers via disulfide bonds allows antigens to be released from the carrier in the reducing environment of cytosol, a property that has been shown to enhance antigen delivery relative to nonreducible bonds (53). Reactive thiol groups were introduced to the model antigen ovalbumin (ova) via modification of lysine primary amines with 2-iminothiolane (approximately five thiols/ova). Conjugates were prepared by adding thiolated ova to polymers in HBG, and reactions were performed at mass ratios of 0.35, 0.175, and 0.0875 mg ova/mg polymer. While ∼25-nm particle sizes were observed for all architectures at the 0.35 ratio, significant precipitate formation was observed for the hyperbranched carrier at ratios of 0.175 and 0.0875 and for the cross-linked architecture at 0.0875 (Fig. 5a). We postulate that this colloidal instability was mediated by cross-linking of polymers/nanoparticles at decreasing concentrations of ova, which bears multiple thiol groups. To allow for size-independent comparison between the three architectures, the 0.35 w/w conjugates were selected for all subsequent studies. Conjugation efficiencies were determined at 0.35 w/w loading by nonreducing SDS polyacrylamide gel electrophoresis using conjugates prepared with fluorescently labeled ova. Complete conjugation of ova was observed for all architectures, as indicated by the disappearance of the free protein band and a shift to higher molecular weights, which largely did not migrate on the gel (Fig. 5b). A control reaction of polymers and non-thiolated ova was performed and resulted in a very small amount of antigen conjugation (Fig. 5c), likely due to native and largely inaccessible thiol groups present on ova. Incubation of conjugates with a reducing agent (20 mM TCEP) for 1 h at room temperature regenerated the free protein band, demonstrating disulfide bond reversibility and protein release (Fig. 5b).
Fig. 5.
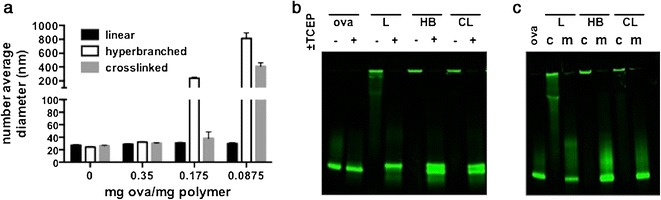
Antigen conjugation to pH-responsive polymer nanoparticles via disulfide exchange reaction. a Size of nanoparticles (number average diameter) by dynamic light scattering after reaction with thiolated ovalbumin (ova-SH) at 0, 0.35, 0.175, and 0.0875 mg ova/mg polymer. b SDS-PAGE of fluorescently labeled ovalbumin (ova) and ova-nanoparticle conjugates (0.35 mg ova/mg polymer) prepared using linear (L), hyperbranched (HB), or cross-linked (CL) polymers. Incubation of conjugates with TCEP liberates ova from the carrier. c SDS-PAGE of fluorescently labeled ova and ova-nanoparticle conjugates (c) and a physical mixture (m) of polymers and non-thiolated ova
In Vitro MHC-I Antigen Presentation
To compare the cross-presentation delivery efficiencies of different polymeric nanoparticles, we utilized an in vitro B3Z T cell antigen presentation assay. DC2.4 cells, a murine dendritic cell line, were incubated with free ova, polymer-ova conjugates, or a physical mixture of ova and polymer. The DC2.4 cells were subsequently co-cultured with a B3Z T cell hybridoma, which produces β-galactosidase upon recognition of ova257–264 (SIINFEKL) complexed with the murine MHC-I haplotype H-2Kb. Studies were performed at fixed antigen loading (35 wt% ova), and ova concentration was varied between 7.7 and 245 μg/mL (21–700 μg/mL polymer). As shown in Fig. 6a, at these concentrations, the linear diblock carrier did not enhance MHC-I antigen presentation relative to the free antigen with both eliciting negligible differences in class I presentation relative to background. Antigen delivery using both the hyperbranched and cross-linked architectures resulted in increased MHC-I antigen presentation levels, with the hyperbranched system eliciting a four- to fivefold increase in the response relative to the cross-linked system. Consistent with our previous findings (11,12), a physical mixture of ova and all polymers tested did not increase antigen presentation levels (Fig. S17A), indicating the need for covalent antigen conjugation. Notably, polymers of analogous hyperbranched and cross-linked architecture that contained a lower mass fraction of the pH-responsive element (∼30–40%) were not effective at enhancing antigen presentation (Fig. S17B). The activity of carriers is thus dependent on polymer architecture and the relative amount of pH-responsive, endosomal-releasing segments in the polymer chain.
Fig. 6.
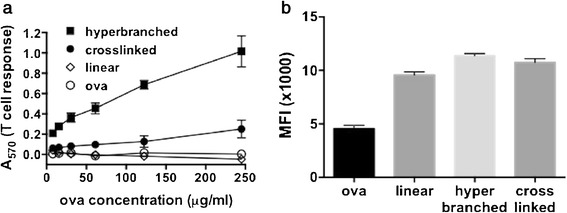
a Effect of polymer architecture on MHC class I presentation in an in vitro co-culture model. Murine dendritic cells (DC2.4) were incubated with free antigen (ova) or conjugates prepared using polymers of different architecture and subsequently co-cultured with B3Z T cells which produce β-galactosidase in response to antigen presentation on MHC-I. Data are from a single representative experiment conducted in quadruplicate (mean ± standard deviation). b Uptake of Alexa Fluor 488-labeled ova measured by DC2.4 cells after 5 h at 37°C as measured by flow cytometry (MFI median fluorescent intensity). Data are from a single experiment conducted in triplicate (mean ± standard deviation)
Antigen delivery with nanoparticles can enhance intracellular antigen uptake, potentially resulting in an increase in antigen cross-presentation (39,54). To determine if observed differences in antigen presentation between architectures were a consequence of antigen uptake, flow cytometry was used to determine the relative amount of fluorescently labeled ovalbumin endocytosed by DC2.4 cells after 5 h. As shown in Fig. 6b, conjugation of antigen to polymeric carriers enhanced antigen uptake by over twofold (2.1-, 2.5-, and 2.4-fold for linear, hyperbranched, and cross-linked, respectively). Differences in antigen uptake between the linear diblock, the hyperbranched, and the cross-linked architecture were modest, however, suggesting that changes in MHC-I presentation are likely a consequence of polymer architecture and membrane-destabilizing activity rather than a simple association with increased antigen uptake. This is in accordance with our previous work (12) demonstrating that equivalent antigen uptake of analogously structured but non-pH-responsive polymeric carriers does not enhance class I antigen presentation.
DISCUSSION
The architecture of drug carriers can play a critical role in determining delivery efficiency, and therefore, control of polymer architecture is an essential tool in the optimization of drug delivery platforms. We have previously demonstrated that polymeric antigen carriers with pH-responsive endosomal escape activity can significantly enhance MHC-I antigen presentation and attendant CD8+ T cell responses (11,12,55). These carriers were linear diblock copolymers composed of a hydrophilic segment with pendant thiol-reactive groups for antigen conjugation and a pH-responsive and endosomolytic terpolymer segment composed of DMAEMA, propylacrylic acid (PAA), and BMA. Herein, we describe a related but new family of architecturally distinct antigen carriers that utilize a pH-responsive DEAEMA-co-BMA element, and compare the membrane-destabilizing activity and antigen delivery capabilities of micellar nanoparticles assembled with linear, hyperbranched, or cross-linked polymers.
Consistent with our previous studies describing linear di- or triblock delivery platforms using DEAEMA-co-BMA polymers as the pH-responsive segment (42,43), we found that addition of a hydrophilic block, here DMA, induces polymer self-assembly into micellar nanoparticles that undergo a pH-dependent micelle-to-unimer transition driven by protonation of DEAEMA residues (12,43). We have previously observed that this transition correlates strongly with pH-dependent membrane-destabilizing activity in an erythrocyte lysis assay (43). Here, we found that the hyperbranched architecture displayed a more gradual transition with a midpoint that was shifted to lower pH values relative to the linear or cross-linked architecture (∼pH 6.4 vs. 6.8). Interestingly, the hyperbranched carrier displayed strong membrane-destabilizing activity at pH 6.6, suggesting that endosomolytic segments are able to interact with erythrocyte membranes, while chains are in a micellar conformation in the bulk. Despite different micelle-to-unimer transitions, both the hyperbranched and cross-linked architectures demonstrated comparable pH-dependent membrane-destabilizing activity that was substantially greater than the linear diblock architecture. While a direct comparison between the membrane-disruptive potency of these carriers and our previously published carriers was not performed, the degree of hemolysis (ca. 80%) observed at pH < 6.6 at only 1.25 μg/mL is substantially greater than what we have observed with other diblock polymers bearing neutral coronas at these concentrations (unpublished observations).
In contrast to our previous reports describing significant enhancements in class I antigen presentation using linear diblock copolymer micelles with a pH-responsive core composed of DMAEMA, PAA, and BMA (11,12), the linear diblock copolymer used in this work did not enhance MHC-I presentation. It is conceivable that the BMA-co-DEAEMA segment may be intrinsically less effective at enhancing antigen delivery to the MHC-I processing pathway or that the DMA corona-forming block used here interferes with activity to a greater extent than the DMAEMA and HPMA segments used previously. Significantly, the increased membrane-destabilizing activity of the hyperbranched and cross-linked architectures resulted in MHC-I presentation, with the hyperbranched carrier facilitating the most efficient antigen delivery to the MHC-I processing pathway. While the relatively low hemolytic activity of the linear carrier provides a reasonable explanation for its inefficient MHC-I presentation, the mechanism by which the hyperbranched architecture enhances MHC-I presentation over the cross-linked carrier is less clear. The hyperbranched polymer architecture exhibited slightly higher hemolytic activities (Fig. 4a) and transitioned at lower pH values to unimers (Fig. 4b). These properties and the underlying differences in endosomal membrane activity are correlated with higher cross-presentation activities. While more fundamental studies aimed at elucidating this mechanism are necessary, these studies clearly demonstrate the superior capacity of the hyperbranched system to enhance presentation of protein antigen on MHC-I.
CONCLUSION
We have utilized RAFT polymerization techniques to synthesize pH-responsive endosomolytic diblock copolymers with three distinct architectures—linear, core-crosslinked, and hyperbranched—that self-assemble into micellar nanoparticles. Micellar nanoparticles bearing pyridyl disulfide groups enabled efficient covalent loading of a protein antigen via a reducible linkage, and by controlling the polymer to antigen ratio, equivalently sized antigen nanocarriers composed of architecturally distinct polymers were generated. Cross-linked and hyperbranched architectures demonstrated more potent pH-dependent membrane-destabilizing activity than their linear counterpart, and both significantly enhanced MHC-I antigen presentation relative to free antigen or the linear variant, with the hyperbranched structure yielding the most efficient antigen cross-presentation of the polymers explored herein. While further study is necessary to elucidate the mechanisms that underlie this observation, this work highlights the potential importance of architecture in enhancing cytosolic delivery through the use of pH-responsive endosomolytic carriers. Additionally, the antigen nanocarriers described herein offer a promising strategy for enhancing CTL responses to protein antigens and merit further investigation as vaccines.
Electronic Supplementary Material
(DOCX 5195 kb)
Acknowledgments
This research is supported by the Science and Industry Endowment Fund, the National Institutes of Health (R01EB002991 and R21EB014572), the Washington State Life Science Discovery Fund (Grant No. 2496490), the National Science Foundation Graduate Research Fellowship under Grant DGE-1256082 (S.K.), and the Irvington Institute Fellowship Program of the Cancer Research Institute (J.T.W.).
Footnotes
John T. Wilson and Almar Postma are equally contributing authors.
Contributor Information
John Chiefari, Phone: +61 3 9545 2508, Email: john.chiefari@csiro.au.
Patrick S. Stayton, Phone: (206) 685-8148, Email: stayton@u.washington.edu
References
- 1.Foged C, Hansen J, Agger EM. License to kill: formulation requirements for optimal priming of CD8(+) CTL responses with particulate vaccine delivery systems. Eur J Pharm Sci. 2012;45(4):482–491. doi: 10.1016/j.ejps.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell JW. Designing CD8+ T cell vaccines: it’s not rocket science (yet). Current opinion in immunology. Elsevier Ltd; 2010 Jun 1;22(3):402–10. [DOI] [PMC free article] [PubMed]
- 3.Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19(12):1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 4.Flatz L, Hegazy AN, Bergthaler A, Verschoor A, Claus C, Fernandez M, et al. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat Med. 2010;16(3):339–345. doi: 10.1038/nm.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AV. Viral vectors as vaccine platforms: deployment in sight. Curr Opin Immunol. 2011;23(3):377–382. doi: 10.1016/j.coi.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Liu MA. Immunologic basis of vaccine vectors. Immunity. 2010;33(4):504–515. doi: 10.1016/j.immuni.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462(7272):449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- 8.Black M, Trent A, Tirrell M, Olive C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev Vaccines. 2010;9(2):157–173. doi: 10.1586/erv.09.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24(28):3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed SS, Plotkin SA, Black S, Coffman RL. Assessing the safety of adjuvanted vaccines. Sci Transl Med. 2011;3(93):93rv2. doi: 10.1126/scitranslmed.3002302. [DOI] [PubMed] [Google Scholar]
- 11.Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee C-C, Opara C, et al. pH-responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano. 2013;7(5):3912–3925. doi: 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller S, Wilson JT, Patilea GI, Kern HB, Convertine AJ, Stayton PS. Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8(+) T cell responses. J Control Release. 2014;191:24–33. doi: 10.1016/j.jconrel.2014.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moon JJ. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nature Materials. Nature Publishing Group; 2011 Feb 20;10(3):243–51. [DOI] [PMC free article] [PubMed]
- 14.Nembrini C, Stano A, Dane KY, Ballester M, van der Vlies AJ, Marsland BJ, et al. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc Natl Acad Sci U S A. 2011;108(44):E989–E997. doi: 10.1073/pnas.1104264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee I-H, Kwon H-K, An S, Kim D, Kim S, Yu MK, et al. Imageable antigen-presenting gold nanoparticle vaccines for effective cancer immunotherapy in vivo. Angew Chem Int Ed. 2012;51:8800–8805. doi: 10.1002/anie.201203193. [DOI] [PubMed] [Google Scholar]
- 16.Sahdev P, Ochyl LJ, Moon JJ. Biomaterials for nanoparticle vaccine delivery systems. Pharm Res. 2014; 31: 2563-82. [DOI] [PMC free article] [PubMed]
- 17.Joshi VB, Geary SM, Salem AK. Biodegradable particles as vaccine delivery systems: size matters. AAPS J. 2013;15(1):85–94. doi: 10.1208/s12248-012-9418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat Mater. 2011;10(3):243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuba E, Kojima C, Harada A, Tana, Watarai S, Kono K. pH-sensitive fusogenic polymer-modified liposomes as a carrier of antigenic proteins for activation of cellular immunity. Biomaterials. 2010;31(5):943–951. doi: 10.1016/j.biomaterials.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Andrews CD, Huh M-S, Patton K, Higgins D, Van Nest G, Ott G, et al. Encapsulating immunostimulatory CpG oligonucleotides in listeriolysin O-liposomes promotes a Th1-type response and CTL activity. Mol Pharm. 2012;9(5):1118–1125. doi: 10.1021/mp2003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duewell P, Kisser U, Heckelsmiller K, Hoves S, Stoitzner P, Koernig S, et al. ISCOMATRIX adjuvant combines immune activation with antigen delivery to dendritic cells in vivo leading to effective cross-priming of CD8+ T cells. J Immunol. 2011;187(1):55–63. doi: 10.4049/jimmunol.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou W, Moguche AO, Chiu D, Murali-Krishna K, Baneyx F. Just-in-time vaccines: biomineralized calcium phosphate core-immunogen shell nanoparticles induce long-lasting CD8(+) T cell responses in mice. Nanomedicine. 2014;10(3):571–578. doi: 10.1016/j.nano.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Wu L, Meng F, Wang Z, Deng C, Liu H, et al. pH and reduction dual-bioresponsive polymersomes for efficient intracellular protein delivery. Langmuir. 2012;28(4):2056–2065. doi: 10.1021/la203843m. [DOI] [PubMed] [Google Scholar]
- 24.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA, Hubbell JA. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials. 2012;33(26):6211–6219. doi: 10.1016/j.biomaterials.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 25.Sheng KC, Kalkanidis M, Pouniotis DS, Esparon S, Tang CK, Apostolopoulos V, et al. Delivery of antigen using a novel mannosylated dendrimer potentiates immunogenicity in vitro and in vivo. Eur J Immunol. 2008;38(2):424–436. doi: 10.1002/eji.200737578. [DOI] [PubMed] [Google Scholar]
- 26.Eby JK, Dane KY, O’Neil CP, Hirosue S, Swartz MA, Hubbell JA. Polymer micelles with pyridyl disulfide-coupled antigen travel through lymphatics and show enhanced cellular responses following immunization. Acta Biomater. 2012;8(9):3210–3217. doi: 10.1016/j.actbio.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Procko E, Berguig GY, Shen BW, Song Y, Frayo S, Convertine AJ, et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014;157(7):1644–1656. doi: 10.1016/j.cell.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chu DSH, Schellinger JG, Shi J, Convertine AJ, Stayton PS, Pun SH. Application of living free radical polymerization for nucleic acid delivery. Acc Chem Res. 2012;45(7):1089–1099. doi: 10.1021/ar200242z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moad G, Rizzardo E, Thang SH. Living radical polymerization by the RAFT process - A third update. Aust J Chem. 2012;65:985–1076.
- 30.Moad G, Rizzardo E, Thang SH. Living radical polymerization by the RAFT process - A second update. Aust J Chem. 2009;62:1402–72.
- 31.Moad G, Rizzardo E, Thang SH. Living radical polymerization by the RAFT process - A first update. Aust J Chem. 2006;59:669–92.
- 32.Moad G, Rizzardo E, Thang SH. Living radical polymerization by the RAFT process. Aust J Chem. 2005;58:379–410.
- 33.Moad G, Rizzardo E, Thang SH. RAFT polymerization and some of its applications. Chem Asian J. 2013;8:1634–44. [DOI] [PubMed]
- 34.Moad G. RAFT-Crosslinking (co)polymerization of multi-olefinic monomers to form polymer networks. Polym Int. 2015;60:15–24.
- 35.Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: the RAFT process. Macromolecules. 1998;31(16):5559–5562. doi: 10.1021/ma9804951. [DOI] [Google Scholar]
- 36.Gregory A, Stenzel MH. Complex polymer architectures via RAFT polymerization: from fundamental process to extending the scope using click chemistry and nature’s building blocks. Prog Polym Sci. 2012;37(1):38–105. doi: 10.1016/j.progpolymsci.2011.08.004. [DOI] [Google Scholar]
- 37.Gao H, Matyjaszewski K. Synthesis of functional polymers with controlled architecture by CRP of monomers in the presence of cross-linkers: from stars to gels. Prog Polym Sci. 2009;34(4):317–350. doi: 10.1016/j.progpolymsci.2009.01.001. [DOI] [Google Scholar]
- 38.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 39.De Temmerman M-L, Rejman J, Demeester J, Irvine DJ, Gander B, De Smedt SC. Particulate vaccines: on the quest for optimal delivery and immune response. Drug Discovery Today. Elsevier Ltd; 2011 Jul 1;16(13–14):569–82. [DOI] [PubMed]
- 40.Galloway AL, Murphy A, DeSimone JM, Di J, Herrmann JP, Hunter ME, et al. Development of a nanoparticle-based influenza vaccine using the PRINT technology. Nanomedicine. 2013;9(4):523–531. doi: 10.1016/j.nano.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Yoo J-W, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nature Publishing Group; 2011;10:521-535. [DOI] [PubMed]
- 42.Cheng C, Convertine AJ, Stayton PS, Bryers JD. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials. 2012;33(28):6868–6876. doi: 10.1016/j.biomaterials.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manganiello MJ, Cheng C, Convertine AJ, Bryers JD, Stayton PS. Diblock copolymers with tunable pH transitions for gene delivery. Biomaterials. 2012;33(7):2301–2309. doi: 10.1016/j.biomaterials.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Brien N, McKee A, Sherrington DC, Slark AT, Titterton A. Facile, versatile and cost effective route to branched vinyl polymers. Polymer. 2000;41(15):6027–6031. doi: 10.1016/S0032-3861(00)00016-1. [DOI] [Google Scholar]
- 45.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release. 2009;133(3):221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crownover EF, Convertine AJ, Stayton PS. pH-responsive polymer–antigen vaccine bioconjugates. Polym Chem. 2011;2(7):1499–504.
- 47.Wei Z, Hao X, Kambouris PA, Gan Z, Hughes TC. One-pot synthesis of hyperbranched polymers using small molecule and macro RAFT inimers. Polymer. 2012;53:1429–1436. doi: 10.1016/j.polymer.2012.02.011. [DOI] [Google Scholar]
- 48.Murthy N, Robichaud JR, Tirrell DA, Stayton PS, Hoffman AS. The design and synthesis of polymers for eukaryotic membrane disruption. J Control Release. 1999;61(1–2):137–143. doi: 10.1016/S0168-3659(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 49.Poly J, Wilson DJ, Destarac M, Taton D. A comprehensive investigation into “controlled/living” chain growth crosslinking copolymerization including a back to basics modeling. J Polym Sci A Polym Chem. 2009;47(20):5313–5327. doi: 10.1002/pola.23580. [DOI] [Google Scholar]
- 50.Wang R, Luo Y, Li B-G, Zhu S. Modeling of branching and gelation in RAFT copolymerization of vinyl/divinyl systems. Macromolecules. 2009;42(1):85–94. doi: 10.1021/ma802006c. [DOI] [Google Scholar]
- 51.Bannister I, Billingham NC, Armes SP, Rannard SP, Findlay P. Development of branching in living radical copolymerization of vinyl and divinyl monomers. Macromolecules. 2006;39(22):7483–92.
- 52.Rosselgong J, Armes SP, Barton W, Price D. Synthesis of highly branched methacrylic copolymers: observation of near-ideal behavior using RAFT polymerization. Macromolecules. 2009;42(16):5919–5924. doi: 10.1021/ma900958a. [DOI] [Google Scholar]
- 53.Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: cross-presentation and T cell activation. Vaccine. Elsevier Ltd; 2010 Nov 23;28(50):7897–906. [DOI] [PubMed]
- 54.Swartz MA, Hirosue S, Hubbell JA. Engineering approaches to immunotherapy. Sci Transl Med. 2012;4(148):148rv9. doi: 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
- 55.Foster S, Duvall CL, Crownover EF, Hoffman AS, Stayton PS. Intracellular delivery of a protein antigen with an endosomal-releasing polymer enhances CD8 T-cell production and prophylactic vaccine efficacy. Bioconjug Chem. 2010;21(12):2205–2212. doi: 10.1021/bc100204m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 5195 kb)