

Abstract
The quantification of drug-metabolizing enzymes and transporters is important for in vitro-in vivo extrapolation (IVIVE) of xenobiotic clearance, which has become an integral part of drug development. There are different mass spectrometry-based techniques used for quantitative proteomics, and as more laboratories are opting for the use of these methods, selecting the most appropriate tool is becoming a concern. For the first time, we attempt to determine the significance of cost of different LC-MS methods of quantitative analysis of these proteins and to present a framework to objectively assess the choice of the techniques. Based on our analysis, quantification using labeled internal standards is more expensive per sample but provides higher quality data than label-free quantification. Quantification using absolute quantification synthetic peptides is the approach of choice for analyzing less than nine proteins, whereas when quantifying a defined set of proteins (10–50), such as enzymes, in a reasonably large number of samples (20–100), the quantification concatemer technique is more economical, followed by label-free quantification. When analyzing proteomes or sub-proteomes (≥500 proteins), label-free quantification is more cost-effective than the use of labeled internal standards. A cost-benefit approach is described to assess the choice of the most appropriate mass spectrometry-based approach for the quantification of proteins relevant to IVIVE.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9712-6) contains supplementary material, which is available to authorized users.
KEY WORDS: cost, drug-metabolizing enzymes, LC-MS, performance, transporters
INTRODUCTION
Mass spectrometry (MS) is a powerful technique for the qualitative analysis of peptides and proteins in complex mixtures. It is both extremely sensitive, detecting peptides down to the attomole range, and extremely selective, allowing peptides differing in molecular weight by less than 1 Da to be easily distinguished. Quantitative analysis by mass spectrometry is, however, much more challenging; the size of a signal in mass spectrometry depends on the concentration of an analyte and numerous other factors (its gas-phase basicity (1) and the ionizing conditions, for example).
In recent years, the importance of quantitative measurements in proteomics has driven the development of novel MS-based quantitative methods, and this development has been reflected in the rise in the number of publications on its application in different fields. A simple literature search, covering the last 5 years, using the words “quantitative proteomics” and “mass spectrometry” in different databases of articles shows a rapid increase in the number of publications (see Fig. 1). For example, a PubMed search showed a fourfold increase in publication “hits” using these terms in the last 5 years (2009–2013). As more laboratories are opting for the use of these methods to measure enzymes and transporters, they have to decide on the best option that matches the project objectives (e.g., the number of samples to analyze and the number of target proteins to measure).
Fig. 1.
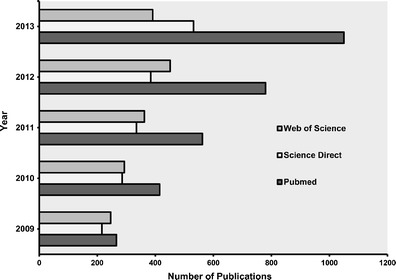
Results of the literature search for publication trends on mass spectrometry quantitative proteomics in the last 5 years in three databases
MS-based quantification methods used in proteomics are generally described as either “relative” or “absolute” (2). Relative quantification compares the quantity of the same protein in different samples (inter-sample relative quantification) and the results are defined in terms of fold change. An example of this type of quantification is comparing the fold change of protein expression in samples from patients (disease state) relative to that in reference samples (healthy state). Absolute quantification, by contrast, establishes the exact concentration of a protein in a mixture, and results are expressed in units such as copies per cell, or picomole per gram of tissue. More recently, another level of quantification has been recognized. It may be important to quantify proteins in a single sample relative to one another. This intra-sample relative quantification might conveniently be termed stoichiometric quantification (3) and requires the same methodology as absolute quantification, rather than inter-sample relative quantification. Stoichiometric quantification can also refer to quantification of different forms of the same protein in a sample, for example, phosphorylated and non-phosphorylated forms of the same protein. Protein stoichiometry determined from proteins absolute quantification results can give insights into the regulation of protein abundances within a sample (4, 5).
MS-based absolute quantification normally relies on analysis of proteotypic peptides using chemically or biologically synthesized stable isotope-labeled peptides as internal standards. Absolute quantification may be done by introducing a defined amount of chemically synthesized standard peptides to the sample prior to LC-MSMS analysis (6) and these peptides are sometimes known as absolute quantification (AQUA) peptides. An alternative approach is to synthesize the standards biologically, the quantification concatemer (QconCAT) approach. QconCATs are artificial proteins made of concatenated peptides, and each peptide is an internal standard (surrogate) that represents a specific protein. The construct is expressed using an artificial gene in Escherichia coli grown in heavy-isotope-enriched medium (7–9). Protein standards for absolute quantification (PSAQ) are isotopically labeled, recombinantly expressed analogues of analyte proteins used at a known concentration. AQUA and QconCAT techniques rely on the use of surrogate peptide standards for the quantification process whereas PSAQ are proteins that conserve the native context in which the quantified peptides exist; therefore, any differences between analyte and standard in proteolytic cleavage and procedural losses are minimized. This technique can therefore yield very high quality quantification (10, 11). All these methods are rather restricted in scope, and it is not surprising that researchers have sought to quantify complex samples using label-free methods. Essentially, there are two main approaches to label-free quantification of proteins. One is through the measurement of peak intensities of peptide ions (12–14) and the other is to count the number of peptides observed for a particular protein and to compare this with the theoretical number of observable peptides (15). Non-labeled protein standards at a known concentration are added to the assay and quantified simultaneously in order to obtain absolute measurements for analyte proteins (16). MS-based absolute quantification methods have been extensively reviewed in the literature (17–24). For an illustrative chart of these quantitative methods, please refer to Supplementary Fig. 1.
Proteomic techniques for quantifying drug-metabolizing enzymes and transporters are laborious, may require specialized skills and complex steps, are costly, and involve time-consuming data analysis. Complex biological samples have a higher dynamic range of protein abundance than most available analytical methods can cover. Consequently, robust and reproducible sample preparation is crucial to the accuracy and reproducibility of quantitative results. The selection of a suitable quantitative technique depends on the purpose of the experiment, especially whether relative or absolute quantification is needed. Other factors that should be considered include the biological origin of the sample (e.g., tissues, cell lines, primary cell cultures, body fluids, plants, bacteria, or viruses), the number of samples, the availability of instruments, and the associated cost and time.
On the basis of the information provided above, it is evident that cost analysis of these techniques is one of the fundamental pieces of information required prior to the implementation of mass spectrometry-based proteomics. In this report, we describe a framework developed for assessing and choosing different LC-MS quantitative methods based on their advantages, limitations, and cost implications.
MATERIALS AND METHODS
Comparison of Different Quantitative Techniques Used for Absolute Quantitative Proteomics
The literature contains no systematic and objective assessment of the strengths and weaknesses of different quantification techniques. Therefore, three independent researchers with technical and theoretical experience of different quantitative techniques assessed the advantages and the disadvantages of four different absolute quantification techniques: AQUA, QconCAT, PSAQ, and label-free quantification. In addition to cost, the following criteria were assessed: reproducibility, accuracy, precision, time required for the experiment, number of proteins that can be analyzed, and discrimination between isoforms and post-translational modifications. The performance of the techniques according to these criteria was scored by the researchers using a four-point Likert scale and the overall score is presented as an average of three individual scores.
Cost Analysis of Different Quantitative Techniques
This cost analysis is valid for laboratories equipped with instruments necessary for proteomics techniques including SDS-PAGE, ultra-centrifugation, and LC-MSMS. The cost values in this paper are based on the quantification of the abundance of cytochrome P450 (P450) and uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes and drug transporters in human tissue with QconCAT, AQUA, and label-free methods (7, 25–32). The cost of comprehensive method development and validation of the techniques was not included in the analysis as the time needed and validation methods used vary considerably between laboratories. Quantification of transporters requires additional sample preparation steps to extract the trans-membrane domains, and these steps should be included if the intention is to measure transporters. The cost of all the materials and consumables used was calculated initially in British pounds sterling then converted to US dollars. The costs of all consumable materials used per sample were incorporated in the analysis. The cost takes into account two possible options in sample preparation: in-gel digestion and in-solution digestion; however, the main values in this article are based on in-solution digestion. For a list of consumables used in this cost analysis, please refer to Supplementary Table 1. The proteolytic enzymes considered were a combination of trypsin and Achromobacter endopeptidase Lys-C. Instrument time cost is established on two platforms: nano-HPLC connected to electrospray ionization quadrupole time of flight (QTOF) MS and nano-HPLC connected to electrospray ionization triple quadrupole MS.
Cost analysis of absolute quantification using different proteomic methods was based on the smallest number of technical replicates that can provide reliable data; there is a lack of consensus in the literature on the number of required replicates. In this study, the costing of label-free analysis was carried out based on three technical replicates and that of quantification using labeled internal standards was based on two technical replicates as explained in the “RESULTS.”
Development of a Generic Cost-Benefit Framework
Incorporation of the results from the cost analysis and the performance comparison of different quantitative techniques by variation of the sample size and the number of the target proteins to be measured presented a generic framework for the selection of the most appropriate method on an informed basis. The framework considers the impact of the cost and performance in different scenarios in the scoring of each quantitative technique used for absolute quantification. The selected scenarios represent some of the common applications of interest of absolute quantification methods.
RESULTS
Comparison of the Performance of Different Quantitative Techniques in Absolute Quantitative Proteomics
The information in Table I compares four quantitative methods in terms of performance, time, and applications. The performance of the techniques is scored as an average of three scores and further displayed in a bar chart in Fig. 2. These are, of course, subjective scores. There was overall agreement between the three sets of scores and therefore only the average score is presented. AQUA peptides scored lowest in protein sequence coverage and the ability to discriminate isoforms. This is explained by the fact that the length of AQUA peptides is constrained to 15 amino acids, and it is usual to use only a single peptide to quantify each protein. This method scored the highest in quantifying post-translational modifications as post-translationally modified, e.g., phosphorylated, peptides can be synthesized and used for direct comparison. QconCAT displayed an overall satisfactory score especially in quantification of stoichiometry where it had the highest score. PSAQ analyzes data from every detectable proteolytic peptide and therefore had the highest scores for protein sequence coverage; because so many measurements based on different peptides are made, it is also best for accuracy of results and isoform discrimination. However, it scored lowest in post-translational modifications and stoichiometry applications. PSAQ proteins are expressed recombinantly and post-translational modifications, if any, would not necessarily reflect biology. Stoichiometric quantification requires quantification of several proteins necessitating the use of a matching number of PSAQ standards making it a laborious and expensive process.
Table I.
Assessment of Different Quantitative Techniques Used for Absolute Quantitative Proteomics
| AQUA | QconCAT | PSAQ | Label-free | |
|---|---|---|---|---|
| Availability of commercial isotope-labeled standards | Available | Available | Available | N/A |
| Isotopes used | 13C, 15N, D | 13C, 15N, D | 13C, 15N, D | N/A |
| Evaluation of digestion | Required | Required | Not requireda | N/A |
| Number of proteins | 1/standard | 50/standard | 1/standard | ≥1,000 |
| Number of replicates | 6 (3 biological, 2 technical) |
6 (3 biological, 2 technical) |
6 (3 biological, 2 technical) |
9 (3 biological, 3 technical)b |
| Sequence coverage | 1.3 ± 0.6 | 2.3 ± 0.6 | 4.0 ± 0.00 | 3.0 ± 0.0 |
| Reproducibility | 2.7 ± 0.6 | 3.0 ± 0.0 | 3.3 ± 0.6 | 2.3 ± 0.6 |
| Results accuracy | 2.7 ± 0.6 | 3.0 ± 0.0 | 4.0 ± 0.0 | 2.0 ± 0.0 |
| Results precision | 3.7 ± 0.6 | 3.7 ± 0.6 | 3.3 ± 0.6 | 2.3 ± 0.6 |
| Isoforms discrimination | 1.3 ± 0.6 | 1.7 ± 0.6 | 3.0 ± 1.0 | 1.3 ± 0.6 |
| Stoichiometry | 2.7 ± 0.6 | 3.0 ± 1.0 | 1.7 ± 1.2 | 2.0 ± 0.6 |
| Post-translational modifications | 3.3 ± 0.6 | 1.3 ± 0.6 | 1.0 ± 0.0 | 1.7 ± 0.6 |
| Duration of experiment | 2 days | 2 days | 2 days | 3 days |
| Instrument time | 1–2 h/sample | 1–2 h/sample | 1–2 h/sample | 1–6 h/sample |
| MS data size per sample | Large | Large | Large | Very large |
Performance of the techniques is scored based on a four-point Likert scale by three researchers as follows; poor = 1; fair = 2: good = 3; excellent = 4 to provide an average score ± standard deviation
aUnless variations in protein folding are present between standard and sample
bAdditional replicates required for label-free quantification to achieve the same level of precision
Fig. 2.
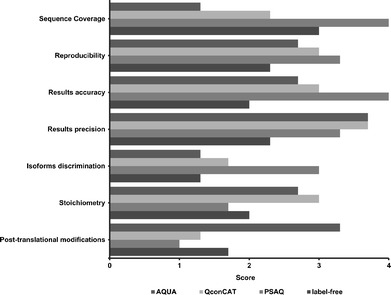
Differences in the performance of quantitative techniques: AQUA, QconCAT, PSAQ, and label-free in absolute quantitative proteomics applications. The bar chart is created based on the average scores shown in Table I
Concerns were expressed about the precision and accuracy of label-free methods as they do not use labeled internal standards for the process of quantification. They also displayed the lowest scores in reproducibility and ability to discriminate isoforms. All the techniques involving isotope standards require similar amounts of time to complete the quantitative experiments, whereas label-free techniques involve quicker sample preparation but a longer time for data acquisition and analysis.
Cost Analysis for Different Absolute Quantification Techniques
The cost for each step of sample analysis including sample preparation and LC-MS analysis was calculated and the results are presented in Table II. Expenses for tissue homogenization and protein digestion are optimized to fit all the previously mentioned quantitative techniques. PSAQ is excluded from the cost analysis as it is relatively new and applications to quantification of drug-metabolizing enzymes have not yet been reported. In sample preparation, in-solution digestion costs $3 less than in-gel digestion per sample. The cost of a QconCAT standard is based on the MetCAT (31), which is designed to quantify 25 drug-metabolizing enzymes with two peptides per protein. The capital cost of QconCAT is presented in Table II and is estimated at $4,400 of which the cost of QconCAT consumables is estimated to be around $450. The capital cost of the standard is the major contributor to the overall cost. Label-free methodology saves around 70 to 95% of the cost of quantifying one protein in one replicate compared to using AQUA or QconCAT standards. However, the cost of running nine replicates, three technical replicates for three biological replicates per sample, using label-free quantification is comparable to AQUA. The expense of analyzing one replicate with AQUA and QconCAT standards differs by approximately $3,800. However, the cost of analyzing one protein remains comparable to the cost of analyzing 50 proteins with QconCAT, which makes this technique more than fivefold cheaper than AQUA when analyzing 50 distinct proteins in the same sample, as presented in Table III and Fig. 3. It is conventional in techniques that use isotope-labeled standards to run six replicates (three biological, two technical) and nine replicates (three biological, three technical) for label-free methods to achieve an acceptable precision of quantitative data (CV of <20%). When nine replicates are run, the cost of label-free measurement is seven times higher compared with a single measurement. In the case of AQUA and QconCAT measurements, the corresponding increase is less than twofold.
Table II.
Cost of Consumable Materials, Sample Preparation, and LC-MS Analysis Based on Different Quantitative Techniques (AQUA, QconCAT, Label-Free)
| Process | Capital cost | Cost/replicate |
|---|---|---|
| Preparation of tissue sample | ||
| Homogenization reagents for 1 g of tissue | $198 | $1 |
| Microsomal protein concentration (Bradford assay) in triplicate | $218 | $1 |
| In-solution digestion reagents | $874 | $6 |
| In-gel digestion reagents | $1,363 | $9 |
| Total sample prep. cost with in-solution digestion | $1,290 | $8 |
| Total sample prep. cost with in-gel digestion | $1,779 | $11 |
| Cost of standards | ||
| AQUA per peptide | $524 | $0.02 |
| QconCAT (50 peptides) | $4,358 | $0.00 |
| Protein standard for label-free | $95 | $0.01 |
| Cost of instrument timea | ||
| QTOF or triple quadrupole connected to nano-HPLC | – | $128 |
| SRM method development for (1–10 peptides) | – | $128 |
| Personnel time cost for SRM method development | – | $47 |
| Label-free data analysisb | – | $95 |
| Total cost | ||
| Total sample cost of label-free quantification | – | $326 |
| Total sample cost of AQUA quantification | – | $834 |
| Total sample cost of QconCAT quantification | – | $4,669 |
aInstrument time cost values are applied as a 2-h run per sample
bThe value used to represent the cost of label-free data analysis does not take in account the cost of bioinformatics tools such as the software and the server
Table III.
The Effect of the Numbers of Replicates on the Cost of Absolute Quantification Proteomics Analysis of Human Tissue Samples
| Number and type of replicate | Label-free | AQUA | QconCAT |
|---|---|---|---|
| 3 biological replicates for the analysis of 1 protein | $788 | $1,106 | $4,941 |
| Technical replicates for each biological replicatea | $709 | $497 | $1,755 |
| Biological × technical replicates for the analysis of 1 protein | $2,126 | $1,490 | $5,325 |
| Biological × technical replicates for the analysis of 50 proteins | $2,126 | $27,841 | $6,025 |
| Biological × technical replicates for the analysis of 500 proteins | $2,126 | $271,282 | $31,908 |
aThe values are based on two technical replicates for AQUA and QconCAT, and three technical replicates for label-free methods
Fig. 3.
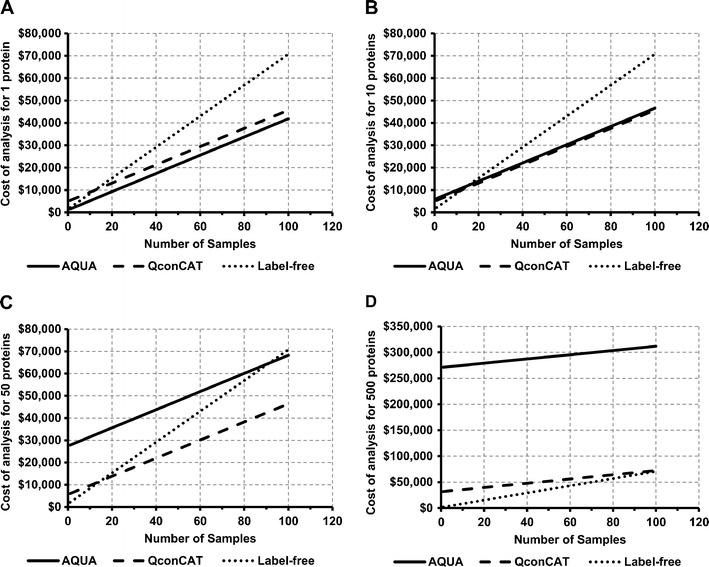
Simulated data based on the developed framework of cost analysis showing the effect of the number of samples and the number of proteins quantified using different absolute quantitative techniques evaluated in four different scenarios. To achieve the same level of precision, two technical replicates are used for isotope-labeled standards-based methods and three technical replicates are used for label-free methods. The first scenario (a) shows AQUA as the ideal method for quantification of one protein in a large number of samples (screening). AQUA and QconCAT present a comparable choice for routine quantification of about 10 proteins in different numbers of samples (b). In the third scenario (c) QconCAT is the cheapest choice for quantification of a defined set of proteins (such as enzymes and transporters) in different numbers of samples. Label-free and QconCAT are the most cost-effective methods for quantification of 500 proteins or more (whole or sub-proteome) in different numbers of samples (d)
In Fig. 3, the effect of the number of samples and the number of proteins quantified using different absolute quantitative techniques is evaluated in four different scenarios using the results of the cost analysis. The presented results are based on the use of six replicates (three biological, two technical) for isotope-labeled standards methods and nine replicates (three biological, three technical) for label-free methods. The first scenario (Fig. 3a) describes the cost of quantifying one protein in different numbers of samples. Accordingly, AQUA is the ideal choice for absolute quantification of one protein in a large number of samples. Routine analysis of ten proteins or less is represented in the second scenario (Fig. 3b), wherein AQUA and QconCAT are comparable in terms of cost for the quantification of ten proteins in different numbers of samples. The cost of quantifying more than ten different proteins using AQUA is more than $10,000, whereas the increase in the cost of quantifying 10 to 50 proteins using QconCAT is minimal representing the increase in instrument time. Quantification of a defined set of proteins (50 proteins) in a large number of samples shows the greatest cost saving with QconCAT (Fig. 3c). Figure 3d shows the fourth scenario where a whole proteome or a large sub-proteome (ca. 500 proteins) is quantified in different numbers of samples. Label-free quantification is the most cost-effective method for this scenario and QconCAT can be a good alternative to label-free methods as the number of samples reaches 100; however, the quantification of more than 150 distinct proteins using QconCAT can be very expensive costing more than $10,000.
The Choice of Different Quantitative Techniques Based on Cost and Performance
The previous results (Fig. 3) presented the most cost-effective quantitative method for absolute quantification. On the other hand, a framework assessing the best quantitative technique to select for absolute quantification ought to consider both cost and performance. The choice of quantitative technique for absolute quantification is mainly determined by the application of interest, the number of samples, and the number of proteins. These determine both the suitability and affordability of the method.
Figure 4 presents an evaluation of the best quantitative technique to select in three different scenarios based on the results of Tables I, II, and III and considering performance and cost of each technique. The scores on the y-axis (out of four) are an average score of performance and cost evaluation. In the first scenario, the target of absolute quantification is one protein in 20 samples and the use of AQUA or PSAQ is the best choice. Secondly, if the aim is to quantify 30 proteins in ten samples, then QconCAT is the ideal choice followed by AQUA and label-free techniques. PSAQ is not the best choice for quantifying multiple proteins in the same sample. Finally, in the case of quantifying more than 100 proteins in one sample, label-free would be the obvious choice followed by QconCAT.
Fig. 4.
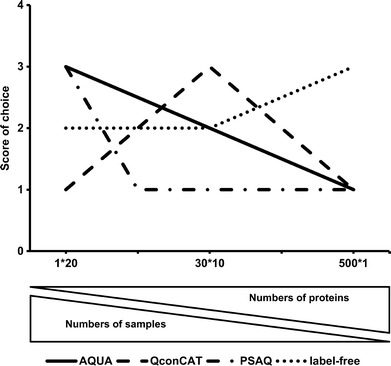
The effect of the number of samples and the number of proteins in each sample on the quantitative technique used for absolute quantification (x-axis: number of proteins * number of samples). The graph displays implications of the cost analysis information shown in Tables II and III and the performance analysis shown in Table I
In essence, based on the present cost-benefit analysis, AQUA is more desirable than QconCAT when fewer than nine proteins are to be quantified in less than 30,000 samples. However, if the number of proteins to be quantified exceeds nine proteins, QconCAT is the best choice regardless of the number of samples. QconCAT, however, becomes prohibitively expensive when the number of proteins per sample to be quantified exceeds 100, and label-free quantification becomes the best choice.
DISCUSSION
Abundances of drug-metabolizing enzymes, such as P450s and UGTs, play an important role as scaling factors in the process of in vitro-in vivo extrapolation of pharmacokinetic data. These abundance values are also required in physiologically based pharmacokinetic (PBPK) modeling to predict drug metabolic clearance. Abundance values of different enzymes in various tissues have become more available because of the use of LC-MS-based quantitative proteomics. The work in this paper presents a cost analysis of quantitative proteomic strategies for absolute quantification of P450s and UGTs in human liver tissue. These drug-metabolizing enzymes are an appropriate choice for comparative cost analysis as they represent a relatively small set of proteins relevant to drug pharmacokinetics. The framework developed for these enzymes was then extended to describe more comprehensive protein quantification scenarios and other tissues.
Prior to MS analysis, samples are subjected to cell lysis, protein extraction, solubilization, reduction and denaturation, alkylation, and digestion. The compatibility of the sample preparation method with the nature of the sample and the analytical technique is important for the reliability of the results. The total cost of the sample preparation step is less than 4% of total analysis cost for label-free and less than 1% for isotope-labeled standards methods. Even though the capital cost for sample preparation is moderately high, it is an essential cost to initiate the work and it decreases with the total number of samples to be analyzed. For example, tiny amounts of proteolytic enzymes, such as trypsin and endoproteinase Lys-C, are required in each digestion. The limited half-life of trypsin means that one purchase essentially digests however many samples are to be analyzed, and the cost of analysis per sample decreases with increasing numbers of samples. Trypsin is a common choice for digestion in proteomics because of its specific cleavage of peptide C-terminal arginine and lysine residues. The combination of trypsin with other proteases such as endoproteinase Lys-C is commonly used to enhance protein sequence coverage (33). Lys-C enzymes are tolerant to denaturants and only one labeled amino acid in isotope-labeled quantification experiments is required. Lys-C from Achromobacter is much cheaper than Lys-C from Lysobacter (34), making the use of sequential digestion of sample proteins with Lys-C then trypsin an attractive digestion strategy. In-gel protein digestion is more expensive and requires more time and sample handling steps when compared to in-solution digestion. However, in-gel digestion is more attractive when large amounts of detergents are used as it offers a cleaner analyte solution for LC-MS analysis.
The AQUA method is very popular in proteomics because isotope-labeled synthetic peptides are commercially available and the process can be performed in an easy, direct way. The AQUA strategy can be applied to assess post-translational modifications such as phosphorylation and ubiquitinylation (35, 36). The application of AQUA can be complex, time-consuming, and expensive for quantification of large number of proteins since standard peptides need to be synthesized and assessed independently. The use of AQUA for up to ten proteins is reasonable in terms of both cost and time of the experiments. The cost of running samples with AQUA to quantify several proteins in a set of samples is very high. It costs more than $10,000 to quantify 20 or more different proteins. By contrast, the amount used from an AQUA standard to quantify one peptide is extremely small; hence, one standard can be used to quantify one protein in thousands of samples. Despite cost implications, AQUA peptides have been applied extensively to generate quantitative data on several drug-metabolizing enzymes and transporters (29, 37, 38).
The main highlight of QconCAT is the ability to apply multiplexed protein quantification, where several proteins are quantified at the same time for the same overall cost. QconCAT can increase protein quantification scale, where up to 50 proteotypic peptides (size of artificial protein standard 50–150 kDa) of different target proteins can be included in a QconCAT construct; thus, the cost-effectiveness and the robustness of absolute quantification is improved. The cost of absolute quantification with QconCAT is almost the same for 1 to 50 proteins (one QconCAT can contain up to 50 standard peptides) with a slight increase in cost after ten proteins as the cost of instrument time for SRM method development starts to rise. Quantification of different proteins in the same sample becomes cheaper with QconCAT in comparison to AQUA for about ten or more distinct proteins in the same sample. Similarly to AQUA, the cost of the amount of QconCAT used, as a standard, to quantify a host of proteins in the same sample is insignificant. Hence, the produced QconCAT standard covers thousands of samples with the same-targeted proteins. The use of QconCAT to quantify more than 150 different proteins becomes very expensive costing more than $10,000, as at least three individual QconCAT standards would be needed. In addition, QconCAT is a remarkable method to obtain stoichiometric quantification of multiple proteins since QconCAT peptides are released in strictly equimolar stoichiometry (4, 39). The Census Of the Proteome of Yeast (COPY) project is worth mentioning as an example of the application of QconCAT to obtain whole proteome absolute quantification of a minimum of 4,000 proteins in the yeast Saccharomyces cerevisiae (40). In addition, QconCAT was successfully used to quantify drug-metabolizing enzymes in the human liver (32). The cost analysis results display QconCAT as a very cost-effective choice for the absolute quantification of several proteins.
The possibility of AQUA and QconCAT failure to express the stable isotope-labeled peptides selected in silico needs to be taken in consideration in the total cost. The cost of $523 for a new AQUA peptide would be the added for a failed experiment for the absolute quantification with AQUA peptide standard. In the case of QconCAT standards, failure to express would require a new QconCAT gene, which costs around $2,000. If the first attempt at using a QconCAT fails to express, there are reasonably well-documented protocols to employ (31).
The PSAQ method provides superior quantification accuracy compared to the previously mentioned methods. PSAQ avoids the effect of variations in protease digestion efficiency and loss of proteins during protein pre-fractionation on the accuracy of the results. Theoretically, all proteotypic peptides of the target protein can be observed with PSAQ providing the highest sequence coverage (41). In theory, PSAQ is the ideal choice for absolute quantification of proteins; however, PSAQ, a biological labeled standard, needs to be produced for every protein under study, with all the attendant cost and time implications. Heterologous expression of PSAQ standard proteins can lead to variations in the folding and post-translational modifications in comparison with the proteins of interest. Thus, the resultant peptides might display differences in stoichiometry when comparing the standards with sample peptides (42).
The exclusion of labeled standards from label-free quantification simplifies the technique and decreases sample preparation steps. The literature suggests using three technical replicates per biological sample is sufficient to obtain precision of less than 20% (CV) with label-free quantification methods (43–45). This level of precision is achieved with two technical replicates when isotope-labeled internal standards, such as QconCAT (32), are used. The results present label-free analysis as the cheapest choice for absolute quantification of a large number of proteins as the cost remains constant regardless of the number of proteins in one sample. Therefore, the possibility of high throughput analysis makes label-free quantification a very appealing quantification approach. Quantification of a small number of proteins with label-free methods is not the cheapest option when analyzing multiple (>20) samples because the cost of label-free quantification increases with the numbers of samples due to the effect of the large number of replicates and data analysis cost. In addition, quantitative measurements provided by label-free strategies are generally considered less reliable compared to labeled standard-based methods. Reliable quantification with label-free techniques requires validation and assurance of results reproducibility along the whole experimental workflow. The use of label-free approaches to quantify low-abundance proteins and to assess stoichiometry of proteins within the same sample (intra-sample relative quantification) is challenging. However, in occasions where there are significant similarities between the quantified proteins in terms of size and sequence homology (e.g., cytochrome P450 enzymes), intra-sample protein quantification is especially feasible (28).
The choice of the instrument used for the analysis is crucial in label-free quantification. Control over the performance of the mass spectrometer, the ionization efficiency, and the quality of chromatography is important to ensure reproducibility of the results. The cost-effective MSMS platform for routine label-free quantification using MSE experiments (label-free quantification based on intensity of peptide ion signals) (12) would be a quadrupole-time of flight mass spectrometer. Triple quadrupole and Orbitrap-based instruments can equally be used for selected reaction monitoring (SRM)-based assays involved in labeled standard (AQUA/QconCAT)-based quantification. However, quadrupole-based instruments tend to be more cost effective than Orbitrap mass analyzers. Mass spectrometry is a fast evolving technology and the development of quadrupole-Orbitrap instruments operating in PRM mode (parallel reaction monitoring) shows improved selectivity of measurements in comparison to SRM using triple quadrupole mass analyzers (46, 47). However, newly developed instruments are less cost effective.
CONCLUSION
The proposed cost-benefit analysis framework is applied to the absolute quantification of drug-metabolizing enzymes in human liver tissue, but it is also valid for other routine applications. As the cost of sample preparation is negligible, other factors such as the cost of LC-MS analysis, the number of samples, the number proteins quantified, and the capital cost of standards need to be considered in the quantification process. This work provides objective guidelines for the assessment of different quantitative proteomics techniques in terms of cost, performance, and applications.
Electronic supplementary material
(DOCX 4010 kb)
{kind=link}
(GIF 327 kb)
Acknowledgments
The authors thank Manchester Pharmacy School, the University of Manchester, for financial support, Zubida Al-Majdoub for participating in the assessment of different quantitative techniques used for absolute quantification, David Knight and Stacy Warwood from the Faculty of Life Sciences at the University of Manchester for advice on instrument time cost, and Eleanor Savill for assisting in the preparation of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
- P450
Cytochrome P450
- UGT
Uridine 5′-diphospho-glucuronosyltransferase
- QconCAT
Quantification concatemer
- AQUA
Absolute quantification
- PSAQ
Protein standards for absolute quantification
- SRM
Selected reaction monitoring
- LC-MS
Liquid chromatography in conjunction with mass spectrometry
- MSMS
Tandem mass spectrometry
References
- 1.Couto N, Barber J, Gaskell SJ. Matrix-assisted laser desorption/ionisation mass spectrometric response factors of peptides generated using different proteolytic enzymes. J Mass Spectrom. 2011;46(12):1233–40. doi: 10.1002/jms.2009. [DOI] [PubMed] [Google Scholar]
- 2.Ong S-E, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1(5):252–62. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 3.Al-Majdoub ZM, Carroll KM, Gaskell SJ, Barber J. Quantification of the proteins of the bacterial ribosome using QconCAT technology. J Proteome Res Am Chem Soc. 2014;13(3):1211–22. doi: 10.1021/pr400667h. [DOI] [PubMed] [Google Scholar]
- 4.Kito K, Ota K, Fujita T, Ito T. A synthetic protein approach toward accurate mass spectrometric quantification of component stoichiometry of multiprotein complexes. J Proteome Res. 2007;6(2):792–800. doi: 10.1021/pr060447s. [DOI] [PubMed] [Google Scholar]
- 5.Ding C, Li Y, Kim B-J, Malovannaya A, Jung SY, Wang Y, et al. Quantitative analysis of cohesin complex stoichiometry and SMC3 modification-dependent protein interactions. J Proteome Res Am Chem Soc. 2011;10(8):3652–9. doi: 10.1021/pr2002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100(12):6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat Protoc Nat Publ Group. 2006;1(2):1029–43. doi: 10.1038/nprot.2006.129. [DOI] [PubMed] [Google Scholar]
- 8.Carroll KM, Simpson DM, Eyers CE, Knight CG, Brownridge P, Dunn WB, et al. Absolute quantification of the glycolytic pathway in yeast: deployment of a complete QconCAT approach. Mol Cell Proteomics. 2011;10(12):M111.007633. doi: 10.1074/mcp.M111.007633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat Methods. 2005;2(8):587–9. doi: 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- 10.Brun V, Dupuis A, Adrait A, Marcellin M, Thomas D, Court M, et al. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol Cell Proteomics. 2007;6(12):2139–49. doi: 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Dupuis A, Hennekinne J-A, Garin J, Brun V. Protein standard absolute quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics. 2008;8(22):4633–6. doi: 10.1002/pmic.200800326. [DOI] [PubMed] [Google Scholar]
- 12.Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5(1):144–56. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Silva JC, Denny R, Dorschel C, Gorenstein MV, Li G-Z, Richardson K, et al. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol Cell Proteomics. 2006;5(4):589–607. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 15.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4(9):1265–72. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Mayr BM, Kohlbacher O, Reinert K, Sturm M, Gröpl C, Lange E, et al. Absolute myoglobin quantitation in serum by combining two-dimensional liquid chromatography-electrospray ionization mass spectrometry and novel data analysis algorithms. J Proteome Res Am Chem Soc. 2006;5(2):414–21. doi: 10.1021/pr050344u. [DOI] [PubMed] [Google Scholar]
- 17.Kito K, Ito T. Mass spectrometry-based approaches toward absolute quantitative proteomics. Curr Genomics. 2008;9(4):263–74. doi: 10.2174/138920208784533647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langenfeld E, Meyer HE, Marcus K. Quantitative analysis of highly homologous proteins: the challenge of assaying the “CYP-ome” by mass spectrometry. Anal Bioanal Chem. 2008;392(6):1123–34. doi: 10.1007/s00216-008-2407-z. [DOI] [PubMed] [Google Scholar]
- 19.Brun V, Masselon C, Garin J, Dupuis A. Isotope dilution strategies for absolute quantitative proteomics. J Proteomics. 2009;72(5):740–9. doi: 10.1016/j.jprot.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Rodríguez-Suárez E, Whetton AD. The application of quantification techniques in proteomics for biomedical research. Mass Spectrom Rev. 2012;32(1):1–26. doi: 10.1002/mas.21347. [DOI] [PubMed] [Google Scholar]
- 21.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404(4):939–65. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 22.Ahrné E, Molzahn L, Glatter T, Schmidt A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics. 2013;13(17):2567–78. doi: 10.1002/pmic.201300135. [DOI] [PubMed] [Google Scholar]
- 23.Villanueva J, Carrascal M, Abian J. Isotope dilution mass spectrometry for absolute quantification in proteomics: concepts and strategies. J Proteomics. 2014;96:184–99. doi: 10.1016/j.jprot.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Prasad B, Unadkat JD. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J. 2014;16(4):634–48. doi: 10.1208/s12248-014-9602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang MZ, Wu JQ, Dennison JB, Bridges AS, Hall SD, Kornbluth S, et al. A gel-free MS-based quantitative proteomic approach accurately measures cytochrome P450 protein concentrations in human liver microsomes. Proteomics. 2008;8(20):4186–96. doi: 10.1002/pmic.200800144. [DOI] [PubMed] [Google Scholar]
- 26.Langenfeld E, Zanger UM, Jung K, Meyer HE, Marcus K. Mass spectrometry-based absolute quantification of microsomal cytochrome P450 2D6 in human liver. Proteomics. 2009;9(9):2313–23. doi: 10.1002/pmic.200800680. [DOI] [PubMed] [Google Scholar]
- 27.Seibert C, Davidson BR, Fuller BJ, Patterson LH, Griffiths WJ, Wang Y. Multiple-approaches to the identification and quantification of cytochromes P450 in human liver tissue by mass spectrometry. J Proteome Res. 2009;8(4):1672–81. doi: 10.1021/pr800795r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Achour B, Barber J, Rostami-Hodjegan A. Cytochrome P450 Pig liver pie: determination of individual cytochrome P450 isoform contents in microsomes from two pig livers using liquid chromatography in conjunction with mass spectrometry [corrected] Drug Metab Dispos. 2011;39(11):2130–4. doi: 10.1124/dmd.111.040618. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami H, Ohtsuki S, Kamiie J, Suzuki T, Abe T, Terasaki T. Simultaneous absolute quantification of 11 cytochrome P450 isoforms in human liver microsomes by liquid chromatography tandem mass spectrometry with in silico target peptide selection. J Pharm Sci. 2011;100(1):341–52. doi: 10.1002/jps.22255. [DOI] [PubMed] [Google Scholar]
- 30.Ohtsuki S, Schaefer O, Kawakami H, Inoue T, Liehner S, Saito A, et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab Dispos. 2012;40(1):83–92. doi: 10.1124/dmd.111.042259. [DOI] [PubMed] [Google Scholar]
- 31.Russell MR, Achour B, Mckenzie EA, Lopez R, Harwood MD, Rostami-Hodjegan A, et al. Alternative fusion protein strategies to express recalcitrant QconCAT proteins for quantitative proteomics of human drug metabolizing enzymes and transporters. J Proteome Res. 2013;12(12)):5934–42. doi: 10.1021/pr400279u. [DOI] [PubMed] [Google Scholar]
- 32.Achour B, Russell MR, Barber J, Rostami-Hodjegan A. Simultaneous Quantification of the Abundance of Several Cytochrome P450 and Uridine 5’-Diphospho-Glucuronosyltransferase Enzymes in Human Liver Microsomes Using Multiplexed Targeted Proteomics. Drug Metab Dispos 2014;42(4):500–10. [DOI] [PubMed]
- 33.Choudhary G, Wu S-L, Shieh P, Hancock WS. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J Proteome Res. 2003;2(1):59–67. doi: 10.1021/pr025557n. [DOI] [PubMed] [Google Scholar]
- 34.Achour B, Barber J. The activities of Achromobacter lysyl endopeptidase and Lysobacter lysyl endoproteinase as digestive enzymes for quantitative proteomics. Rapid Commun Mass Spectrom. 2013;27(14):1669–72. doi: 10.1002/rcm.6612. [DOI] [PubMed] [Google Scholar]
- 35.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35(3):265–73. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 36.Kettenbach AN, Rush J, Gerber SA. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat Protoc Nat Publ Group. 2011;6(2):175–86. doi: 10.1038/nprot.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fallon JK, Harbourt DE, Maleki SH, Kessler FK, Ritter JK, Smith PC. Absolute quantification of human uridine-diphosphate glucuronosyl transferase (UGT) enzyme isoforms 1A1 and 1A6 by tandem LC-MS. Drug Metab Lett. 2008;2(3):210–22. doi: 10.2174/187231208785425764. [DOI] [PubMed] [Google Scholar]
- 38.Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–45. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 39.Johnson H, Eyers CE, Eyers PA, Beynon RJ, Gaskell SJ. Rigorous determination of the stoichiometry of protein phosphorylation using mass spectrometry. J Am Soc Mass Spectrom. 2009;20(12):2211–20. doi: 10.1016/j.jasms.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Brownridge P, Holman SW, Gaskell SJ, Grant CM, Harman VM, Hubbard SJ, et al. Global absolute quantification of a proteome: challenges in the deployment of a QconCAT strategy. Proteomics. 2011;11(15):2957–70. doi: 10.1002/pmic.201100039. [DOI] [PubMed] [Google Scholar]
- 41.Janecki DJ, Bemis KG, Tegeler TJ, Sanghani PC, Zhai L, Hurley TD, et al. A multiple reaction monitoring method for absolute quantification of the human liver alcohol dehydrogenase ADH1C1 isoenzyme. Anal Biochem. 2007;369(1):18–26. doi: 10.1016/j.ab.2007.06.043. [DOI] [PubMed] [Google Scholar]
- 42.Simpson DM, Beynon RJ. QconCATs: design and expression of concatenated protein standards for multiplexed protein quantification. Anal Bioanal Chem. 2012;404(4):977–89. doi: 10.1007/s00216-012-6230-1. [DOI] [PubMed] [Google Scholar]
- 43.Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li G-Z, et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem Am Chem Soc. 2005;77(7):2187–200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 44.Vissers JPC, Langridge JI, Aerts JMFG. Analysis and quantification of diagnostic serum markers and protein signatures for Gaucher disease. Mol Cell Proteomics. 2007;6(5):755–66. doi: 10.1074/mcp.M600303-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Patel VJ, Thalassinos K, Slade SE, Connolly JB, Crombie A, Murrell JC, et al. A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J Proteome Res. 2009;8(7):3752–9. doi: 10.1021/pr900080y. [DOI] [PubMed] [Google Scholar]
- 46.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11(11):1475–88. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallien S, Duriez E, Demeure K, Domon B. Selectivity of LC-MS/MS analysis: implication for proteomics experiments. J Proteomics. 2013;81:148–58. doi: 10.1016/j.jprot.2012.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 4010 kb)
(GIF 327 kb)