

Abstract
ABT-639 is a selective T-type calcium channel blocker with efficacy in a wide range of preclinical models of nociceptive and neuropathic pain. In the current first-in-human (FIH) study, the pharmacokinetics, tolerability, and safety of ABT-639 after single- (up to 170 mg) and multiple doses (up to 160 mg BID) were evaluated in healthy volunteers in a randomized, double-blinded, placebo-controlled manner. ABT-639 demonstrated acceptable safety and pharmacokinetic profiles in human. Results from assessment of the routine laboratory variables showed an unexpected statistically significant and clinically relevant decrease in blood uric acid with the increase in ABT-639 dose, which is possibly due to inhibition in URAT1 transporter. Pharmacokinetic/pharmacodynamic models were constructed to characterize the relationship between ABT-639 exposure and uric acid response. The final model was a mechanism-based indirect response pharmacodynamic model with the stimulation of uric acid elimination by ABT-639. The model estimated Kin values in males and females were 10.2 and 7.13 μmol/h, respectively. The model estimated Kout was 0.033 1/h. ABT-639 concentration that can produce 50% stimulation in uric acid elimination was estimated to be 8,070 ng/mL. Based on the final model, further simulations were conducted to predict the effect of ABT-639 on uric acid in gout patients. The simulation results indicated that, if the urate-lowering response to ABT-639 in gout patients is similar to that in healthy subjects, ABT-639 BID doses of 140 mg or higher would be expected to provide clinically meaningful lowering of blood uric acid levels below the 380 μmol/L solubility limit of monosodium urate.
KEY WORDS: ABT-639, exposure-response analyses, uric acid, PK-PD modeling, population pharmacokinetics, URAT1 inhibition
INTRODUCTION
ABT-639 is a selective T-type calcium channel blocker that has little or no activity at other calcium channels, such as L-type, P-type, N-type, and R-type calcium channels. ABT-639 produced robust analgesic activities following oral administration across a wide range of preclinical models of nociceptive and neuropathic pain in rodents—these effects are suggestive of broad-spectrum analgesic efficacy for chronic pain (1). Nonclinical experiments indicated favorable safety profile at doses that provide ABT-639 exposures equivalent to the estimated efficacious exposures in humans (1).
As part of the clinical development of ABT-639, the pharmacokinetics, tolerability, and safety of ABT-639 after single- and multiple doses were evaluated in healthy volunteers in a phase 1 first-in-human (FIH) study. In this study, results from assessment of the routine laboratory variables showed an unexpected statistically significant decrease in blood uric acid with the increase in ABT-639 dose. Additional in vitro studies investigating the mechanism underlying this interesting phenomenon revealed that ABT-639 has potent inhibitory effect on URAT1, a urate transporter known to play a key role in uric acid reabsorption (2). To characterize ABT-639 pharmacokinetic profile and its effect on uric acid, a population PK/PD (uric acid) model was constructed based on the observed data that were collected in the FIH clinical trial. This model was then used to perform simulations to predict the change in blood uric acid under different ABT-639 dosing regimens.
SUBJECTS AND METHODS
Subjects
Male and female subjects between the ages of 18 and 55 years (parts I to III) or at least 65 years (part IV) were eligible for inclusion into the study. All subjects were in good health, as determined by medical history, physical examination, vital signs, electrocardiography, and clinical laboratory measurements. The study protocol was approved by the ethics committee and all subjects provided written informed consent to participate. The study was conducted at the Abbott Clinical Pharmacology Research Unit, at Vista Medical Center (Waukegan, IL, USA). The study was carried out in accordance with the protocol, International Conference on Harmonization Good Clinical Practice guidelines, and applicable local regulatory requirements and laws.
Study Design
This phase 1 single- and multiple-dose study consisted of four parts:
-
Part I (Single-Dose Study in Healthy Adults)
Single-dose pharmacokinetics and tolerability was assessed using a double-blind, randomized, placebo-controlled, escalating-dose study design. ABT-639 was administered orally under fasting condition. Forty subjects participated in this study in five dose groups (10, 30, 60, 100, and 170 mg of ABT-639). In each group, eight subjects were randomized in a 3:1 ratio such that six subjects received ABT-639 and two subjects received placebo. Venous blood samples were collected within 10 min prior to oral dosing (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 16, 24, 36, 48, 60, 72, 95, and 120 h after oral dosing.
-
Part II (Food Effect)
Effect of food on ABT-639 pharmacokinetics was investigated in a randomized, single-dose, open-label, and two-period crossover manner. Twelve subjects participated in this study. ABT-639 was administered at a dose of 50 mg in each period. Blood samples were collected using the same sampling scheme as that in part I.
-
Part II0049 (Multiple-Dose Study in Healthy Adults)
Multiple-dose pharmacokinetics and tolerability was assessed by a double-blind, randomized, placebo-controlled, escalating-dose approach. ABT-639 was administered orally 30-min after breakfast. Forty-eight subjects participated in this study in four dose groups (10, 40, 100, and 160 mg of ABT-639 administered twice daily [BID]). In each group, 12 subjects were randomized in a 3:1 ratio such that 9 subjects received ABT-639 and 3 subjects received placebo for 7 days (10, 40, and 100 mg BID dose groups) or 14 days (160 mg BID dose group). For the 10, 40, and 100 mg BID dose groups, blood samples were collected within 10 min prior to morning dosing on study day 1 at 0 h (morning dose) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, and 12 h; on study days 5 and 6 at 0 h (prior to morning dose); and on study day 7 at 0 h (prior to morning dose), and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12 (prior to evening dose), 12.5, 13, 13.5, 14, 15, 16, 18, 21, 24, 36, 48, 60, 72, and 96 h. For the 160-mg BID dose group, in addition to the above-mentioned samples, blood samples were collected on study days 12 and 13 at 0 h (prior to morning dose); and on study day 14 at 0 h (prior to morning dose) and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12 (prior to evening dose), 12.5, 13, 13.5, 14, 15, 16, 18, 21, 24, 36, 48, 60, 72, and 96 h.
-
Part IV (Multiple-Dose Study in Healthy Elderly Subjects)
Multiple-dose pharmacokinetics were assessed in healthy elderly subjects in a double-blind, randomized, placebo-controlled manner. Twenty-four subjects participated in this study in two dose groups (40 and 160 mg of ABT-639 BID). In each group, 12 subjects were randomized in a 2:1 ratio such that 8 subjects received ABT-639 and 4 subjects received placebo for 7 days. Blood samples were collected using the same sampling scheme as that in part III.
In addition, urine samples for assay of ABT-639 were collected in parts III and IV. Urine samples were collected over the following intervals: prior to morning dose, 0 to 6, 6 to 12, 12 to 18, and 18 to 24 h after morning dose on day 7 (all groups in parts III and IV) and day 14 (160 BID group in part III).
Analytical Methods
ABT-639 plasma and urine concentrations were determined using a validated liquid chromatography method with tandem mass spectrometric detection at AbbVie. A-1145843 d4 was used as an internal standard. The lower limit of quantitation (LLOQ) value was 1.1 ng/mL for ABT-639. The analytical method was validated over the concentration range of 1.11 to 3,450 ng/mL for the plasma analyses and 1.12 to 1,230 ng/mL for the urine analyses. All calibration curves had r2 values >0.993. The coefficient of variation (CV) values ranged from 2.3 to 26.8% for the plasma samples and 2.1 to 5.2 for the urine samples. The mean bias values ranged from −4.5 to 5.8% for the plasma samples and −2.5 to 2.4% for the urine samples. Samples with concentrations above the upper limit of quantitation for any given run were diluted with blank matrix and re-assayed. Values that were less than the LLOQ for a given run were reported as zero. The plasma samples were stored at −20°C or lower. The storage stability was 154 days. All samples were analyzed within the validated storage stability.
Pharmacokinetic Analysis
Plasma pharmacokinetic parameters of ABT-639 were estimated using non-compartmental analysis (NCA) with WinNonlin Professional software (version 5.2, Pharsight Corporation, Mountain View, California). The estimated pharmacokinetic parameters included the maximum observed plasma concentration (Cmax), time to Cmax (tmax), the apparent terminal phase elimination rate constant (λ), terminal phase elimination half-life (t1/2), and the apparent oral clearance (CL/F). For the single dose pharmacokinetic study, the area under the plasma concentration-time curve (AUC) from time 0 to infinity (AUCinf) was calculated using the linear trapezoidal rule. For the multiple-dose pharmacokinetic study, the AUC for the dosing intervals (AUC0–12) on days 1, 7, and 14 (160 mg dose only) was calculated. ABT-639 trough concentrations were determined for the 12-h interval on day 7 or day 14. The accumulation ratio (Rac) was calculated as the ratio of AUC0–12 on day 7 or day 14 to AUC0–12 on day 1. The fraction of unchanged ABT-639 recovered in urine (fe) and renal clearance (CLR) were determined based on the urine results obtained in parts III and IV.
Statistical Analysis
Statistical analyses were performed using SAS software (Version 8.2; SAS Institute, Inc, Cary, North Carolina). Pharmacokinetic linearity and dose proportionality were assessed by ANCOVA of ABT-639 dose-normalized Cmax, AUCinf, tmax, and λ following administration of single doses and dose-normalized Cmax, AUC0-12 and Ctrough, tmax, and λ following administration of multiple doses. Attainment of steady state for ABT-639 was assessed by repeated measures analysis of dose-normalized, pre-morning dose ABT-639 plasma concentrations for study days 5, 6, and 7 for multiple-dose regimens evaluated. In addition, one-way ANCOVA of study day 7 morning dose (0–12 h) was performed for dose-normalized Cmax, AUC0–12, and Ctrough to compare the exposure of ABT-639 in healthy adults and elderly subjects at comparable dose levels (40 and 160 mg BID for elderly subjects versus 40 and 160 mg BID for healthy adults).
Pharmacokinetic/Pharmacodynamic Modeling of Uric Acid
Population Model-Building
Population pharmacokinetic and pharmacodynamic analyses were conducted using the nonlinear mixed-effects modeling software NONMEM, Version 7.2 (Icon Development Solutions, Ellicott City, Maryland). Single- and multiple-dose ABT-639 pharmacokinetic data in healthy adults and elderly subjects were compiled and analyzed simultaneously. The corresponding final post-hoc Bayes estimates of ABT-639 pharmacokinetic parameters for each subject were then fixed during the development of pharmacodynamic model of uric acid. Actual sampling times were used in the analysis. The first-order conditional estimation method with the interaction (FOCEI) and a user-defined subroutine (ADVAN6) were used to estimate the typical population parameters, random inter-individual variability, and residual variability between observed and individually predicted plasma ABT-639 concentrations and blood uric acid concentrations. All inter-individual variability was assumed to be log-normally distributed (mean 0, variance ω2) and described by an exponential model. The residual error model was described by combined additive and exponential random effects model. Model development was guided by the objective function value, graphical goodness-of-fit analysis, the precision of parameter estimation, the plausibility of the estimated parameters, and the stability of the model. The likelihood ratio test was used for comparing nested models where a drop of at least 3.84 in the objective function (corresponding to p < 0.05 for one-parameter difference, assuming χ2 distribution) was considered as significant.
Pharmacokinetic Model
Based on the observed bi-exponential decline of ABT-639, a two-compartment model served as the starting pharmacokinetic model. Single-dose pharmacokinetic evaluation was conducted under fasting conditions and the multiple-dose evaluation was conducted under non-fasting conditions. Non-compartmental analysis of the data showed that ABT-639 tmax was considerably delayed and ABT-639 Cmax was lower when ABT-639 was administered with food, indicating different absorption rates between fasting and non-fasting conditions. Therefore, different absorption rates (ka1 and ka2) were assigned for the single-dose and morning multiple-dose data, respectively. In the multiple-dose study, exposure following the morning dose was higher than that in the evening which is probably due to heavier evening meal. To account for the different food effect, ka3 was used for the evening dose of ABT-639.
Pharmacodynamic Model
In the absence of ABT-639, the physiological process of production and elimination of uric acid can be described by the following equation:
| 1 |
where kin represents the zero-order constant for production of the uric acid and kout represents the first-order constant for uric acid elimination. R is the response variable representing uric acid.
According to this basic indirect-response model, ABT-639’s dose-dependent decrease in blood uric acid concentration could be caused either by an inhibition of kin or a stimulation of kout. Results from the in vitro study revealed that ABT-639 inhibited URAT1, a transporter which plays a key role in uric acid reabsorption. Based on this mechanism of action, stimulation of kout by ABT-639 was considered to be appropriate to describe ABT-639 pharmacodynamics. In the presence of ABT-639, the differential equation for the response (i.e., uric acid) is:
| 2 |
with
| 3 |
Where S(C) is the stimulation function, Cp is the concentration of ABT-639 in plasma, Smax is the maximum stimulation capacity, and SC50 represents ABT-639 concentration producing 50% stimulation.
The schematic of the final pharmacokinetic/pharmacodynamic model of ABT-639 on uric acid is shown in Fig. 1.
Fig. 1.
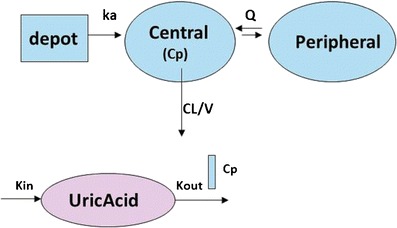
Pharmacokinetic/pharmacodynamic model of ABT-639’s effect on uric acid
Model Simulation
Based on the estimated pharmacokinetic and pharmacodynamic parameters from the final model, simulations were performed to predict the urate-lowering effect of ABT-639 for different ABT-639 dosing regimens. The dosing regimens evaluated in the simulation include 40, 100, 120, 140, 160, 180, and 200 mg BID. The urate-lowering effect of ABT-639 was predicted in both healthy volunteers and gout patients. The urate-lowering behavior of ABT-639 was assumed to be similar between the two populations. Based on the information collected from the current study and the literature data (3,4), the baseline values of blood uric acid in healthy volunteers and gout patients were assumed to be 300 and 520 μmol/L, respectively.
RESULTS
One hundred and twenty-four subjects entered the study and received study medications. There was no premature discontinuation during the study. A summary of subject demographics is presented in Table I.
Table I.
Demographic Summary for All Subjects (Parts I Though IV)
| Demographic | Mean ± SD | Min ∼ max |
|---|---|---|
| Part I (N = 40) | ||
| Age (years) | 26.4 ± 8.3 | 19–48 |
| Weight (kg) | 75.4 ± 9.4 | 56–92 |
| Height (cm) | 175 ± 6 | 165–185 |
| Sex | 40 males (100%), 0 females (0%) | |
| Race | 34 White (85.0%), 5 Other (12.5%), 1 Asian (2.5%) | |
| Part II (N = 12) | ||
| Age (years) | 29.4 ± 13.7 | 18–52 |
| Weight (kg) | 73.6 ± 7.9 | 63–90 |
| Height (cm) | 183 ± 8 | 172–194 |
| Sex | 10 males (83.3%), 2 females (16.7%) | |
| Race | 12 White (100%) | |
| Part III (N = 48) | ||
| Age (years) | 34.0 ± 11.8 | 20–56 |
| Weight (kg) | 75.5 ± 11.0 | 57–98 |
| Height (cm) | 178 ± 8 | 164–195 |
| Sex | 46 Males (95.8%), 2 females (4.2%) | |
| Race | 43 White (89.6%), 3 Black (6.3%), 1 Asian (2.1%), 1 American Indian/Alaska Native (2.1%) | |
| Part IV (N = 24) | ||
| Age (years) | 69.3 ± 2.7 | 66–76 |
| Weight (kg) | 73.4 ± 11.9 | 54–95 |
| Height (cm) | 170 ± 11 | 151–188 |
| Sex | 14 males (58.3%), 10 females (41.7%) | |
| Race | 22 White (91.7%), 2 Asian (8.3%) | |
ABT-639 Pharmacokinetics
The mean ± SD pharmacokinetic parameters of ABT-639 after single and multiple dose regimens in healthy adults are presented in Tables II and III, respectively. The harmonic mean terminal half-life of ABT-639 ranged from 5 to 7 h, on average, across all dose regimens evaluated in healthy adults. Food delayed ABT-639 Tmax by 3 h and resulted in 27% decrease in Cmax. Food had no effect on ABT-639 AUC. After single-dose administration, ABT-639 Cmax was dose-proportional in the 10 to 170 mg dose range. ABT-639 AUC was dose-proportional over the 10 to 100 mg dose range but less than dose-proportional at 170 mg. Consistent with the single dose data, after multiple-dose administration, ABT-639 exposure increased approximately dose-proportionally over the 10 to 100 mg dose range, with less than dose-proportional increase at higher doses. As the solubility of ABT-639 is low, this less than dose-proportional increase in ABT-639 exposure in the highest dose group may be explained by the decreased solubility and consequently decreased bioavailability at higher doses. ABT-639 steady state was achieved by day 5 following multiple doses and the steady state exposures of ABT-639 were 1.1- to 1.4-fold of the first dose exposure. Compared with the morning dose, lower Cmax and higher Ctrough were observed after the evening dose of ABT-639, indicating a diurnal effect on ABT-639 exposure, which may be caused by food. The excretion of ABT-639 as unchanged drug through urine was minimal. Compared with the exposure in healthy adults, ABT-639 exposures in elderly subjects were slightly higher (10–30%) with a slightly longer terminal half-life (7–9 vs. ∼6 h). However, no statistically significant difference was found between elderly and adults in any of the pharmacokinetic parameters evaluated.
Table II.
Pharmacokinetic Parameters of ABT-639 Following Single Dose Administration
| Part I, fasting | Part II | ||||||
|---|---|---|---|---|---|---|---|
| Pharmacokinetic parameters | 10 mg | 30 mg | 60 mg | 100 mg | 170 mg | 50 mg fasting | 50 mg non-fasting |
| C max (ng/mL) | 420 ± 77 | 1,310 ± 217 | 2,600 ± 522 | 5,080 ± 1,230 | 7,430 ± 1,440 | 2,140 ± 236 | 1,570 ± 161 |
| T max (h) | 1.25 ± 0.27 | 1.67 ± 1.17 | 1.58 ± 0.8 | 1.00 ± 0.63 | 1.50 ± 0.77 | 1.17 ± 0.75 | 4.25 ± 0.87 |
| t ½ a (h) | 6.34 ± 1.14 | 6.01 ± 1.48 | 6.29 ± 1.5 | 5.63 ± 0.59 | 6.56 ± 0.88 | 6.44 ± 1.08 | 6.21 ± 0.91 |
| AUC∞ (μg·h/mL) | 4.31 ± 1.17 | 11.7 ± 3.7 | 23.0 ± 7.1 | 34.1 ± 6.6 | 48.0 ± 10.4 | 17.4 ± 2.9 | 16.6 ± 2.4 |
| CL/F (L/h) | 2.46 ± 0.62 | 2.77 ± 0.82 | 2.90 ± 1.18 | 3.04 ± 1.66 | 3.67 ± 0.73 | 2.94 ± 0.40 | 3.07 ± 0.41 |
Mean ± standard deviation reported for all parameters except harmonic mean and pseudo standard deviation for half-life
Table III.
Mean ± SD Pharmacokinetic Parameters of ABT-639 Following Multiple Doses
| Part III, healthy adults | Part IV, elderly subjects | ||||||
|---|---|---|---|---|---|---|---|
| Pharmacokinetic parameters | Units | 10 mg BID (N = 9) | 40 mg BID (N = 9) | 100 mg BID (N = 9) | 160 mg BID (N = 9) | 40 mg BID (N = 9) | 160 mg BID (N = 9) |
| Day 7 | Day 14 | Day 7 | |||||
| C max | ng/mL | 598 ± 204 | 2,070 ± 242 | 5,140 ± 968 | 7,370 ± 1,650 | 2,270 ± 713 | 8,620 ± 1,790 |
| T max | h | 2.56 ± 0.53 | 2.44 ± 0.53 | 2.83 ± 0.50 | 2.17 ± 0.66 | 2.25 ± 0.65 | 1.88 ± 0.58 |
| C trough | ng/mL | 151 ± 80 | 494 ± 210 | 1,080 ± 502 | 1,240 ± 520 | 693 ± 606 | 1,610 ± 608 |
| AUC0–12 a | μg·h/mL | 3.97 ± 1.5 | 13.5 ± 3.1 | 30.7 ± 8.3 | 42.5 ± 11.8 | 15.9 ± 8.2 | 52.0 ± 13.6 |
| AUC12–24 a | μg·h/mL | 3.45 ± 1.40 | 11.9 ± 2.8 | 26.6 ± 7.5 | 39.7 ± 9.7 | 14.7 ± 7.7 | 46.7 ± 10.8 |
| CL/F | L/h | 2.82 ± 0.98 | 3.10 ± 0.64 | 3.46 ± 0.86 | 4.04 ± 1.17 | 2.94 ± 0.95 | 3.28 ± 0.91 |
| t ½ b | h | 5.52 ± 1.2 | 5.80 ± 1.7 | 6.56 ± 1.5 | 6.44 ± 2.24 | 7.40 ± 2.32 | 8.77 ± 2.91 |
| AUC R ac c | 1.36 [1.13–1.75] | 1.12 [1.07–1.45] | 1.16 [1.02–1.47] | 1.23 [1.08–1.40] | 1.25 [1.10–1.60] | 1.13 [0.97–1.29] | |
Mean ± standard deviation reported
aAUC value on study day 7
bHarmonic mean and pseudo standard deviation
cMedian (range)
Assessment of Uric Acid
It has been reported that the body pool of urate is higher in males than that in females, with the value ranging from 800 to 1,500 mg in the former population and 500 to 1,000 mg in the latter (5,6). Consistent with the literature report, in the current study, the mean baseline blood uric acid concentrations in male and female subjects were 317 ± 55 and 221 ± 72 μmol/L, respectively. As shown in Table IV, the mean blood uric acid baseline ranged from 290 to 340 μmol/L in healthy adults and 250 to 300 μmol/L in elderly subjects. It should be noted that in the current study, majority of the population in the healthy adults were male (40/40 in part I, 10/12 in part II, and 46/48 in part III), whereas only 58.3% (14/24) of the subjects in the elderly subjects (part IV) were male. Therefore, the relatively higher blood uric acid baseline values observed in the healthy adults compared with elderly subjects can be attributed to higher percentage of males in the former group.
Table IV.
Uric Acid Before and After ABT-639 Administration
| Dose regimen | Uric acid Day 1 |
Uric acid Day 8 |
% Change in blood uric acid | Proportion of subjects with blood uric acid | |
|---|---|---|---|---|---|
| [μmol/L] | [μmol/L] | >20% decrease | >30% decrease | ||
| Healthy adults | |||||
| 0 mg, BID | 293 ± 35 | 292 ± 58 | 1.30% ↓ | 0% | 0% |
| 10 mg, BID | 303 ± 73 | 291 ± 56 | 2.19% ↓ | 0% | 0% |
| 40 mg, BID | 336 ± 61 | 296 ± 36 | 10.4% ↓ | 11.1% | 11.1% |
| 100 mg, BID | 286 ± 96 | 216 ± 57 | 22.0% ↓ | 66.6% | 22.2% |
| 160 mg, BID | 330 ± 54 | 227 ± 35 | 30.9% ↓ | 88.9% | 44.4% |
| Elderly subjects | |||||
| 0 mg, BID | 291 ± 42 | 306 ± 35 | 5.83 ↑ | 0% | 0% |
| 40 mg, BID | 288 ± 90 | 261 ± 81 | 8.48% ↓ | 0% | 0% |
| 160 mg, BID | 254 ± 77 | 200 ± 63 | 21.3% ↓ | 62.5% | 12.5% |
Dose-dependent decrease in blood uric acid with the increase in ABT-639 dose was observed in both ABT-639 single- and multiple-dose studies. When ABT-639 was administered in healthy adults at doses ≥40 mg BID, the mean decrease in blood uric acid relative to baseline on study day 8 approached 10.4% in the 40 mg dose group, 22.0% in the 100 mg dose group, and 30.9% in the 160 mg dose group. Following ABT-639 multiple dose regimens, the dose-dependent decrease in blood uric acid was detected on study day 2 and the trend of decreasing values remained throughout the dosing period. In addition, uric acid values in all three high multiple dose regimens returned to baseline levels 3 days post-treatment. Similarly, statistically significant decreases in uric acid values were observed in healthy elderly subjects who received multiple doses of 40 or 160 mg BID ABT-639 compared with placebo, with the mean decrease in blood uric acid relative to baseline on study day 8 around 8.5% in the 40 mg dose group and 21.3% in the 160 mg dose group in elderly subjects.
Safety Summary
Fifty-two (52/94, 55%) subjects who received ABT-639 and 18 (18/30, 60%) subjects who received placebo reported at least one treatment-emergent adverse event. None of the adverse events appeared to be dose-dependent. All adverse events were assessed as mild except one event. The majority of adverse events were considered by the investigator as not related or probably not related to study drug. No deaths occurred, no serious adverse events were reported, and no subject discontinued in any part of the study. No consistent and clinically significant mean changes were observed in pulse rate, systolic or diastolic blood pressure, ECG parameters (heart rate, PR, QRS, QT, and QTc), or orthostatic vital signs following a single dose or multiple doses of ABT-639 compared to placebo in both healthy adult and elderly subjects. Except for uric acid, no clinically relevant abnormal findings were observed in other laboratory results, including hematology, clinical chemistry, and urinalysis tests. The safety and tolerability data from elderly subjects were consistent with that from younger subjects. All clinical laboratory tests, including hematology, clinical chemistry, and urinalysis tests, were analyzed in a central lab. No investigations were done to verify that there is no interference in the uric acid assay by ABT-639.
Overall, the safety profile of ABT-639 was acceptable in both single- and multiple-dose studies. The maximum tolerated dose (MTD) was not established as the dose escalation was limited by the safety margin defined based on the preclinical toxicological finding.
Population Pharmacokinetic and Exposure-Response Modeling on Uric Acid
Final pharmacokinetic model parameters are presented in Table V. Initially three distinct absorption rate constants (ka) values were assigned to account for potentially different absorption rates in the fasting, non-fasting morning, and non-fasting evening conditions. As the model estimated ka value in the fasting condition was very close to that in the non-fasting morning condition, in the final model only one ka value was assigned for these two conditions. The model estimated ABT-639 absorption rate was 2.01 h−1 in fasting and non-fasting conditions following the morning dose and 0.91 h−1 in the non-fasting condition after the evening dose. The time course of observed versus population-predicted (PRED) plasma concentrations of ABT-639 after single-dose in healthy adults, multiple-dose in healthy adults, and multiple-dose in elderly subjects are presented in Figs. 2a, and 3a, b, respectively. The proposed pharmacokinetic model overall well characterized the concentration-time profiles of ABT-639 obtained from both single- and multiple-dose investigations.
Table V.
Estimated Population Pharmacokinetic Parameters for ABT-639 from the Final Pharmacokinetic Model
| Parameter | Estimate (RSE) | Parameter | Estimate (RSE) |
|---|---|---|---|
| Pharmacokinetic parameters | |||
| CL (L/h) | 2.5 (5.9%) | ω 2 CL | 0.074 (27.5%) |
| Vc (L) | 18.8 (3.0%) | ω 2 Vc | 0.014 (63.0%) |
| Vp (L) | 4.12 (10.4%) | ω 2 P | 0.201 (179%) |
| Q (L/h) | 0.35 (21.2%) | ω 2 Q | 0.245 (10.4%) |
| k a (h−1) under fasting and non-fasting morning dose conditions | 2.01 (7.2%) | ω 2 ka | 1.55 (7.8%) |
| k a (h−1) under non-fasting evening dose condition | 0.91 (27.3%) | σ 1 2 (proportional residual error) | 0.026 (13.0%) |
| Pharmacodynamic parameters | |||
| K in in male subjects (μmol/hr) | 10.2 (13.3%) | ω 2 Kin | 0.000004 (36.4%) |
| K in in female subjects (μmol/hr) | 7.13 (16.3%) | ω 2 Kout | 0.0323 (8.0%) |
| k out (h−1) | 0.0327 (13.1%) | ω 2 SC50 | 0.354 (7.7%) |
| S max | 1 | ||
| SC50 (ng/mL) | 8,070(5.2%) | σ 3 2 (additive residual error) | 274 (5.2%) |
Fig. 2.

Time courses of observed (mean ± SD) and model predicted (PRED) plasma concentrations of ABT-639 following single escalating doses. Symbols represent observed data. Solid lines represent the predicted results obtained by simultaneous fitting of all available ABT-639 pharmacokinetic data
Fig. 3.
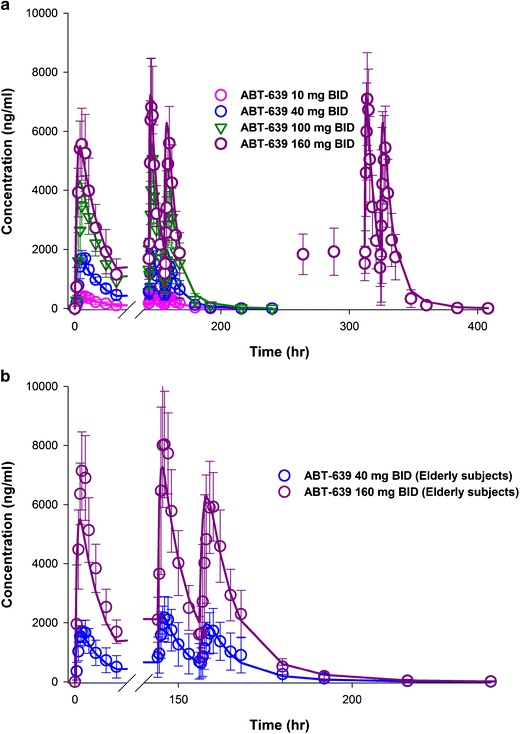
Time courses of observed (mean ± SD) and model predicted (PRED) plasma concentrations of ABT-639 following multiple BID doses in a healthy adults and b elderly subjects. Symbols represent observed data. Solid lines represent the predicted results obtained by simultaneous fitting of all available ABT-639 pharmacokinetic data
Final uric acid model parameters are presented in Table V. Blood uric acid concentration is known to be higher in males than that in females and this trend was also observed in the current study, with the blood uric acid baseline value in women was about two third of the value observed in man. To account for this sex effect in body pool of urate, different input rates (Kin) were assigned in male and female subjects. The model estimated Kin values in males and females were 10.2 and 7.13 μmol/h, respectively. The model estimated Kout was 0.033 1/h. ABT-639 concentration that can produce 50% stimulation in uric acid elimination was estimated to be 8,070 ng/mL. The time course of observed versus population-predicted (PRED) blood uric acid concentrations after multiple-dose administration of ABT-639 in healthy adults and elderly subjects is presented in Fig. 4. As shown in Fig. 4, this mechanism-based indirect response pharmacodynamic model was able to adequately capture the uric acid concentration-time profiles following various ABT-639 dose regimens.
Fig. 4.

Time courses of observed (mean ± SD) and model predicted (PRED) blood uric acid concentrations following ABT-639 BID doses in healthy adults and elderly subjects. Symbols represent observed data. Solid lines represent the predicted results obtained by simultaneous fitting of all available uric acid data
Model Simulation
After the final PK/PD model was constructed, further simulations were performed to predict the effect of ABT-639 on uric acid at various doses in both healthy subjects and gout patients. The dosing regimens evaluated in the simulations included 40, 100, 120, 140, 160, 180, and 200 mg BID for 14 days. The simulated blood uric acid concentration-time profiles in healthy volunteers and gout patients following different ABT-639 dosing regimens are presented in Fig. 5. These simulations predicted that ABT-639 BID doses of 140 mg or higher would be expected to provide clinically meaningful lowering of blood uric acid levels below the 380 μmol/L solubility limit of monosodium urate.
Fig. 5.
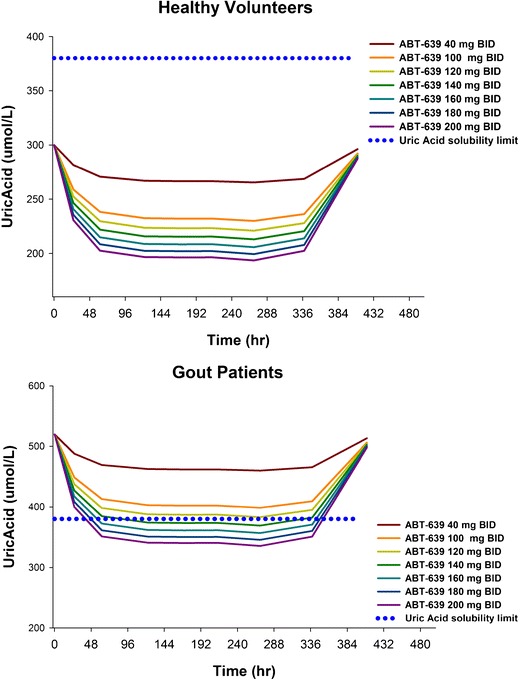
Predicted blood uric acid concentration-time profiles in healthy volunteers (top) and gout patients (bottom) following different ABT-639 dose regimens
DISCUSSION
ABT-639 is a novel selective T-type calcium channel blocker. ABT-639 has demonstrated broad-spectrum analgesic efficacy for chronic pain in the preclinical studies and has favorable preclinical safety profile. Based on these preclinical findings, the development of ABT-639 was moved forward into the clinical stage. The current FIH study was conducted to evaluate the pharmacokinetics, tolerability, and safety of ABT-639 after single dose (up to 170 mg) and multiple-dose (up to 160 mg BID) in healthy volunteers. ABT-639 showed an acceptable safety profile in all evaluated dose regimens and demonstrated favorable pharmacokinetic profiles in human. In this study, blood uric acid concentrations statistically significantly decreased when ABT-639 was administered and this decrease was ABT-639 dose-dependent. This unexpected and interesting phenomenon indicated that ABT-639 may represent a potential candidate not only in the treatment of chronic pain but also in the treatment of hyperuricemia and its complications, such as chronic Gout.
Gout is the most prevalent inflammatory arthritis and it is caused by deposition of monosodium urate crystals within joints after chronic hyperuricemia (7). In gout patients, the rise in the urate pool is generally as a result of impaired renal uric acid excretion, with overproduction of uric acid representing the cause in less than 10% gout patients (8). The primary goal in the treatment of chronic gout is to lower the blood uric acid concentration to less than 6.0 mg/dL (i.e., 360 μmol/L) (9,10), a value below the saturation point of urate (380 μmol/L) in most biological fluids. The most common urate-lowering strategies include reducing urate production with xanthine oxidase inhibitors, such as allopurinol and febuxistat (11,12), and enhancing uric acid urinary excretion with uricosuric agents, such as probenecid, sulfinpyrazone, and benzbromarone. The percentage of reduction from initial blood urate is around 31.7% for allopurinol (4), 44.7–51.5% for febuxistat (11), 58.3% for benzbromarone (4), and 23% for probenecid (13). It should be noted that, for all of the urate-lowering compounds mentioned above, their use is limited due to their less favorable safety or efficacy profiles. For example, allopurinol, the current first-line choice in urate-lowering therapy, can cause life-threatening hypersensitivity reactions (14,15). Febuxistat has been reported to have side effects including elevated liver enzyme activity and a small increase in the rate of serious cardiovascular events (9). Regarding benzbromarone, albeit it is more active than allopurinol, its side effect of hepatotoxicity precludes its wide use. Benzbromarone is not marketed in USA and only has very limited use in Europe due to its severe liver toxicity. In addition to benzbromarone, other uricosuric agents currently available are generally ineffective and potentially nephrotoxic (8,16). In the current study, ABT-639 clearly demonstrated statistically and clinically significant urate-lowering effect in healthy volunteers. In addition, ABT-639 has a favorable safety profile in humans compared to current therapies. Considering the marked rise in prevalence of gout during the past a few decades and the limitation of current urate-lowering therapies, ABT-639 may represent a novel and promising urate-lowering agent in the treatment of hyperuricemia due to its acceptable safety and efficacy profiles.
Results from the in vitro mechanism study indicated that ABT-639 acts through inhibition of URAT1 transporter to enhance uric acid urinary excretion (IC50 4.52 μmol/L, in house data). Therefore, ABT-639 is a uricosuric agent. In addition to URAT1, several other transporters, including OAT, MRP, and UAT, have also been reported to be involved in the transport of uric acid (3,17). The effect of ABT-639 on these transporters is unknown. Further investigations are warranted to fully unravel the mechanism behind the urate-lowering effect of ABT-639.
Based on the observed ABT-639 and uric acid data, we constructed a pharmacokinetic/pharmacodynamic model to characterize the relationship between ABT-639 exposure and uric acid response. The final model was a mechanism-based indirect response pharmacodynamic model with the stimulation of uric acid elimination by ABT-639. Our model was able to characterize both ABT-639 and blood uric acid concentration-time profiles in both single- and multiple-dose evaluations. We also performed simulations to predict the urate-lowering effect of ABT-639 under different ABT-639 dosing regimens in healthy volunteers and gout patients. Based on the literature information, the baseline of blood uric acid in gout patients was set to 520 μmol/L (4), a value well above the solubility limit of monosodium urate. As it is important for gout patients to maintain uric acid concentration below the saturation point for monosodium urate, based on our simulation results, ABT-639 BID doses of 140 mg or higher would be expected to provide clinically meaningful lowering of blood uric acid levels below the 380 μmol/L solubility limit of monosodium urate. It should be noted that in these simulations the urate-lowering behavior of ABT-639 was assumed to be similar between healthy subjects and gout patients after accounting for differences in baseline. Additionally, the uric acid-lowering effect of ABT-639 was assumed to increase in a dose-dependent manner above the studied dose range. Further investigations are warranted to verify these assumptions. In addition, our model estimated SC50, which represents ABT-639 concentration associated with a 50% stimulation of uric acid elimination, is around 8,070 ng/mL. It should be noted that this value is higher than the observed plasma concentration range. Therefore, it should be interpreted with caution. Further investigation is warranted to validate this parameter.
In the current study, the highest dose evaluated in the single- and multiple-dose studies was 170 and 160 mg, respectively. The safety margin of ABT-639 Cmax at these doses was 5.1-fold. Based on our simulations, ABT-639 Cmax safety margins are predicted to be 4.2-fold for the 180 mg BID dose and 3.8-fold for the 200 mg BID dose. Compared to the urate-lowering effect of ABT-639 at 160 BID dose, the simulation results showed a further 4.2% decrease in blood uric acid following 200 mg BID dose regimen. As ABT-639 demonstrated acceptable safety profile in humans and the maximum tolerated dose was not established in the current study, the urate-lowering effect of ABT-639 at doses higher than 160 mg BID may be considered for future investigations.
In summary, ABT-639 showed an acceptable safety profile across all dose regimens evaluated in the current study and demonstrated favorable pharmacokinetic profiles in human. ABT-639 showed statistically significant and clinically relevant urate-lowering effect in healthy volunteers—in vitro studies identified inhibition of the URAT1 transporter as a possible mechanism. A mechanism-based pharmacokinetic/pharmacodynamic model was constructed that adequately captured the time courses of observed ABT-639 plasma concentrations and blood uric acid concentrations in both single-dose and multiple-dose studies. Future investigations, including evaluation of the urate-lowering effect of ABT-639 in gout patients, evaluation of the combination of ABT-639 with compounds that have different mode of action (such as xanthine oxidase inhibitors), and evaluation of the urate-lowering effect of ABT-639 at doses higher than 160 mg BID, are warranted.
Acknowledgments
AbbVie contributed to the study design, research, and interpretation of data, writing, reviewing, and approving the publication. Wei Liu, Rachel Duan, Walid Awni, and Sandeep Dutta are AbbVie employees and may hold AbbVie stocks or options. Guohua An and Wolfram Nothaft are former AbbVie employees and have no further conflicts of interest to disclose.
References
- 1.Jarvis MF, Scott VE, McGaraughty S, Chu KL, Xu J, Niforatos W, et al. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochemical pharmacology. 2014;89(4):536–44. doi: 10.1016/j.bcp.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 2.Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417(6887):447–52. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 3.Anzai N, Kanai Y, Endou H. New insights into renal transport of urate. Curr Opin Rheumatol. 2007;19(2):151–7. doi: 10.1097/BOR.0b013e328032781a. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, Garcia-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57(9):545–9. doi: 10.1136/ard.57.9.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley WN. Current therapy of gout and hyperuricemia. Hosp Pract. 1976;11(5):69–76. doi: 10.1080/21548331.1976.11706934. [DOI] [PubMed] [Google Scholar]
- 6.Benedict JD, Forsham PH, Stetten D., Jr The metabolism of uric acid in the normal and gouty human studied with the aid of isotopic uric acid. J Biol Chem. 1949;181(1):183–93. [PubMed] [Google Scholar]
- 7.Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 63(10):3136–41. doi: 10.1002/art.30520. [DOI] [PubMed]
- 8.Terkeltaub R, Bushinsky DA, Becker MA. Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res Ther. 2006;8(Suppl 1):S4. doi: 10.1186/ar1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richette P, Bardin T. Gout. Lancet. 2010;375(9711):318–28. doi: 10.1016/S0140-6736(09)60883-7. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Doherty M, Bardin T, Pascual E, Barskova V, Conaghan P, et al. EULAR evidence based recommendations for gout. Part II: management. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics (ESCISIT) Ann Rheum Dis. 2006;65(10):1312–24. doi: 10.1136/ard.2006.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker MA, Schumacher HR, Jr, Wortmann RL, MacDonald PA, Eustace D, Palo WA. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450–61. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 12.Hair PI, McCormack PL, Keating GM. Febuxostat. Drugs. 2008;68(13):1865–74. doi: 10.2165/00003495-200868130-00006. [DOI] [PubMed] [Google Scholar]
- 13.Ogryzlo MA, Harrison J. Evaluation of uricosuric agents in chronic gout. Ann Rheum Dis. 1957;16(4):425–37. doi: 10.1136/ard.16.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76(1):47–56. doi: 10.1016/0002-9343(84)90743-5. [DOI] [PubMed] [Google Scholar]
- 15.Fam AG. Difficult gout and new approaches for control of hyperuricemia in the allopurinol-allergic patient. Curr Rheumatol Rep. 2001;3(1):29–35. doi: 10.1007/s11926-001-0048-8. [DOI] [PubMed] [Google Scholar]
- 16.Emmerson BT. The management of gout. N Engl J Med. 1996;334(7):445–51. doi: 10.1056/NEJM199602153340707. [DOI] [PubMed] [Google Scholar]
- 17.Anzai N, Enomoto A, Endou H. Renal urate handling: clinical relevance of recent advances. Curr Rheumatol Rep. 2005;7(3):227–34. doi: 10.1007/s11926-996-0044-0. [DOI] [PubMed] [Google Scholar]